
ABSTRACT
Mitochondrial damage triggers mitochondrial quality control pathways, which act to ensure the health of the mitochondrial network. The turnover of damaged mitochondria by mitophagy is initiated by the Parkinson disease-linked genes PRKN and PINK1, and we recently investigated the role that interorganellar contact sites between the endoplasmic reticulum (ER) and the outer mitochondrial membrane (OMM) play in this pathway. In this punctum, we summarize our findings that show that the ER-OMM tether MFN2 acts as a suppressor of mitophagy through its ability to link the OMM to the ER, potentially limiting the accessibility of other ubiquitination substrates to PINK1 and PRKN. PINK1, PRKN and the AAA-ATPase VCP disrupt contact between mitochondria and the ER via MFN2 ubiquitination, retrotranslocation and turnover from the mitochondrial membrane. Our study provides insight into the role of OMM remodeling in mitophagy.
Mitochondrial quality control pathways curb mitochondrial dysfunction by targeting and eliminating damaged organelles and components of the mitochondrial population, and have the potential to become therapeutic avenues to rout aging and neurodegeneration. In the case of macroautophagy of damaged mitochondria (a process termed mitophagy), many of the genetic actors – PINK1, PRKN, VCP, TBK1, SQSTM1 and OPTN – are accordingly mutated in hereditary forms of neurodegenerative disease. During mitophagy, the ubiquitin (Ub) kinase PINK1 accumulates on the OMM, where it recruits and activates PRKN, a cytosolic Ub ligase, at mitochondria by a mechanism involving the respective phosphorylation of Ub (already conjugated to OMM proteins) on serine 65 (S65) and the PRKN Ub-like domain on the analogous residue. Phosphorylated S65 Ub (p-Ub) directly binds PRKN and thus Ub phosphorylation creates mitochondrial binding sites for the ligase, leading to a feed-forward cycle of PRKN-dependent ubiquitination.
We recently investigated the involvement of membrane contact sites between the endoplasmic reticulum (ER) and mitochondria in mitophagy [Citation1]. As these contacts serve as a platform for a myriad of signaling, metabolic and catabolic functions, we reasoned that crosstalk between these interorganellar junctions and the mitophagic cascade likely existed. Indeed, several PRKN ubiquitination substrates, such as MFN2, RHOT1 and the VDACs, had already been shown to localize to contacts between the ER and mitochondria. We accordingly uncovered a mechanism by which the ubiquitination and degradation of the mitochondria-ER tether MFN2 acts as a molecular gate in the mitophagy pathway, demonstrating that remodeling of outer mitochondrial membrane (OMM) connections is an important aspect of mitochondrial turnover via autophagy.
We first addressed the dynamics of ER-mitochondria contacts during mitophagy by analyzing the ultrastructure of these contacts, where we triggered mitophagy via buildup of PINK1 on the OMM by treating cells with the uncoupling agent CCCP. We observed that CCCP treatment severely reduces the amount of ER that is in contact with the OMM in a PRKN- and PINK1-dependent manner, in both U2OS cells overexpressing PRKN and human dopaminergic neurons derived from induced pluripotent stem cells. Thus, PINK1 and PRKN act to uncouple the ER from mitochondria during mitophagy; notably, this is in contrast to starvation-induced autophagy, wherein ER-mitochondria contacts act as the site of autophagosomal biogenesis. This discrepancy may stem from the fact that PINK1-PRKN mitophagy and starvation-induced autophagy have distinct triggers, i.e. mitochondrial damage and a dearth of nutrients, respectively.
We then addressed the mechanism by which PINK1 and PRKN dissociate the ER from mitochondria during mitophagy. We focused on a potential role for MFN2 in this process, which had been identified as a mitochondria-ER tethering protein. We found that cells lacking MFN2, but not the orthologous MFN1 (which does not function in tethering), have an increased rate of mitophagy, as measured by a variety of methods. Importantly, the effect of MFN2 depletion on mitophagy can be replicated by depleting other factors involved in mediating ER-OMM connections, demonstrating that MFN2 inhibits mitophagy through its ability to tether mitochondria to the ER.
During mitophagy, ubiquitination of the MFNs is coupled to their proteasomal degradation. We found that ubiquitination of MFN2 by PRKN occurs rapidly at the onset of mitophagy compared to other PRKN substrates on the OMM, an effect that we are able to recapitulate in a cell-free mitochondrial ubiquitination (‘in organello’) assay involving purified mitochondria, recombinant PRKN and Ub assay components. Indeed, we found that ubiquitinated MFN2 is phosphorylated on its Ub moieties by PINK1 during mitophagy, allowing PRKN to directly interact with p-Ub-MFN2 and catalyze its further ubiquitination. Whereas MFN2 is ubiquitinated by PRKN on several lysine residues, we identified 3 lysines in the first heptad repeat domain (HR1) of MFN2 – K406, K416 and K420 – as being important for PRKN-catalyzed ubiquitination, as mutation of all 3 lysines to arginine (a construct we refer to as MFN2HR1) severely reduces MFN2 ubiquitination by PRKN and represses the clearance of depolarized mitochondria.
We found that ubiquitination of these residues is important for MFN2 to be degraded by the proteasome in a manner dependent on its retrotranslocation from the OMM by the AAA-ATPase VCP/p97. MFN2 ubiquitination on its own is not sufficient for its degradation; we can block the turnover of MFN2 oligomers by inhibiting VCP, and also directly assay MFN2 retrotranslocation from the OMM in organello using recombinant VCP. ER-OMM uncoupling by PINK1-PRKN is accordingly blocked by treating cells with inhibitors of either the proteasome or VCP. Further investigation of the MFN2-VCP relationship led us to find that the presence of MFN2 sensitizes mitophagy to VCP inhibition, describing an epistatic relationship between the facilitating role of VCP on mitophagy and MFN2-dependent ER-OMM tethering.
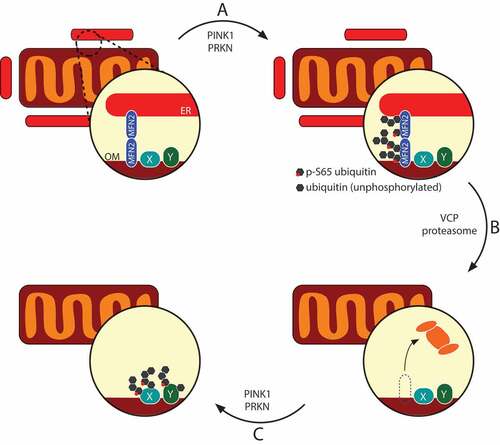
How might this epistasis manifest itself biochemically? We found that, while MFN2 is turned over quite rapidly by the proteasome, the ubiquitination of VDAC1, another contact site protein, occurs over a longer period of time. Moreover, VCP inhibition stabilizes MFN2-Ub conjugates, whereas it suppresses VDAC1 ubiquitination. Notably, VDAC1 ubiquitination becomes insensitive to VCP inhibition when ER-OMM tethering is reduced, and we can stimulate VDAC1 ubiquitination in organello by the addition of recombinant VCP or purified cytosol. This led us to postulate that the ER, via MFN2 tethers at ER-OMM junctions, sterically shields certain substrates from PRKN. Access of PRKN to these substrates necessitates the destruction of contact sites during mitophagy, allowing PRKN and/or PINK1 access to these ‘shielded’ substrates – such as VDAC1, and likely others – and enabling the feed-forward mechanism of PRKN activation to proceed and initiate destruction of the organelle (see ). In this manner, the destruction of MFN2 tethers may gate the availability of stable p-Ub conjugates of OMM proteins such as VDAC1, thus regulating the number of PRKN binding sites on mitochondria. This highlights a link between ER-OMM contacts and mitophagy that warrants further investigation. Indeed, PINK1 may localize to these junctions, implicating these sites as important regulators of mitophagy induction. Our study provides a tractable, molecular framework with which to study how the OMM is remodeled at the onset of mitophagy.
Figure 1. The mitophagy machinery uncouples mitochondria from the ER via MFN2 destruction. MFN2 oligomers bridge the ER and the outer membrane (OM), and are one of the first targets of PINK1-PRKN-mediated (phospho-)ubiquitination (A). Ubiquitinated MFN2 complexes are recognized and retrotranslocated by VCP and fed to the proteasome (B), allowing the PINK1-PRKN system access to more ubiquitination substrates (proteins X and Y) (C). This ‘second-tier’ of PRKN substrates may be resistant to retrotranslocation, thus stably tethering PRKN to mitochondria during mitophagy
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Related Research Data
References
- McLelland GL, Goiran T, Yi W, et al. Mfn2 ubiquitination by PINK1/parkin gates the p97-dependent release of ER from mitochondria to drive mitophagy. eLife. 2018;e32866. PMID:29676259.