
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
Cells challenged by photosensitized oxidations face strong redox stresses and rely on autophagy to either survive or die. However, the use of macroautophagy/autophagy to improve the efficiency of photosensitizers, in terms of inducing cell death, remains unexplored. Here, we addressed the concept that a parallel damage in the membranes of mitochondria and lysosomes leads to a scenario of autophagy malfunction that can greatly improve the efficiency of the photosensitizer to cause cell death. Specific damage to these organelles was induced by irradiation of cells pretreated with 2 phenothiazinium salts, methylene blue (MB) and 1,9-dimethyl methylene blue (DMMB). At a low concentration level (10 nM), only DMMB could induce mitochondrial damage, leading to mitophagy activation, which did not progress to completion because of the parallel damage in lysosome, triggering cell death. MB-induced photodamage was perceived almost instantaneously after irradiation, in response to a massive and nonspecific oxidative stress at a higher concentration range (2 µM). We showed that the parallel damage in mitochondria and lysosomes activates and inhibits mitophagy, leading to a late and more efficient cell death, offering significant advantage (2 orders of magnitude) over photosensitizers that cause unspecific oxidative stress. We are confident that this concept can be used to develop better light-activated drugs.
Abbreviations: ΔΨm: mitochondrial transmembrane inner potential; AAU: autophagy arbitrary units; ATG5, autophagy related 5; ATG7: autophagy related 7; BAF: bafilomycin A1; BSA: bovine serum albumin; CASP3: caspase 3; CF: carboxyfluorescein; CTSB: cathepsin B; CVS: crystal violet staining; DCF: dichlorofluorescein; DCFH2: 2ʹ,7ʹ-dichlorodihydrofluorescein; DMMB: 1,9-dimethyl methylene blue; ER: endoplasmic reticulum; HaCaT: non-malignant immortal keratinocyte cell line from adult human skin; HP: hydrogen peroxide; LC3B-II: microtubule associated protein 1 light chain 3 beta-II; LMP: lysosomal membrane permeabilization; LTG: LysoTracker™ Green DND-26; LTR: LysoTracker™ Red DND-99; 3-MA: 3-methyladenine; MB: methylene blue; mtDNA: mitochondrial DNA; MitoSOX™: red mitochondrial superoxide probe; MTDR: MitoTracker™ Deep Red FM; MTO: MitoTracker™ Orange CMTMRos; MT-ND1: mitochondrially encoded NADH:ubiquinone oxidoreductase core subunit 1; MTT: methylthiazolyldiphenyl-tetrazolium bromide; 1O2: singlet oxygen; OH. hydroxil radical; PRKN/parkin: parkin RBR E3 ubiquitin protein ligase; PBS: phosphate-buffered saline; PI: propidium iodide; PDT: photodynamic therapy; PS: photosensitizer; QPCR: gene-specific quantitative PCR-based; Rh123: rhodamine 123; ROS: reactive oxygen species RTN: rotenone; SQSTM1/p62: sequestosome 1; SUVs: small unilamellar vesicles; TBS: Tris-buffered saline
Introduction
Autophagy is a crucial metabolic pathway that can sustain cellular homeostasis in health and play either a protective or a destructive role in diseases [Citation1]. The original machinery of autophagy promotes degradation and recycling of proteins, lipids, and even entire organelles such as the endoplasmic reticulum, mitochondria, and Golgi apparatus [Citation2]. The role of autophagy goes beyond the maintenance of the metabolic homeostasis, since mitophagy, for example, is essential to keep a stable genome [Citation3–Citation7]. Recent progress in the understanding of the molecular basis of autophagy has uncovered promising strategies to treat difficult diseases, such as cancer and neurologic pathologies [Citation1,Citation3,Citation8].
Photodynamic therapy (PDT), which is a clinical modality used to treat a variety of diseases, can provide a tool to modulate autophagy [Citation9–Citation11]. PDT is based on the light-induced generation of cytotoxic oxidant species, such as singlet oxygen (1O2) and hydroxyl radical (OH∙), by light activation of a photosensitizer (PS) [Citation12,Citation13]. Increasing PS efficiency requires allowing photodynamic damage in deeper and/or hidden sites, which usually holds PDT-escaping cells, responsible for the recurrence of diseases [Citation13–Citation16].
Autophagy was shown to play either a prosurvival or a prodeath role in response to PDT [Citation10,Citation17–Citation21]. Although at low PDT doses autophagy usually offers protection, this metabolic pathway can serve as an alternate death mode when the PDT dose increases [Citation20]. Especially in apoptosis-deficient cells, PDT-induced cell death relies on autophagy, showing robust time-related accumulation of MAP1LC3-II regardless of ATG7 (autophagy related 7) expression [Citation22]. However, it is still unclear how autophagy can be useful to increase PS efficiency.
One of the advantages of photosensitized oxidations is their potential to specifically target tissues or even intracellular organelles [Citation10,Citation13,Citation23–Citation25]. This is because reactive species cause damage within the nanometer vicinity of the PS [Citation26,Citation27]. Indeed, the efficiency of photoinduced cell death depends not only on the amount of oxidant species, but more importantly on the specific location of their generation [Citation13,Citation25]. However, this target-specific knowledge has rarely been used to develop improved PDT PSs [Citation13,Citation28]. To mention 2 of the few exceptions, Kessel et al. report that low level lysosomal photodamage markedly enhances subsequent photokilling by mitochondria-targeted PDT and attribute this phenomenon to an enhancement of the proapoptotic signal(s) resulting from mitochondrial photodamage [Citation29]. A recent report compared the consequences of damaging either lysosomes or mitochondria and concludes that damaging lysosomes is ~5 times more prone to induce autophagy-associated cell death compared with causing a similar damage in mitochondria [Citation30].
Mitophagy (mitochondrial autophagy), which removes oxidized, depolarized, worn out and superfluous mitochondria, has several distinct variants (i.e. type 1, 2 and 3 as reviewed by Lemasters [Citation31]) and prevents release of proapoptotic proteins, generation of toxic reactive oxygen species (ROS) and futile hydrolysis of ATP [Citation3,Citation6,Citation32]. Mitophagy also eliminates mitochondria during cytoplasmic remodeling and degrades mitochondrial DNA (mtDNA), including damaged and mutated mtDNA promoting mitochondrial dysfunction and disease [Citation33,Citation34]. In type 1 mitophagy occurring during nutrient deprivation, preautophagic structures grow into cup-shaped phagophores that surround and sequester individual mitochondria into mitophagosomes, a process requiring the class III phosphatidylinositol-3-kinase (PtdIns3K) [Citation31]. Specifically after mitochondrial depolarization, type 2 mitophagy is stimulated [Citation31]. By triggering photodamage to single mitochondria, depolarization may occur with recruitment of autophagic LC3-containing structures on mitochondrial surfaces followed by vesicular acidification. Under mitochondrial depolarization there is a PINK1-dependent recruitment of PRKN/parkin to the mitochondrial outer membrane [Citation31,Citation35,Citation36]. PRKN is an E3 ubiquitin ligase that ubiquitinates outer membrane proteins to target mitochondria for mitophagy [Citation36]. In the type 3 mitophagy, or micromitophagy, mitochondria-derived vesicles (MDV) containing oxidized mitochondrial proteins bud off from mitochondria and then become internalized into multivesicular bodies [Citation37]. Multivesicular bodies subsequently fuse with lysosomes to complete hydrolytic degradation of the mitochondrial fragments [Citation31,Citation38]. Although MDV formation and transit to lysosomes occurs independently of the autophagic proteins ATG5 and LC3, this form of self-eating of mitochondria may require PINK1 and PRKN proteins, perhaps because MDV are depolarized [Citation39]. Therefore, PDT likely may activate all 3 types of mitophagy: Type 3 mitophagy with mild oxidative stress and types 1 and 2 mitophagy with more severe oxidative stress that cause mitochondrial depolarization and ATP depletion [Citation31].
Here, we compared the exclusive photodamage in both mitochondria and lysosomes, with the condition of a less specific damage, in which a widespread redox misbalance ends up killing the cells. Autophagy is one of the main causes of the cell resistance to the oxidative effects of PDT [Citation10,Citation17,Citation40,Citation41]. We aimed to test the hypothesis that the parallel damage in the membranes of these organelles could activate and inhibit autophagy concomitantly, leading to a very efficient way to cause cell death in mammalian cells. We used 2 positively-charged phenothiazinium salts, methylene blue (MB) and 1,9-dimethyl methylene blue (DMMB), as tools to induce specific photoinduced damage to these organelles () [Citation42–Citation46]. By investigating the molecular responses of cells challenged by photosensitization with DMMB (using photosensitization with MB as control), we uncovered a robust paradigm relating organelle-targeted damage, autophagy modulation and the efficiency of photoinduced cell death.
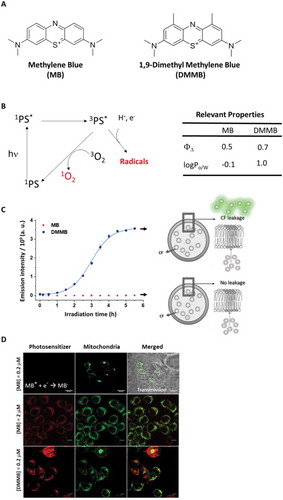
Figure 1. The capability of interacting and damage membranes. (a) Chemical structure of MB and DMMB cations. (b) Simplified Jablonski diagram of photosensitization. At right, table with relevant properties: 1O2 and log Po/w. (c) Membrane permeabilization assay using DOPC:CL liposomes (15% CL) with entrapped CF. Samples contained 15 μM MB or DMMB and were irradiated with a 631 nm LED, with 72 W m−2 irradiance. The average emission intensity at 517 nm (excitation at 480 nm, for CF fluorescence), accompanied by standard deviation, is plotted as a function of irradiation time. (d) Colocalization of DMMB and MB with mitochondria stained by Rh123. At the indicated concentrations, DMMB and MB accumulated at same level in mitochondria (33.4 ± 11% and 44.5 ± 4%), respectively. Scales bar MB (0.2 µM): 20 µm; MB (2 µM) or DMMB (0.2 µM): 10 µm.
Results
Intrinsic properties of DMMB and MB and their interactions with membranes and cells
Before exploring the cytotoxicity mechanisms of DMMB and MB, it was imperative to define some properties of these molecules and their interactions with cells and membranes, to subsequently correlate their intracellular sites of damage with the resultant mechanisms of cell death. Both MB and DMMB are efficient PSs (), generating 1O2 with high generation quantum yields (>0.5) and also engaging in type I reactions () [Citation43]. However, these compounds are considerably different regarding (i) their efficiency of interacting and damaging membranes and (ii) their main sites of intracellular action.
The 2 extra methylene groups in DMMB increase considerably its partition in nonpolar environments. In fact, logPo/w increases from −0.1 in MB to 1.0 in DMMB () [Citation43]. In accordance, our molecular dynamics simulations showed that DMMB is not only bound more efficiently and tighter to membranes, but also inserted itself deeper in the bilayer (see supplementary material, Fig. S1). Consequently, photo-irradiation of DMMB (and not of MB) caused a strong release of carboxyfluorescein (CF) from small unilamellar vesicles (SUVs), indicating membrane permeabilization ().
Both MB and DMMB are positively charged dyes and consequently are likely to accumulate both in mitochondria and in lysosomes of mammalian cells [Citation13,Citation25]. Accumulation in lysosomes occurs because the cellular incorporation of these dyes usually happens via endocytic pathways [Citation25]. In fact, both dyes accumulated in the lysosomes of HaCaT cells (Fig. S2). Positively charged dyes also typically concentrate in mitochondria, because of the negatively-charged electrochemical transmembrane potentials of breathing mitochondria. However, MB is a lot more easily reduced than DMMB [Citation47] and, consequently, MB (and not DMMB) is chemically bleached in mitochondria, leading to the formation of the colorless leuco-MB [Citation25,Citation48]. In accordance, colocalization studies with MB and Rh123-stained mitochondria revealed that MB’s fluorescence at low concentration was either inexistent or faint compared with that of DMMB ().
MB and DMMB photosensitization in mammalian cells
At the concentration regimes used at concentration range (10–20 nM) or (1–3 µM), neither DMMB nor MB induced dark toxicity, respectively (). Nevertheless, after photoexcitation both dyes significantly decreased the viability of HaCaT cells and, interestingly, the photocytotoxicity of DMMB was significantly larger than that of MB (). Note that the IC50 (obtained 48 h after irradiation) in DMMB-treated cells was ~ 200 fold smaller (10 nM) compared with that of MB-treated cells (2 µM). We remark that at these conditions, the amount of 1O2 molecules generated per second in DMMB-treated cells is around 150 fold smaller than that generated in MB-treated cells (see material and methods) [Citation25,Citation43].
Figure 2. Analysis of biological effects after irradiation using HaCaT cells. (a) Cellular survival 48 h after irradiation with a 633 nm LED (46 W m−2 irradiance) as a function of DMMB and MB concentrations, as measured by CVS and MTT assays. (b) DCFH oxidation 3 h after irradiation using DMMB and MB as indicated. (c) DCFH oxidation and cell survival measured by CVS assay 3 h after irradiation using DMMB (0 to 20 nM) and MB (0 to 3 µM). (d) Analysis of amplified long mitochondrial DNA target (mtDNA) and short mtDNA target (MT-ND1) after photosensitization of HaCaT cells preloaded with 20 nM DMMB and MB. Relative amplification was determined by the ratio between long mitochondrial DNA target (mtDNA) and short mtDNA target (MT-ND1). Mean ± standard error of 3 independent experiments are shown. The significance levels are indicated as *0.05, **P < 0.01, ***P < 0.001.
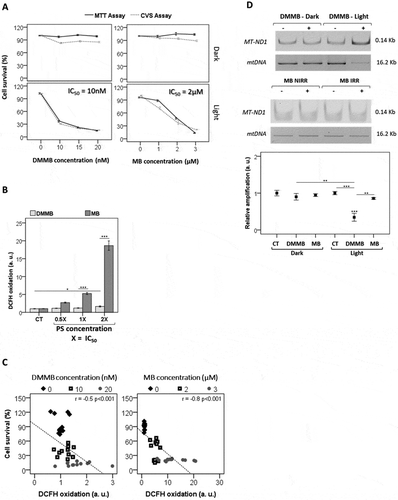
The photocytotoxicity of MB (and not of DMMB) correlated with the induction of a redox misbalance sensed in the whole cell (). Note that the increase in MB concentration, expressed in multiples of IC50, correlated well with the amount of 2ʹ,7ʹ-dichlorodihydrofluorescein (DCFH2) oxidation and formation of emissive dichlorofluorescein (DCF) (r = 1.0, P < 0.0001), while in the case of DMMB there was almost no change in the level of DCF emission (). Consequently, for DMMB the correlation between DCFH2 oxidation and the decrease in cell viability was very weak (r = −0.5, P < 0.0001), while for MB there is a strong and significant correlation (r = −0.8, P < 0.001) (). DCFH2 does not have a specific intracellular location and reacts with oxidants (peroxides and 1O2) generated throughout the cell [Citation49]. Because MB (in the micromolar concentration) generates a lot more oxidant species than DMMB, which are also not as well localized as those generated by DMMB in the nanomolar concentration, DCFH2 can only be considerably oxidized in MB-treated cells.
Although DMMB did not significantly oxidize DCFH2 (), it was capable of generating oxidizing species in key intracellular locations even in the nanomolar regime. Indeed, MitoSOX™ fluorescence was remarkably higher in DMMB-treated cells compared with control cells, which is a direct consequence of the generation of oxidizing species within mitochondria (Fig. S3A). The fluorescence intensity from MitoTracker™ Deep Red FM (MTDR; a probe for mitochondrial mass) was also significantly decreased in DMMB-treated cells, which was also in agreement with mitochondrial damage.
When both PSs were compared at the same concentration, we found out that photosensitization with DMMB (and not with MB) severely damaged mitochondrial DNA (mtDNA) (). Mitochondrial photodamage led to DNA lesions capable of blocking the progression of DNA polymerase on a long template, resulting in lower amplification of the target long DNA (16.2 kb). Note that PCR failed on the amplification of long mtDNAs, while the amplification of a short DNA target MT-ND1 (0.14 kb) increased as mitochondria accumulated due to impaired mitophagy. Note also that only DMMB significantly decreased the mitochondrial transmembrane inner potential (ΔΨm), as indicated by the smaller incorporation of Rh123 (). By being reduced and inactive, MB at nanomolar concentrations hardly caused any damage to mitochondria, while DMMB was able to severely harm this organelle, even at low concentration ( and (a)). Therefore, at the nanomolar level, only DMMB efficiently caused oxidative injury in mitochondria. By increasing MB concentration to the micromolar range, we could observe ΔΨm impairment () and generation of oxidizing species within mitochondria (Fig. S3A) at similar levels to those observed in cells treated with DMMB at nanomolar concentrations [Citation25].
Figure 3. Analysis of biological effects after irradiation using HaCaT cells. (a) Mitochondrial inner transmembrane potential (ΔΨm), measured by Rh123 fluorescence intensity relative to control (100%), using cytofluorometric analysis 30 min after photosensitization with DMMB and MB (20 nM). (b) ΔΨm determined by fluorescence microscopy and cytofluorometric analysis after photosensitization with DMMB (10 nM) and MB (2 µM). The decrease in ΔΨm was measured in terms of Rh123 fluorescence intensity relative to control (100%). All next analyses were performed in HaCaT cells pretreated with DMMB (10 nM) and MB (2 µM) and irradiated with a 633 nm LED (46 W m−2 irradiance), as done for control cells without photosensitizer. (c) After the indicated times, cytofluorometric analysis of cells stained with LysoTracker™ Green DND-26 (LTG). The decrease in lysosomal stability was measured in terms of LTG fluorescence intensity relative to control (100%). (d) 3 h after irradiation, the CTSB activity from cytosol fraction was measured in presence (+) or absence (-) of CA-074 (10 µM). Mean ± standard error of 3 independent experiments are shown. The significance levels were indicated as *P < 0.05, **P < 0.01, ***P < 0.001. Scale bars: 10 µm.
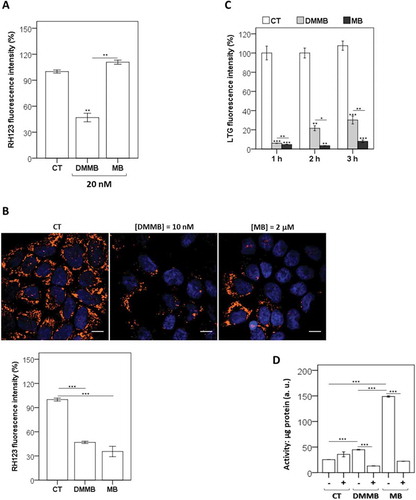
We also asked if the irradiation of cells previously treated with these dyes would permeabilize lysosomal membranes, since they both localized within lysosomes (Fig. S2). Following lysosomal membrane permeabilization (LMP), cells treated with acidophilic probes such as LysoTracker™ Green (LTG) become weakly fluorescent [Citation50]. Both phenothiazines induced significant decrease in LTG fluorescence intensity compared with the control, although LTG fluorescence decrease was more intense and resilient in MB-treated cells (). In order to confirm this observation, we also tested protease activities in cytosolic extracts with cathepsin-specific substrates such as amino-4-trifluoromethyl coumarin. There was a significant increase in the CTSB (cathepsin B) activity after photoactivation of both PSs, which were lessened by CTSB selective inhibitor CA-074 [Citation51] (). Note also that CTSB activity increased a lot more in MB-treated cells (). It is evident that photoactivation of both dyes damaged lysosomes. However, the injury by MB was more severe and resilient, which is a clear consequence of the different concentration regimes used (200-fold larger concentration for MB).
Generic mechanisms of photoinduced cell death
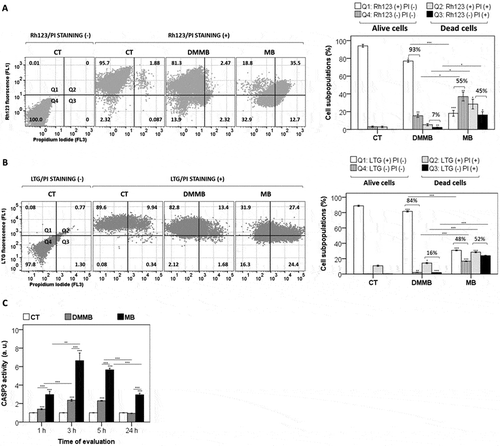
Despite the evident damage that DMMB caused both in mitochondria and lysosomes, 3 h after initial photodamage few DMMB-treated cells (13 ± 2%) showed features of cell death, which was a response similar to that of control cells (9 ± 1%, p = 0.15) (). Meanwhile about half of the cells treated with MB were already dead (propidium iodide positive [PI+], ), with ~15% of cells showing depolarized mitochondria (PI+ and rhodamine 123 negative [Rh123−]) () and ~20% showing significant signs of LMP (PI+ and LTG−) (). Note also that MB was capable to trigger cell death in ~30% of cells without modulation of mitochondrial membrane permeabilization or MMP () or LMP (), which is a consequence of the widespread and unspecific oxidative damage induced by MB (). Interestingly, even though DMMB-treated cells showed to suffer lysosomal photodamage, there was a reduced amount of CASP3 (caspase 3) activation (Caspase 3 Fluorometric Protease Assay), indicating smaller levels of apoptotic activation compared with MB regardless of the time of evaluation (). Therefore, the lysosomal photodamage triggered by DMMB did not release factors capable of activating the intrinsic apoptotic program, as demonstrated for other PSs [Citation27,Citation52]. In agreement with the stronger role played by apoptosis in the cell death mechanism induced by MB [Citation53], there was a substantial larger increase in CASP3 activation (). It has been suggested that lysosomal photodamage can activate calpain, which in turn cleaves the autophagy-associated protein ATG5 to a truncated form that potentiates an acute initiation (i.e. 2 h after irradiation) of the intrinsic apoptotic caspase-dependent pathway [Citation52]. However, ATG5-silenced cells are still responsive as wild-type cells to engage in cell death through apoptosis after lysosomal photodamage, indicating that other factors play a role in facilitating lysosomal photodamage, such as ATG7 [Citation27]. In the case of DMMB, it is likely that its lysosomal triggered-photodamage is so subtle that it cannot directly activate this lysosomal-dependent (via calpain cleavage) apoptotic caspase-dependent mechanism [Citation52].
Figure 4. Cell death efficiency and organelle specific photodamage. All analyses were performed in HaCaT cells pretreated with DMMB (10 nM) and MB (2 µM) and irradiated with a 633 nm LED (46 W m−2 irradiance), as done for control cells without photosensitizer. (a) FACS scatter plots gating cells according to 2 parameters (ΔΨm and cell death), for cells stained with Rhodamine 123 (Rh123) and PI right after irradiation. Right: bars show the mean values of cell subpopulations. (b) FACS scatter plots gating cells according to 2 parameters (LMP and cell death), for cells stained with LysoTracker™ Green DND-26 (LTG) and PI right after irradiation. Right: bars show the mean values of cell subpopulations. (c) Fluorometric analysis of CASP3 activity from cytosol fraction 1, 3, 5, and 24 h after irradiation. The CASP3 activity was compared to a control following normalization by total protein concentration and expressed as arbitrary units (a. u.). Mean ± standard error of 3 independent experiments are shown. The significance levels were indicated as *P < 0.05, **P < 0.01, ***P < 0.001.
DMMB did not induce measurable cell death neither just after () nor 3 h after photodamage ( and S3B), in spite of its effects on mitochondria and lysosomes (). Hence, we wondered if its effect on cells would be more evident in later times after photosensitization. Indeed, cells incubated with DMMB (10 nM) showed similar death-response 24 h after irradiation if compared with MB in shorter times (3 h) (Fig. S3B and ). However, notable consequences of DMMB photosensitization were observed 48 h after irradiation (). Note that DMMB photosensitization induced a remarkable cell death associated with accumulation of acidic vacuoles (stained by acridine orange), which was completely absent in MB-treated cells (). Incidentally, vacuolization increased remarkably the cell size of DMMB-treated cells compared with MB-treated cells and to the control (Fig. S4A). Note also that only in DMMB-treated vacuolated cells (upper right quadrant), the enhanced and significant lysosomal accumulation was associated with significant increase of the relative PI incorporation, which is an indicative of cell death (Fig. S4B).
Figure 5. Long-term evaluation of cell death. For analysis performed in A to D, HaCaT cells were pretreated with DMMB (10 nM) and MB (2 µM) and irradiated with a 633 nm LED (46 W m−2 irradiance), as done for control cells without photosensitizer. (a) Cytometry analysis for cells stained with PI 24 h and 48 h after irradiation. Bars show the ponderation of PI fluorescence compared to control and are represented as arbitrary units. (b). Fluorescence microscopy images for cells stained with acridine orange 48 h after irradiation. (c) Fluorescence microscopy images for cells double-stained with LysoTracker™ Red DND-99 (lysosomes, red) and MitoTracker™ Deep Red FM (mitochondrial mass, green) 24 h after irradiation. Below: profile plots showing colocalization of mitochondria and lysosome staining. Right: surface plots of mitochondria and lysosome staining. (d) Fluorescence microscopy images for cells double-stained with LysoTracker™ Red DND-99 (lysosomes, red) 120 h after irradiation. (e) MTT response and lysosomal accumulation determined by AAU 48 h after irradiation. For the human tumor cells HepG2, SKMEL-25 2 µM MB and 10 nM DMMB were employed, while for HeLa and SKMEL-28 2 µM MB and 20 nM DMMB were employed instead. Mean ± standard error of 3 independent experiments are shown. The significance levels were indicated as *P < 0.05, **P < 0.01, ***P < 0.001. Scale bars: 10 µm.
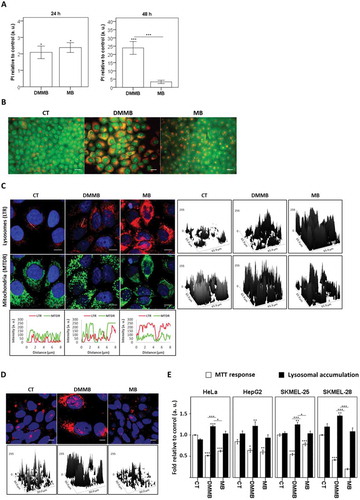
By comparing the cell´s response to LTG and PI fluorescence as a function of time (3 h versus 48 h), we observed distinct effects on lysosomes and homeostasis regarding the type of PS (Fig. S4B). The proportion of dead cells (PI+) showing lysosomal injury (LTG−) was significantly lower upon irradiation with DMMB (1.9 ± 0.1%) compared with MB (24 ± 0.6%) 3 h after irradiation. However, 48 h after irradiation this subpopulation of DMMB-treated cells remarkably augmented (14 ± 1.5%) compared with MB (4 ± 0.6%). Accordingly, the percentage of DMMB-treated cells that remained alive (PI−) with LTG-loaded lysosomes (LTG+) was significantly reduced (37 ± 2%) compared with MB (57 ± 3%) (Fig. S4B). Note also that after irradiation with DMMB there was a significant increase of death associated with vacuolization at longer evaluation times (i.e. 3 h versus 48 h after irradiation).
The correlation of cell death with the accumulation of acidic vacuoles suggests that DMMB-treated cells suffer from autophagy malfunction. Double staining of HaCaT with MTDR and LysoTracker™ Red DND-99 (LTR; lysosomotropic dye) indicated induction of mitophagy, which was nevertheless abrogated 24 h after irradiation (). Note that in the majority of cells irradiated with DMMB, LTR-loaded lysosomes selectively accumulated on regions with higher mitochondrial mass (). Interestingly, in the case of MB, mitophagy caused a decrease in mitochondrial mass, suggesting efficient removal of photodamaged mitochondria (profile plots, ). Note that the accumulation of acidic vacuoles was noticeable even 120 h after irradiation (). We perceive lysosome accumulation as an outcome of the photoactivation of DMMB as a consequence of a typical scenario of inhibition of the autophagic flux, a concept which we aim to prove below.
Another evidence of autophagy malfunction in DMMB-treated cells was obtained by comparing viability measurements that monitor lysosomes (neutral red), mitochondria (MTT) and cell count (CVS). As proposed by Martins et al., these measurements are translated into lysosomal accumulation by calculation of Autophagy Arbitrary Units (AAU) [Citation54]. For all of the studied human tumor cells (HeLa, HepG2, SKMEL-25 and SKMEL-28) we only observed an increase in lysosomal accumulation associated with declined cell viability for DMMB-treated cells (and not in MB treated cells). This indicates that the autophagy-associated cell death triggered by DMMB was not specific of HaCaT cells, but instead involves regulations present in different tumor cell types [Citation55,Citation56] (). Note also that the effect of DMMB (in terms of AAU) on tumor cell lines HeLa and SKMEL-28 (Fig. S5A) was significantly correlated with decrease of cell survival in a dose dependent-manner (Fig. S5B).
Inhibition of prosurvival autophagy
Several reports ascribed to autophagy a cytoprotective role after photodamage to cells [Citation10]. It was suggested that the initiation rate of apoptosis might be a factor related to the role of autophagy on photokilling. Specifically, cells that engage into rapid apoptosis after photodamage might use autophagy as a cytoprotecting pathway [Citation32,Citation57], while cells with a slow or absent apoptotic response could have autophagy as a death pathway after photosensitization [Citation22,Citation30].
In an attempt to address the effect of DMMB on autophagy and correlate this effect with cell death, we evaluated the survival of cells previously treated with autophagic inhibitors 3-methyladenine (3-MA) and bafilomycin A1 (BAF) (). Pretreatment with BAF did not affect the response to DMMB-mediated photodamage, suggesting that DMMB photosensitization itself inhibited the autophagic flux. Interesting, upstream inhibition of autophagy with 3-MA significantly worsen the survival of DMMB-treated cells, suggesting that DMMB photosensitization induced activation of prosurvival autophagy (). Further assays were used to show that DMMB photosensitization indeed induced both mitophagy activation as well as overall autophagy blockage.
Figure 6. Autophagy modulation after photosensitization. For all analyses performed, HaCaT cells were pretreated with DMMB (10 nM) and irradiated with a 633 nm LED (46 W m−2 irradiance), as done for control cells without photosensitizer. (a) Analysis of cell survival regarding autophagy inhibition. After pretreatment for 24 h in presence (+) or absence (-) of BAF (2 nM) and 3-methyladenine, 3-MA (5 mM), cells were washed, treated with DMMB (10 nM) and irradiated. Cell viability was determined by MTT assay 48 h after irradiation. (b) Autophagy inhibition upon treatment in presence (+) or absence (−) of BAF (20 nM), as determined by immunoblotting of LC3B-II 6 h after irradiation. (c) Expression profile of LC3B-II 1 and 6 h after irradiation. (d) Fluorescence microscopy images of cells immunoassayed for LC3B-II (green) 1 h after photosensitization. (e) Fluorescence microscopy images for cells immunoassayed for SQSTM1 (red) 48 h after irradiation. Autophagy inhibition upon treatment in presence (+) or absence (-) of BAF (20 nM). (f) Fluorescence microscopy images immunoassayed for CTSB (green) and SQSTM1 (cyan) just after photosensitization. Bottom: surface plots for CTSB and SQSTM1 fluorescence of DMMB-treated cells. (g) Fluorescence microscopy images for cells stained with MitoTracker™ Deep Red FM (mitochondrial mass, red) following immunoassay for PRKN (green), just after photosensitization. Right: surface plots for parkin fluorescence. Mean ± standard error of 3 independent experiments are shown. The significance levels were indicated as *P < 0.05, **P < 0.01, ***P < 0.001. Scale bars: 10 µm.
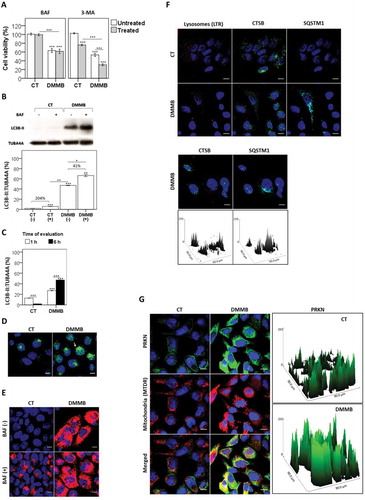
In order to evaluate the status of the autophagic flux we assessed the redistribution of LC3B from a diffuse cytoplasmic localization (LC3B-I) to a characteristic punctate cytoplasmic pattern, which is a consequence of the recruitment of LC3B-II to the autophagosome (AP), by forming LC3–phosphatidylethanolamine conjugates [Citation58]. In HaCaT cells, evaluation of endogenous LC3B-II can be performed directly without any of the biases commonly ascribed to transfection methodologies [Citation58–Citation60]. Immunoblotting results indicated a high level of autophagy activation in DMMB-treated cells 6 h after irradiation (). Note that cell irradiated with DMMB showed a significant increase of LC3B-II (47.2 ± 0.5%) in comparison to the control (1.9 ± 0.2%), P ≤ 0.007 (). This significant accumulation of LC3B-II was slightly increased upon treatment with BAF (20 nM) (41%) in comparison with the 204% increase of LC3B-II under BAF in the control (). Even 1 h after irradiation, immunoblotting quantification () and intracellular staining () showed that DMMB significantly induced accumulation of (LC3B-II) compared with the control without PS, and this scenario did not change significantly in comparison with 6 h after irradiation (). DMMB photosensitization also triggered the accumulation of receptor molecule SQSTM1/p62 [Citation61], with no additional accumulation being induced under BAF (), in agreement with the severe blockage of the autophagic flux by DMMB photosensitization. Note also that SQSTM1 significantly accumulated in cells irradiated with DMMB in which the lysosomes underwent membrane permeabilization with loss of CTSB punctate signal ().
Aiming to show the initial activation of autophagy, we evaluated the selective recruitment of the ubiquitin-ligase PRKN to photodamaged type 2 mitochondria, which is one of the initial steps of mitophagy activation [Citation6,Citation35]. Just after irradiation, PRKN disperses in the cytosol of control cells without PS and accumulated in specific micro-environments in DMMB cells (). Merged images showed that PRKN staining colocalized with mitochondria in DMMB cells, but not in control cells (). Therefore, DMMB photosensitization triggered the translocation of PRKN to depolarized mitochondria, indicating mitophagy activation [Citation62].
Parallel damage in mitochondria and lysosomes: an efficient route to modulate phototoxicity
The case of DMMB at nanomolar concentration illustrates a condition in which a subtle photoinduced challenge (in terms of the total amount of oxidizing species) induces specific damages in mitochondria and lysosomes, driving cells to a situation of initial selective autophagy activation, which is abrogated by a severe autophagy inhibition. Therefore, we wanted to establish the link between the parallel damage in mitochondria and lysosomes and the induction of a regulated mechanism of cell death associated with autophagy impairment. MB-treated cells suffer an oxidative imbalance [Citation13] and undergo the common mechanism of cell death by PDT, which is a combination of necrosis and apoptosis [Citation53,Citation63–Citation66]. Finding a condition in which MB would have a subtle specific damage that would mimic the effect of DMMB, could help to prove our hypothesis that parallel damage in mitochondria and lysosomes modulates autophagy-associated cell death.
In this regard, we irradiated HaCaT cells previously incubated with MB, but at a considerable lower concentration (i.e. 0.5 µM) than in the experiments described before. In this condition, MB is promptly reduced and unable to compromise mitochondria and to decrease cell viability (), despite being able to cause mild damage in lysosomes () [Citation25]. Note that, as lysosomes of MB-treated cells are mildly photodamaged, there was a significant recruitment of SQSTM1 (Surface plots, ), parallel to a diffuse staining of CTSB indicating LMP. Therefore, aiming to cause parallel damage in mitochondria and lysosomes and test our hypothesis, other chemical compounds, which specifically target mitochondria, were employed in combination to photodamage by MB. Our hypothesis being correct, in the presence of these external agents we should observe the accumulation of acidic vacuoles with a significant increase in the photocytotoxicity of MB.
Figure 7. Testing the work main hypothesis. (a) Fluorescence microscopy images of cells stained with LysoTracker™ Red DND-99 of lysosomes (LTR, red), followed by immunoassay for CTSB (green) just after irradiation. Cells sensitized with a noncytotoxic dose of MB (0.5 µM) were irradiated with a 633-nm LED (46 W m−2 irradiance), as done for control cells without photosensitizer. Prior chemical damage in mitochondria triggered by 3 mM hydrogen peroxide (HP) and 1.0 μM rotenone (RTN) for 15 min was followed by irradiation with a 633-nm LED (46 W m−2 irradiance) in the presence (+) or absence (-) of a low dose of MB (0.5 µM): (b) cell viability, as determined by MTT reduction assay; (c) microscopy analysis of acidic vacuoles, as performed by acridine orange staining 48 h after irradiation and (d) autophagy levels, as determined by AAU. Treatment with 60 µM chloroquine (CQ) for 24 h and with 10 nM DMMB were used as positive controls for impaired autophagy condition. (e) Microscopy of cells double-stained with LysoTracker™ Red DND-99 for lysosomes (LTR, red) and MitoTracker™ Deep Red FM for mitochondria (MTDR, green). Prior chemical damage in mitochondria triggered by 3 mM hydrogen peroxide (HP) for 15 min was followed by irradiation with a 633-nm LED (46 W m−2 irradiance) in the presence (+) or absence (−) of a low dose of MB (0.5 µM). (f) FACS analysis of cells double-stained with LysoTracker™ Green DND-26 (LTG) and PI 48 h after irradiation with red light. Representative scatter-plots showing cells gated according to the parameters LTG (FL1) and PI (FL3). Right: bars show the mean values of gated cell subpopulations (Q2 and Q3). Mean ± standard error of 3 independent experiments are shown. The significance levels were indicated as *P < 0.05 **P < 0.01, ***P < 0.001, ****P < 0.0001. Scale bars: 10 µm.
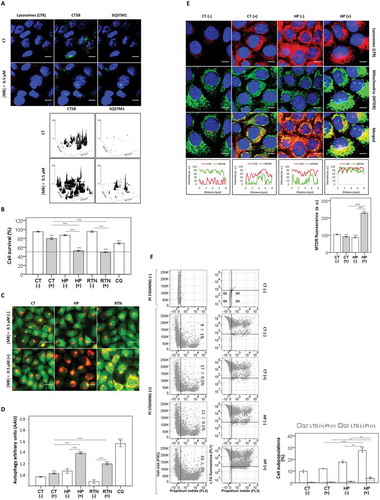
The lysosomal damage observed after irradiation with 0.5 μM MB, which was verified as CTSB leakage (), did not cause a significant decrease in cell viability (80 ± 2.2%), compared with the control (95 ± 1.0%) (). Hydrogen peroxide (HP) and rotenone (RTN) at the concentrations used herein do not cause any cytotoxicity by themselves, but have been shown to activate prosurvival type 2 mitophagy, as a consequence of mitochondrial depolarization associated with mild oxidative stress. Interestingly, by pretreating HaCaT cells with either one of these chemicals followed by MB irradiation (0.5 μM), a significant decrease in the cell viability (by ~50%) was observed (). This fact translates into a 2-fold decrease in IC50 in the presence of an external agent that causes mitochondria damage. Also, HaCaT cells showed a remarkable accumulation of acidic vacuoles (), which is a common hallmark observed after autophagy impairment, and typical characteristic of the effect of DMMB [Citation30,Citation58,Citation62]. The AAU level also significantly increased by pretreating cells with HP and RTN, approaching the AAU values of chloroquine and proving that, after the external damage in mitochondria, autophagy plays a role in the cell death induced by MB photoactivation ().
Next, the degree of mitochondria sequestration by lysosomes was analyzed by performing a double staining protocol using MTDR (in green) and LTR (in red), 24 h after irradiation (). MB photosensitization combined with mitochondria injury by HP led to a significant increase in the levels of LTR-loaded lysosomes, which overlapped with mitochondria (line scans, ). It is interesting that after treatment with MB, autophagy activation caused a significant decrease in mitochondrial mass, suggesting efficient removal of chemically damaged mitochondria (line scans, ). In HaCaT cells previously challenged with HP and subsequently photosensitized with MB, we observed a significant accumulation of mitochondria, suggesting failure in mitophagy efficiency (right panel, ).
Distinct effects on cell homeostasis were observed by comparing the cell´s response to LTG and PI fluorescence as a function of prior mitochondrial damage by HP associated (+) or not (-) with MB-photosensitization (). The damage by HP followed by MB-sensitization significantly increased death in 33 ± 3% of cells compared with MB without HP treatment (11 ± 0.1%), 48 h after irradiation (). Accordingly, after parallel low chemical damage on mitochondria and subtle lysosomal photodamage by MB, cells significantly engaged in death with vacuolization (28 ± 1.5%) compared with control cells (10 ± 1%) (). These findings sustain the main hypothesis raised in this work (i.e. that in presence of parallel damage in mitochondria and lysosomes, prosurvival autophagy fails and leads to cell death).
Discussion
MB and DMMB share similar photodynamic efficiencies in isotropic solution and under the illuminating conditions used herein, each molecule of MB and DMMB generates 1.71 and 2.25 molecules of 1O2 per second, respectively. Therefore, it is remarkable that the IC50 of DMMB is 200-fold lower than that of MB. In other words, under IC50 condition, DMMB kills half of the HaCaT cell population by generating 150 times less 1O2 molecules. By binding significantly more to membranes (as shown by molecular dynamics simulations and indicated by its capability to permeabilize SUVs more efficiently than MB), DMMB photodamages its targets more specifically than the more hydrophilic PS MB. Therefore, each 1O2 generated by DMMB is a lot more effective in causing molecular damage, indicating that they are generated in key locations to compromise cell homeostasis, as shown throughout the results section, activating the lysosomal-mitochondrial axis of cellular stress [Citation43]. We showed that the type 2 mitophagy process occurred too late to be able to prevent initiation of apoptosis after MB photosensitization and, alternatively, in the absence of acute apoptosis as revealed by DMMB plays a role as the major death pathway through a lysosomal-mitochondrial axis of cellular stress.
In general terms, the increased efficiency of DMMB can be attributed to the fact that it damages specific cellular components, hence activating a precise mechanism of autophagy-regulated cell death [Citation10,Citation13,Citation30]. We have not observed a significant involvement of lysosome-dependent apoptosis activation [Citation52], probably due to the low level of lysosomal damage. The efficiency of specific DMMB response in multidrug resistant tumor cells associated with high autophagic indexes and with less response to chemotherapy (e.g. SKMEL-25, SKMEL-28 and HepG2) proves the involvement of autophagy impairment in this cell death mechanism [Citation67–Citation71]. On the contrary, MB has to rely on the nonspecific generation of large amounts of oxidant species, so that some of them end up finding targets that trigger antiproliferative cellular responses. Indeed, we present evidence suggesting that the oxidative damage to the primary sites of localization might be less significant than the damage incurred to the sites to which the PS may redistribute during irradiation, a phenomenon also demonstrated for 5-ethylamino-9-diethyl-aminobenzo[a]phenothiazinium chloride (EtNBS) [Citation72].
Conversely, damage by MB is not specific and activates several cell responses, such as prosurvival autophagy, apoptosis and necrosis [Citation53,Citation63–Citation66]. By decreasing MB concentration to the nonphototoxicity level, we showed the same paradigm observed for DMMB (). MB damages mainly lysosomes and by adding external chemicals, which target mitochondria, we proved the hypothesis that the parallel damage in mitochondria and lysosomes are sufficient and necessary to activate mitophagy and inhibit it, leading to a late but efficient way to kill cells.
Figure 8. Scheme of working hypothesis using HaCaT keratinocytes. Parallel damage to mitochondrial and lysosomal membranes leads to autophagy-associated cell death. This figure was drawn by W. K. M.
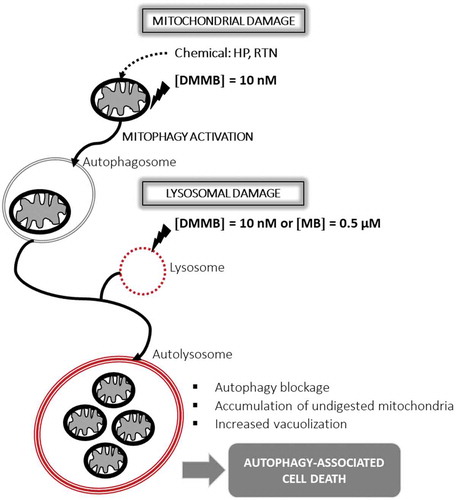
The concept of sequential parallel damages to key intracellular organelles has been previously shown to play an important role in the efficiency of PSs. For instance, the Kessel group shows that lysosomal photodamage potentiates the phototoxic effect of subsequent mitochondrial photodamage [Citation29]. In addition to that, the concept that the parallel damage in mitochondria and lysosomes leads to cell death with autophagy has been shown in other experimental systems [Citation62,Citation73]. DMMB, by being photoactivable, uses the energy of light photons to produce oxidant species catalytically, allowing its effect to occur at the nanomolar level. In a recent publication, Kessel argues that sequential damage in mitochondria and lysosomes mediated by the lysosomal PS chlorin NPe6 and mitochondrial PS benzoporphyrin derivative (BPD) in murine hepatoma 1c1c7 cells causes an acute apoptotic response that eventually evolves into accumulation of vacuolized intracellular vesicles, which were attributed to endoplasmic reticulum (ER) stress [Citation74]. Whether or not DMMB’s and MB’s effects involve a later ER stress remains to be studied. However, we are confident that the platform shown herein can also be used to further elucidate the relationship between autophagy inhibition and ER stress, since the control of intracellular damage attained by DMMB photosensitization is unprecedent.
The use of double-targeting protocols is starting to emerge as a promising tool to improve the efficiency of PSs, with several examples of multitarget PSs having been shown recently [Citation29,Citation75,Citation76]. Autophagy is one of the main causes of the development of resistance in PDT [Citation10,Citation17,Citation40,Citation41] but, to our knowledge, this is the first example of the use of a PS to specifically modulate the mitochondrial-lysosomal axis of cellular stress. We trust that learning to control autophagy will significantly impact the efficiency of PDT protocols. In a broader perspective, modulation of autophagy has been considered by several authors as a very promising strategy to treat oncologic diseases, but attaining autophagy modulation in the tumor tissue has been challenging our community for quite some time [Citation77]. Also, we are sure that using precisely localized PSs to cause specific damages in intracellular organelles will be useful to improve our understanding of autophagy activation and inhibition and its consequences to cell physiology.
Conclusion
Our results indicate a new paradigm concerning the fate of cells challenged by photosensitization. Whether photodamage will cause fast (and drastic) or late cell death depends on the extent of PS interaction with membranes and, in turn, on the level of photoinduced membrane damage. We showed the cross-talk between lysosomes and mitochondria after DMMB photosensitization and its consequences for the fate of cells. We hope that the concept of modulating autophagy by mutually challenging these 2 organelles will allow a better way to control autophagy, helping the development of more efficient drugs against cancer as well as other proliferative diseases.
Material and methods
Materials
Methylene blue (MB, 50484) and 1,9-dimethyl methylene blue (DMMB, 341088) were purchased from Sigma-Aldrich and all stock solutions were prepared in deionized water. The DMMB and MB molar concentration was checked using a Shimadzu UV-2400-PC spectrophotometer (Oregon, USA) by considering the molar extinction coefficient of 78,000 and 81,600 M−1cm−1 at λ = 651 nm, respectively. CA-074 (Sigma-Aldrich, C5732), 3-methyladenine (3-MA, Sigma-Aldrich M9281) and bafilomycin A1 (BAF), Sigma-Aldrich, B1793) were dissolved in deionized water and 100% dimethyl sulfoxide (DMSO; Sigma-Aldrich, D2650) to create 10 mM and 1 mM stock solutions, respectively. The dyes rhodamine 123 (Sigma-Aldrich, R8004), propidium iodide (Sigma-Aldrich, 81845) and acridine orange (AO; Sigma-Aldrich, A6014) were dissolved in deionized water to create 2 mM and 1 mg/mL stock solutions, respectively. 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC, 850375P) and 1ʹ,3ʹ-bis[1,2-dioleoyl-sn-glycero-3-phospho]-sn-glycerol (sodium salt) (CL, 710335) were obtained from Avanti Polar Lipids. The antibodies used included CTSB (cathepsin B) [CA10] (Abcam®, ab58802), LC3B (D11) XP® (Cell Signaling Technology®, 3868), SQSTM1/p62 [D10E10] (Cell Signaling Technology®, 7695) and TUBA4A/tubulin (Sigma-Aldrich, T5169). LysoTracker™ Green DND-26 (L7526) and Red DND-99 (LTR, L7528) or MitoTracker™ dyes Orange CMTMRos (MTO, M-7510) and Deep Red FM (M-22426) were all purchased from (Molecular Probes) and dissolved in DMSO to create 1 mM stock solutions. ProLong™ Gold antifade mountant with DAPI (P36935) was obtained from Molecular Probes. Piperazine-N,N´-bis(2-ethanesulfonic acid) (PIPES, P1851), NaCl (S9888), ethylenediaminetetraacetic acid (EDTA, E9884), sucrose (S0389), 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate hydrate (CHAPS, C3023), Triton™ X-100 (T8787), polyethylene glycol sorbitan monolaurate (Tween® 20, 274348), phenylmethylsulfonyl fluoride (PMSF, 78830), pepstatin A (77170), ethidium bromide (E7637), 5(6)-carboxyfluorescein (CF, C0537), chloroquine (CQ, 50635) and digitonin (D141) were purchased from Sigma-Aldrich.
Cell lines and cell culture
Human nonmalignant immortalized keratinocytes (HaCaT) and malignant human cells A549, HeLa, SKMEL-25 and SKMEL-28 were cultured in Dulbecco Modified Eagle Medium, DMEM (Sigma-Aldrich, D5648) supplemented with 10% (v:v) fetal bovine serum (FBS; Gibco™, 12657029), 100 units/mL of penicillin, 100 μg/mL of streptomycin and 250 ng/mL of amphotericin B in a 37°C incubator under a moist atmosphere of 5% carbon dioxide [Citation55,Citation56,Citation60].
Photosensitization protocol
After seeding (6 × 104 cells cm−2), exponentially growing human cells were irradiated after incubation with different concentrations of DMMB (0 to 20 nM) and MB (0 to 3.0 µM) in phosphate-buffered saline (PBS; 1.76 mM potassium phosphate monobasic, 137 mM sodium chloride, 2.7 mM potassium chloride, 10 mM sodium phosphate dibasic) for 1 h in a 37°C incubator under a moist atmosphere of 5% carbon dioxide. Prior to LED irradiation, MB and DMMB treated and untreated cells were washed twice with PBS. Sample irradiation was carried out with a LED with a maximum emission wavelength at 633 nm. To provide 11 J cm−2, cells were irradiated for 40 min at an irradiance of 46 W m−2. In parallel, nonirradiated cells were treated similarly.
Membrane permeabilization
A leakage assay was used to assess membrane permeabilization of DOPC:CL small unilamellar vesicles (SUVs) by MB or DMMB (15 µM) under irradiation with red light. The fluorescent probe 5-carboxifluorescein (CF) was initially entrapped in the aqueous compartment of the liposomes under a concentration in which fluorescence is self-quenched. Leakage to the outer solution leads to fluorescence intensity increases and indicates that the integrity of the lipid membrane was compromised. 14.2 µM of DOPC and 2.5 µM of CL dissolved in chloroform were used to produce a lipid film that was dried under a nitrogen flux. The film was hydrated with a 50 mM CF solution containing 10 mM Tris (pH = 8). The resulting suspension was extruded (pore diameter = 50 nm) and eluted in a Sephadex® G-50 (Sigma-Aldrich, G50150) column equilibrated with a 300 mM NaCl solution containing 10 mM Tris, pH 8 [Citation43,Citation78]. A suspension in which CF is virtually absent from the surrounding solution, though present in the inner compartment of the liposomes, can be obtained by this procedure. Since CF faces fluorescence quenching at the 50 mM concentration, dilution in the outer solution as a result of membrane permeabilization leads to fluorescence increase [Citation79]. Samples containing 15 µL of SUVs suspension (the final lipid concentration per well was determined as 0.37 mM), 15 µM MB or DMMB and enough Tris-NaCl buffer to complete 300 µL of solution were set in a 96-well fluorescence microplate and were irradiated with a 631-nm LED with 72 W m−2 irradiance at a 20-cm distance [Citation80]. Emission intensity at 517 nm (excitation at 480 nm) was measured using a fluorescence microplate reader (SpectraMax® ie3 Microplate Detection Platform, Molecular Devices, CA, USA) and plotted as an average and standard deviation, as a function of irradiation time.
Molecular dynamics simulations
The hydrated POPC (1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine) bilayer was generated by CHARMM-GUI webserver, containing 64 CHARMM27 lipids and 5787 TIP3P water molecules [Citation81–Citation83]. All simulations were performed in NPT ensemble at 303.15 K and 1 atm using Langevin thermostat with a damping factor of 1 ps−1 and Nosé-Hoover barostat with an oscillation period of 50 fs using 2 fs as timestep and SETTLE algorithm. Lennard-Jones interactions were shifted to zero at 12 Å [Citation84]. For the treatment of long-range electrostatic interactions, the Particle Mesh Ewald (PME) method was used [Citation85]. MD simulations were carried out by NAMD2.8 package [Citation86]. To set the bilayers systems at physiological salt concentration, 17 K+ and 18 Cl− were added to the system. The intermolecular Lennard-Jones parameters and topology were obtained by SwissParam webserver [Citation87]. The partial atomic charges were obtained by the Merz-Singh-Kollman (‘MK) protocol at level HF/6-31G(d) and the ground-state geometry optimizations at the B3LYP/6-31G* level [Citation88,Citation89]. Quantum chemistry calculations were performed using the Gaussian03W program [Citation90]. The preferential orientation for both PSs was obtained by analyzing the tilt angle distributions with respect to the outwardly directed membrane normal (Fig. S1B-ii). Orientation distribution function in terms of the angle between the sulfur-phosphorus (S-N) vector and the outward normal of the bilayer are shown in Fig. S1B i-ii.
Fitted parameters of gaussian deconvolution
Fitted parameters of Gaussian deconvolution for MB and DMMB are described according to Equations (S1) and (S2), whose parameters are presented in Tables 1 and 2, respectively.
Cell survival via MTT reduction and CVS assays
To analyze survival rate rather than short-term cytotoxicity, we allowed human cells to proliferate for 2 population doubling times after irradiation (i.e. 48 h). Next, we performed independently CVS (Crystal Violet Staining) and MTT assays as described [Citation54]. Briefly, after a period of 48 h following photosensitization we washed cells and added medium 1% (v:v) FBS containing Methylthiazolyldiphenyl-tetrazolium bromide (MTT, Sigma-Aldrich, M5655) at 50 µg/mL and incubated for 2 h in a 37°C incubator under a moist atmosphere of 5% carbon dioxide. Next, the reduced-product formazan was solubilized in 0.2 mL DMSO, and absorbance values were read at 550 nm with a wavelength correction set at 800 nm for subtraction of backgrounds, using the Infinite M-200 Tecan microplate reader (Männedorf, Switzerland). Cell survival rates were normalized to the absorbance values of untreated and nonphotosensitized cells.
For the CVS assay, NR-fixed cells (see below) were stained with crystal violet (CV; Sigma-Aldrich, C6158) at 0.02% (w:v) for 5 min at room temperature. After washing, CV was eluted with 0.1 M sodium citrate in 50% (v:v) ethanol, and recorded absorbance values at 585 nm [Citation54].
Autophagy quantification via AAU
It is well known that nonfunctional lysosomes accumulate in cells (HaCaT, HeLa and SKMEL-28) leading to autophagy-associated cell death [Citation58,Citation62,Citation73]. To assess this effect, after photodamage we employed the numeric variable AAU (autophagy arbitrary units) [Citation54]. Briefly, 48 h after photosensitization, cells were incubated with 30 µg/mL neutral red (NR, Sigma-Aldrich, N4638) for 2 h in a 37°C incubator under a moist atmosphere of 5% carbon dioxide. After washing, NR was eluted with an alcoholic-based 1% (v:v) acetic acid fixing solution and measured at 540 nm absorbance as described above. These fixed cells after washing with water were used for CVS assay (above). Finally, to calculate AAU, the mean NRU survival rate was normalized to the mean of the MTT and CVS survival rates according to the function w (x, y, z):
Where x, y, and z were the survival rates measured by NRU, CVS and MTT assays, respectively [Citation73]. Next, the propagated error, σw, of the function w (x, y, z) was determined according to formula below:
1O2 efficiency generation: calculations
The number of 1O2 molecules generated by a PS per unit time (Q) can be calculated by
Where ΦΔ is the 1O2 generation quantum yield of the PS, λi is the irradiation wavelength, Pi is the irradiance of the light source, σ is the absorption cross-section of the PS at the irradiation wavelength, and hc is the product of the Planck constant and the speed of light [Citation91,Citation92]. Using the parameters from Table 3, Q values of 1.71 and 2.25 1O2 molecules per second are obtained for MB and DMMB, respectively. If considered that the concentration of MB is actually 200 times higher than of DMMB (i.e. 2 µM compared to 10 nM), a solution of MB is expected to generate 152 times more 1O2 molecules per second. For the irradiation at 631 nm employed for liposome experiments, the same ratio applies, since for both PSs the absorption cross sections at 631 and 633 nm are very similar.
Immunoassaying and confocal microscopy
Supernatant cells were washed out and adherent cells were immunoassayed after aldehyde fixation 4% (p:v) in PBS pH 7.4, blocking and incubation with primary monoclonal antibodies against LC3B, CTSB and SQSTM1 according to manufacturer’s instructions, and then with goat Alexa Fluor-coupled antibodies against rabbit IgG (H + L) (A-11034, A-11035 and A-21070) or mouse IgG (A-11001 and A-21050) purchased from Molecular Probes. We analyzed the DAPI-counterstained slides using confocal microscope (Zeiss™ Axiovert 200 LSM 510 Laser, Carl Zeiss, Jena, Germany) equipped with a Plan-APOCHROMAT 63X/1.40 oil DIC M27 objective (Zeiss™, Carl Zeiss, Jena, Germany) and determined the overlap between these proteins as described previously [Citation93]. LysoTracker™ Red DND-99 is an acidophilic probe compatible with aldehyde fixation and thus may be combined with the immunofluorescence detection of proteins, such as CTSB, LC3B-II and SQSTM1 [Citation62].
We analyzed the DAPI-counterstained slides as above by using filter sets that provide an excitation of 364, 488, 543 and 633 nm with emission band pass (BP) of 437 to 490 nm, 515 to 534 nm, 565 to 640 nm and 651 to 704 nm to detect the fluorescence of DAPI, Alexa Fluor® 488 (A-11034 and A-11001), LTR or Alexa Fluor® 546 (A-11035) and MTDR or Alexa Fluor® 633 (A-21050 and A-21070), respectively.
Subcellular localization of phenothiazine
Acidophilic lysosomotropic LysoTracker™ Green DND-26 (LTG) and Rhodamine 123 (Rh123) were used as probes to stain lysosomes and mitochondria, respectively. Briefly, after preloading HaCaT cells with DMMB (0.2 µM) and MB (2 µM) for 30 min, LTG (300 nM) or Rh123 (300 nM) were added following further incubation for 30 min in a 37°C incubator under a moist atmosphere of 5% carbon dioxide. After washing, we immediately analyzed live-cells using confocal microscope as described above. Both lysosomes and mitochondria were analyzed under excitation at 488 nm with emission at 515 to 534 nm (green). For phenothiazines, we used an excitation/emission filter set of 633 and BP 704 nm (red). Finally, we measured the percentage of correlation between phenothiazine and organelles after deconvolution using the software MetaMorph 6.3r2 (Molecular Devices).
Labeling of lysosomes and acidic vacuoles in live cells
Both lysosomotropic dyes acridine orange and LTG were used as probes to stain acidic acidophilic autophagic vacuoles, endosomes or lysosomes [Citation58]. Right after indicative times, we incubated HaCaT cells with 200 nM LTG or 1 µg/mL AO in DMEM 1% (v:v) FBS for 15 min in a 37°C incubator with a moist atmosphere of 5% carbon dioxide. After washing, we immediately analyzed live-cells using confocal microscope as described above. Alternatively, we collected at least 30,000 events to further flow cytofluorometric analysis (BD FACS Verse™, Piscataway, NJ, USA) using FlowJo software.
Mitochondrial function and mass
Fluorescent probes were used to examine mitochondrial function, according to manufacturer’s instructions: Rh123 or MitoTracker™ Orange CMTMRos (MTO) and MitoTracker™ Deep Red FM to monitor mitochondrial transmembrane potential (ΔΨm) and mitochondrial mass, respectively [Citation94]. MTDR is a cell-permeant mitochondrial-specific dye that binds to mitochondria regardless of ΔΨm lost that accurately measure mitochondrial mass. After photosensitization, HaCaT cells cultured on coverslips were washed and refreshed with DMEM supplemented with 10% (v:v) FBS, 100 units/mL of penicillin, 100 μg/mL of streptomycin and 250 ng/mL of amphotericin B. After indicative times, the biological effects were evaluated. Following incubation MTDR (50 nM) or MTO (25 nM) in 1% (v:v) DMEM FBS for 30 min in a 37°C incubator with a moist atmosphere of 5% carbon dioxide, we performed fixation with cooled 4% (w:v) formaldehyde pH 7.2 in PBS. After washing, slides were mounted in ProLong™ Gold antifade reagent containing DAPI nuclear stain. We used filter sets that provide an excitation of 364, 543 and 633 nm with emission BP of 437 to 490 nm, 554 to 608 nm, and 661 to 704 nm to detect the fluorescence of DAPI, MTO, and MTDR, respectively. Alternatively, we measured mitochondrial dye-related fluorescence (Rh123) parallel to incorporation with propidium iodide. Briefly, just after the irradiation, supernatant and adherent cells were collected, sedimented by centrifugation (300 g, 5 min, 4°C), washed twice with PBS-EDTA 0.05% (w:v) and resuspended in 980 µL PBS-EDTA 0.05% (w:v). A total of 10 µL of Rh123 (50 µM) and 10 µL of PI (100 µg/mL) were added, and the samples were maintained at room temperature for 15 min in the dark. After washing twice in PBS-EDTA 0.05% (w:v) we quantified the fluorescence emission of Rh123 (FL1) or PI (FL3) by collecting at least 30,000 events using the cytofluorometer (BD FACS Verse™). To analyze the data, the FlowJo software was employed.
Next, Rh123 and PI fluorescence were detected by flow cytometry (BD FACS Verse™) using excitation laser at 488 nm (FL1) and 640 nm (FL3), respectively. At least 30,000 events were collected in each analysis and data was analyzed by FlowJo software.
Mitochondrial ROS determination
Aiming to determine ROS in mitochondria, we measured superoxide radical generation by using MitoSOX™ Red mitochondrial superoxide (Molecular Probes, M36008), a useful indicator for highly selective detection of superoxide in the mitochondria of live cells [Citation95]. Briefly, right after irradiation using DMMB and MB at their IC50 (i.e. 10 nM and 2 µM, respectively), we double-stained of mitochondria with MitoSOX™ (5 µM) and MTDR (0.1 µM) for 30 min in a 37°C incubator under a moist atmosphere of 5% carbon dioxide. Thus, we could restrict the mitochondrial ROS analysis to the mitochondrial mass. Following washing and fixing, slides were mounted in ProLong™ Gold antifade reagent containing DAPI nuclear stain. Next, we analyzed the DAPI-counterstained slides using a confocal microscope (Zeiss™ LSM 510 Axiovert 200M Confocal Laser Scanning Module (Carl Zeiss, Jena, Germany) equipped with a Plan-APOCHROMAT 63X/1.40 oil DIC M27 objective (Zeiss™) and we analyzed with the ImageJ software (National Institutes of Health). We used filter sets that provide an excitation of 364, 488 and 633 nm with emission BP of 437 to 490 nm, 522 to 576 nm and 661 to 704 nm to detect the fluorescence of DAPI, MitoSOX™ and MTDR, respectively.
Mitochondrial DNA damage
The mitochondrial DNA (mtDNA) represents a critical target for ROS damage, and several studies have shown that mtDNA is more susceptible to genotoxic stress and ROS damage than is nuclear DNA [Citation96]. To analyze mitochondrial DNA damage after irradiation with DMMB, we used the gene-specific quantitative PCR-based (QPCR) assay, which measures the DNA integrity of the mitochondrial genome based on amplification of a long DNA target [Citation96]. Briefly, the QPCR assay is based on the ability of different kinds of DNA lesions to block or slow down the progression of DNA polymerase on a long template, resulting in decreased amplification of the target DNA [Citation97,Citation98]. Primers for QPCR were designed based on the human mitochondrion complete sequence (NC_012920.1) in regions that target long (16.2 kb) and short (0.14 kb) DNA fragments. The oligonucleotide sequences of the primers used here to amplify human targets are shown in Table 4. In summary, 30 ng total DNA was applied as template and the QPCRs targeting long mitochondrial DNA fragment were carried out using the AccuPrime™ Taq DNA Polymerase High Fidelity kit (Invitrogen™, 12346086). Amplification conditions were: initial denaturation for 30 s at 94°C followed by 26 cycles of 30 s at 94°C, 30 s at 60°C, and 18 min at 68°C. On the other hand, 3 ng total DNA were applied as template in case of amplification of MT-ND1 (a short target mtDNA, 0.14 kb) using the Taq DNA Polymerase Recombinant kit (Invitrogen™, 10342046). Amplification conditions were: initial denaturation for 3 min at 94°C followed by 22 cycles of 45 s at 94°C, 30 s at 56°C, 30 s at 56°C, and 1 min at 72°C. Long and short PCR products were analyzed on ethidium bromide stained 1% (w:v) agarose and 10% polyacrylamide gels, respectively.
Immunobloting
After photodamage, all cells (supernatant and adherent) were collected and processed for lysis with buffer containing 20 mM PIPES, 100 mM NaCl, 1 mM EDTA, 10% (w:v) sucrose, 0.1% (v:v) CHAPS, 0.1% (v:v) Triton X-100, 1 mM PMSF, 2 µM pepstatin A, 50 µM digitonin. 15 to 20 µg of total proteins were separated in 12% acrylamide gels and transferred to Amersham® Hybond P 0.45 PVDF membranes (GE healthcare Life Sciences, 10600023) using a Mini Trans-Blot® electrophoretic transfer cell (Bio-Rad, 1703930). Membranes were incubated in Tris-buffered saline (TBS; 50 mM Tris-HCl, pH 7.6, 150 mM NaCl) with 5% (w:v) bovine serum albumin (BSA; Sigma-Aldrich, A7906) and 0.1% (v:v) Tween® 20 for 1 h and with primary monoclonal antibodies anti-LC3 and anti-TUBA4A in TBS with 2.5% (w:v) BSA and 0.1% (v:v) Tween® 20 overnight at 4°C. Three washing steps in TBS with 0.1% (v:v) Tween® 20 for 10 min were performed and followed by incubation with anti-rabbit IgG (H + L) peroxidase labeled (KPL) diluted in TBS with 2.5% (v:v) BSA and 0.1% (v:v) Tween® 20 for 1 h. Membranes were washed twice in TBS with 0.1% (v:v) Tween® 20 and once in TBS, 10 min each. After incubation with ECL™ Western blotting detection reagent (GE healthcare Life Sciences, RPN2209) for 5 min, membranes were exposed to CL-XPosure™ Films (ThermoFisher Scientific, 34090). Images were analyzed using ImageJ software and the results were normalized to tubulin band intensities [Citation62].
Assay for measuring of CASP3 activity
At 1, 3, 5 or 24 h after irradiation with DMMB (10 nM) and MB (2 µM), the activity of CASP3 was evaluated using a fluorescence-based assay kit (BioVision Inc., K105-100). The assay is based on detection of the cleavage of the substrate DEVD-AFC (AFC: 7-amino-4-trifluoromethyl coumarin) by CASP3 or related caspases. Briefly, treated-cells were lysed in 50 µL of chilled CB cell lysis buffer. After incubation on ice for 10 min and centrifugation at 10,000 g for 10 min at 4°C, we obtained active CASP3 that is capable of cleaving the synthetic substrate DEVD-AFC to release free AFC (7-amino-4-trifluoromethyl coumarin) whose yellow-green fluorescence could be quantified using 400 nm excitation filter and 505 nm emission filter (Infinite M-200 Tecan microplate reader, Switzerland). 40 µg of protein per sample was used in this enzymatic assay whose concentration was measured using the BRADFORD assay (Quick Start™ Bradford Protein Assay, BIO-RAD, 500–0205). Finally, the CASP3 activity was compared to control following normalization by total protein and expressed as arbitrary units (a. u.).
Assay for measuring of CTSB activity
After indicative times following irradiation with DMMB (10 nM) and MB (2 µM), the activity of cytosolic CTSB was evaluated by using a fluorescence-based assay kit (BioVision Inc., K140-100), which utilizes the preferred CTSB substrate sequence RR labeled with AFC (amino-4-trifluoromethyl coumarin). Briefly, treated cells were lysed in 50 µL of chilled CB cell lysis buffer. After incubation on ice for 10 min and centrifugation at 10,000 g for 10 min at 4°C, we obtained the cytosolic-fraction of CTSB that is capable to cleave the synthetic substrate Ac-RR-AFC to release free AFC. After incubation protected from light at 37°C for 1 h, the released AFC could easily be quantified using a fluorescence plate reader (Infinite M-200 Tecan microplate reader, Switzerland) with Ex/Em 400/505 filter. Protein (15 µg) per sample was used for enzymatic assays whose concentration was quantified as above. Finally, the CTSB activity was normalized by total protein and expressed as arbitrary units (a. u.), as previously proposed [Citation62].
Lysotracker™ green DND-26 and PI double-labeled flow cytometry
The cell death associated with lysosomal accumulation was measured in HaCaT cells treated with DMMB (10 nM) and MB (2 µM) 3 h and 48 h after photosensitization. Two-color flow cytometry was applied to detect lysosomes 200 nM (LTG) and the exclusion of PI (1 µg/mL). The cells positive for LTG and negative for PI represented live-vacuolated cells, whereas the cells positive for both markers represented dead-vacuolated cells. After irradiation, HaCaT cells were washed and incubated 10% (v:v) DMEM FBS for 48 h in a 37°C incubator with a moist atmosphere of 5% carbon dioxide. Next, supernatant and adherent cells were collected, sedimented by centrifugation (300 g, 5 min, 4°C) and washed twice with PBS-EDTA 0.05% (w:v) and resuspended in 180 µL PBS-EDTA 0.05% (w:v). A total of 10 µL of LTG (4 µM) and 10 µL of PI (20 µg/mL) were added, and the samples were maintained at room temperature for 15 min in the dark. After washing twice in PBS-EDTA 0.05% (w:v), we quantified the fluorescence emission of LTG (FL1) or PI (FL3) by collecting at least 30,000 events using the cytofluorometer (BD FACS Verse™). To analyze the data, the FlowJo software was employed.
Statistics
Statistical analysis was performed using IBM® SPSS Statistics version 20. The strength of linear correlation analysis was calculated by the Pearson coefficient (r). To perform comparative statistical analysis, we first analyzed the variance between the groups of samples. Next, we performed one-way analysis of variance (ANOVA) with Dunnett T3 or Bonferroni post-hoc tests, depending on homogeneity of variance. We analyzed data obtained from at least 3 independent experiments and expressed as mean values ± error standard. As statistically significant, we considered P values lower than 0.05. The significance levels relative to control were depicted with asterisks above bars and for multiple or pair comparisons according to what is indicated by lines.
Supplemental Material
Download Zip (3.2 MB)Acknowledgments
The authors are grateful to Alessandra A. Araújo de Sousa, Ingrid Torres Lima, Luana de Souza Barbosa, Laryssa Santos, Larissa N. Xavier de Albuquerque and Edson Alves for technical assistance in cell culture service; and Wilton J. R. Lima for helping with confocal microscopy and flow cytometry. We also thank Dr. Hugo Armelin (Instituto Butantan, Brazil), Dr. Roger Chammas (Instituto do Câncer do Estado de São Paulo, Brazil) and Érico T. Costa (Ludwig Institute for Cancer Research at Centro de Oncologia Molecular, Hospital Sírio Libanês, São Paulo) by supplying HaCaT, SKMEL-28, SKMEL-25 and HepG2, respectively. This work was part of the master thesis of Nayra Fernandes Santos, who was a graduate student supported by CAPES. MSB acknowledges CNPq for his productivity fellowship 304071/2014-5. We also acknowledge support by PNPD (Programa Nacional de Pós Doutorado)/CAPES (Coordenação de Aperfeiçoamento de Pessoal de Nível Superior)/FINEP (Financiadora de Estudos e Projetos) grant number 02533/09-0, Brazil; and by FAPESP (Fundação de Amparo à Pesquisa do Estado de São Paulo) grants 2012/50680-5, 2013/07937-8, 2013/11640-0 and 2016/07642-6.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
Related Research Data
References
- Rubinsztein DC. Levine PC and B. Autophagy modulation as a potential therapeutic target for diverse diseases. Nat Rev Drug Discov. 2012;29:709–730.
- Glick D, Barth S, Macleod KF. Autophagy: cellular and molecular mechanisms. J Pathol. 2010;221:3–12.
- Ding W-X, Yin X-M. Mitophagy: mechanisms, pathophysiological roles, and analysis. Biol Chem. 2012;393:547–564.
- Lemasters JJ, Autophagy, or mitophagy, as a targeted defense against oxidative stress, mitochondrial dysfunction, and aging. Rejuvenation Res. 2005;8:3–5.
- Reggiori F, Komatsu M, Finley K, et al. Selective types of autophagy. Int J Cell Biol. 2012;2012:156272.
- Youle RJ, Narendra DP. Mechanisms of mitophagy. Nat Rev Mol Cell Biol. 2011;12:9–14.
- Gaziev AI, Abdullaev S, Podlutsky A. Mitochondrial function and mitochondrial DNA maintenance with advancing age. Biogerontology. 2014;15:417–438.
- Martins WK, Baptista MS. Autophagy modulation for organelle-targeting therapy. In: Gorbunov Nikolai SM, editor. Autophagy Curr. Trends Cell. Physiol. Pathol.. 1st ed. InTech; 2016. p. 350–390.
- Inguscio V, Panzarini E, Dini L. Autophagy contributes to the death/survival balance in cancer photodynamic therapy. Cells. 2012;464–491.
- Reiners JJ, Agostinis P, Berg K, et al. Assessing autophagy in the context of photodynamic therapy. Autophagy. 2010;6:7–18.
- Mallidi S, Spring BQ, Hasan T. Optical imaging, photodynamic therapy and optically triggered combination treatments. Cancer J. 1974;21:194–205.
- Ochsner M. Photophysical and photobiological processes in the photodynamic therapy of tumours. J Photochem Photobiol B. 1997;39:1–18.
- Bacellar I, Tsubone T, Pavani C, et al. Photodynamic efficiency: from molecular photochemistry to cell death. Int J Mol Sci. 2015;16:20523–20559.
- Spring BQ, Rizvi I, Xu N, et al. The role of photodynamic therapy in overcoming cancer drug resistance. Photochem Photobiol Sci. 2015;14:1476–1491.
- Oleinick NL, Evans HH. The photobiology of photodynamic therapy: cellular targets and mechanisms. Radiat Res. 1998;150:S146–S156.
- Morton CA, Szeimies RM, Sidoroff A, et al. European guidelines for topical photodynamic therapy part 1: treatment delivery and current indications – actinic keratoses, Bowen’s disease, basal cell carcinoma. J Eur Acad Dermatology Venereol. 2013;27:536–544.
- Kessel D, Oleinick NL. Initiation of autophagy by photodynamic therapy. Methods Enzymol. 2009;453:1–16. 1st ed.
- Ji H-T, Chien L-T, Lin Y-H, et al. 5-ALA mediated photodynamic therapy induces autophagic cell death via AMP-activated protein kinase. Mol Cancer. 2010;9:91.
- Separovic D, Kelekar A, Nayak AK, et al. Increased ceramide accumulation correlates with downregulation of the autophagy protein ATG-7 in MCF-7 cells sensitized to photodamage. Arch Biochem Biophys. 2010;494:101–105.
- Kessel D, Reiners JJ. Apoptosis and autophagy after mitochondrial or endoplasmic reticulum photodamage. Photochem Photobiol. 2009;83:1024–1028.
- Panzarini E, Inguscio V, Dini L. Timing the multiple cell death pathways initiated by Rose Bengal acetate photodynamic therapy. Cell Death Dis. 2011;2:e169.
- Xue L-Y, Chiu S-M, Oleinick NL. Atg7 deficiency increases resistance of MCF-7 human breast cancer cells to photodynamic therapy. Autophagy. 2010;6:248–255.
- Jr JJ R, Caruso JA, Mathieu P, et al. Release of cytochrome c and activation of pro-caspase-9 following lysosomal photodamage involves bid cleavage. Cell Death Differ. 2002;9:934–944.
- Kessel D, Reiners JJ. Promotion of proapoptotic signals by lysosomal photodamage. Photochem Photobiol. 2015;91:931–936.
- Oliveira CS, Turchiello R, Kowaltowski AJ, et al. Major determinants of photoinduced cell death: subcellular localization versus photosensitization efficiency. Free Radic Biol Med. 2011;51:824–833.
- Castano AP, Demidova TN, Hamblin MR. Mechanisms in photodynamic therapy: part one – photosensitizers, photochemistry and cellular localization. Photodiagnosis Photodyn Ther. 2004;1(4):279–293.
- Kessel DH, Price M, Reiners JJ. ATG7 deficiency suppresses apoptosis and cell death induced by lysosomal photodamage. Autophagy. 2012;8:1333–1341.
- Robertson CA, Evans DH, Abrahamse H. Photodynamic therapy (PDT): A short review on cellular mechanisms and cancer research applications for PDT. J Photochem Photobiol B Biol. 2009;96:1–8.
- Kessel D, Reiners JJ. Enhanced efficacy of photodynamic therapy via a sequential targeting protocol. Photochem Photobiol. 2014;4:889–895.
- Tsubone TM, Martins WK, Pavani C, et al. Enhanced efficiency of cell death by lysosome-specific photodamage. Sci Rep. 2017;7:6734.
- Lemasters JJ. Variants of mitochondrial autophagy: types 1 and 2 mitophagy and micromitophagy (type 3). Redox Biol Elsevier. 2014;2:749–754.
- Kim I, Lemasters JJ. Mitophagy selectively degrades individual damaged mitochondria after photoirradiation. Antioxid Redox Signal. 2011;14:1919–1928.
- Rodriguez-Enriquez S, Kai Y, Maldonado E, et al. Roles of mitophagy and the mitochondrial permeability transition in remodeling of cultured rat hepatocytes. Autophagy. 2009;5:1099–1106.
- Suen D-F, Narendra DP, Tanaka A, et al. Parkin overexpression selects against a deleterious mtDNA mutation in heteroplasmic cybrid cells. Proc Natl Acad Sci. 2010;107:11835–11840.
- Narendra D, Tanaka A, Suen DF, et al. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol. 2008;183:795–803.
- Narendra DP, Jin SM, Tanaka A, et al. PINK1 is selectively stabilized on impaired mitochondria to activate parkin. Green DR, editor. . PLoS Biol. 2010;8:e1000298.
- Soubannier V, Rippstein P, Kaufman BA, et al. Reconstitution of mitochondria derived vesicle formation demonstrates selective enrichment of oxidized cargo. Shirihai OS, editor. PLoS One. 2012;7:e52830.
- Soubannier V, McLelland GL, Zunino R, et al. A vesicular transport pathway shuttles cargo from mitochondria to lysosomes. Curr Biol. 2012;22:135–141.
- Mclelland G, Soubannier V, Chen CX, et al. Parkin and PINK 1 function in a vesicular trafficking pathway regulating mitochondrial quality control. EMBO J. 2014;33:282–295.
- Casas A, Di Venosa G, Hasan T, et al. Mechanisms of resistance to photodynamic therapy. Curr Med Chem. 2011;18:2486–2515.
- Kessel D. Autophagic death probed by photodynamic therapy. Autophagy. 2015;11:1941–1943.
- Gollmer A, Felgenträger A, Bäumler W, et al. A novel set of symmetric methylene blue derivatives exhibits effective bacteria photokilling – a structure–response study. Photochem Photobiol Sci. 2015;14:335–351.
- Bacellar IOL, Pavani C, Sales EM, et al. Membrane damage efficiency of phenothiazinium photosensitizers. Photochem Photobiol. 2014;90:801–813.
- Walker I, Gorman SA, Cox RD, et al. A comparative analysis of phenothiazinium salts for the photosensitisation of murine fibrosarcoma (RIF-1) cells in vitro. Photochem Photobiol Sci. 2004;3:653–659.
- Ball DJ, Luo Y, Kessel D, et al. The induction of apoptosis by a positively charged methylene blue derivative. J Photochem Photobiol B. 1998;42:159–163.
- Mellish KJ, Cox RD, Vernon DI, et al. In vitro photodynamic activity of a series of methylene blue analogues. Photochem Photobiol. 2002;75:392–397.
- Wainwright M, Phoenix DA, Rice L, et al. Increased cytotoxicity and phototoxicity in the methylene blue series via chromophore methylation. J Photochem Photobiol B. 1997;40:233–239.
- Gabrielli D, Belisle E, Severino D, et al. Binding, aggregation and photochemical properties of methylene blue in mitochondrial suspensions. Photochem Photobiol. 2004;79:227–232.
- Daghastanli NA, Itri R, Baptista MS. Singlet oxygen reacts with 2ʹ,7ʹ-dichlorodihydrofluorescein and contributes to the formation of 2ʹ,7ʹ-dichlorofluorescein. Photochem Photobiol. 2008;84:1238–1243.
- Boya P, Kroemer G. Lysosomal membrane permeabilization in cell death. Oncogene. 2008;27:6434–6451.
- Yamamoto A, Kaji T, Tomoo K, et al. Crystallization and preliminary X-ray study of the cathepsin B complexed with CA074, a selective inhibitor. J Mol Biol. 1992;227:942–944.
- Kessel D, Evans CL. Promotion of proapoptotic signals by lysosomal photodamage: mechanistic aspects and influence of autophagy. Photochem Photobiol. 2016;92:620–623.
- Lim EJ, Oak C-H, Heo J, et al. Methylene blue-mediated photodynamic therapy enhances apoptosis in lung cancer cells. Oncol Rep. 2013;30:856–862.
- Martins WK, Severino D, Souza C, et al. Rapid screening of potential autophagic inductor agents using mammalian cell lines. Biotechnol J. 2013;8:730–737.
- Carey TE, Takahashi T, Resnick LA, et al. Cell surface antigens of human malignant melanoma: mixed hemadsorption assays for humoral immunity to cultured autologous melanoma cells. Proc Natl Acad Sci U S A. 1976;73:3278–3282.
- Scherer WF, Syverton JT, Gey GO. Studies on the propagation in vitro of poliomyelitis viruses. IV. Viral multiplication in a stable strain of human malignant epithelial cells (strain HeLa) derived from an epidermoid carcinoma of the cervix. J Exp Med. 1953;97:695–710.
- Andrzejak M, Price M, Kessel DH. Apoptotic and autophagic responses to photodynamic therapy in 1c1c7 murine hepatoma cells. Autophagy. 2011;7:979–984.
- Klionsky DJ, Abdelmohsen K, Abe A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. 2016;12:1–222. 3rd edition
- Aymard E, Barruche V, Naves T, et al. Autophagy in human keratinocytes: an early step of the differentiation? Exp Dermatol. 2011;20:263–268.
- Boukamp P, Petrussevska RT, Breitkreutz D, et al. Normal keratinization in a spontaneously immortalized aneuploid human keratinocyte cell line. J Cell Biol. 1988;106:761–771.
- Klionsky DJ, Abdalla FC, Abeliovich H, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. 2012:445–544.
- Martins WK, Ét C, Cruz MC, et al. Parallel damage in mitochondrial and lysosomal compartments promotes efficient cell death with autophagy: the case of the pentacyclic triterpenoids. Sci Rep. 2015;5:12425.
- Lu Y, Jiao R, Chen X, et al. Methylene blue-mediated photodynamic therapy induces mitochondria-dependent apoptosis in HeLa cell. J Cell Biochem. 2008;105:1451–1460.
- Chen Y, Zheng W, Li Y, et al. Apoptosis induced by methylene-blue-mediated photodynamic therapy in melanomas and the involvement of mitochondrial dysfunction revealed by proteomics. Cancer Sci. 2008;99:2019–2027.
- Guan J, Lai X, Wang X, et al. Photodynamic action of methylene blue in osteosarcoma cells in vitro. Photodiagnosis Photodyn Ther. 2014;11:13–19.
- Tardivo JP, Del Giglio A, De Oliveira CS, et al. Methylene blue in photodynamic therapy: from basic mechanisms to clinical applications. Photodiagnosis Photodyn Ther. 2005;2:175–191.
- Ma X, Piao S, Wang D, et al. Measurements of tumor cell autophagy preditc invasiveness resistance to chemotherapy, and survival in melanoma. Clin Cancer Res. 2011;17:3478–3489.
- Klose J, Stankov MV, Kleine M, et al. Inhibition of autophagic flux by salinomycin results in anti-cancer effect in hepatocellular carcinoma cells. PLoS One. 2014;9:e95970.
- Du H, Yang W, Chen L, et al. Role of autophagy in resistance to oxaliplatin in hepatocellular carcinoma cells. Oncol Rep. 2012;27:143–150.
- Zhai B, Hu F, Jiang X, et al. Inhibition of Akt reverses the acquired resistance to sorafenib by switching protective autophagy to autophagic cell death in hepatocellular carcinoma. Mol Cancer Ther. 2014;13:1589–1598.
- Xu N, Zhang J, Shen C, et al. Cisplatin-induced downregulation of miR-199a-5p increases drug resistance by activating autophagy in HCC cell. Biochem Biophys Res Commun. 2012;423:826–831.
- Georgakoudi I, Foster TH. Effects of the subcellular redistribution of two nile blue derivatives on photodynamic oxygen consumption. Photochem Photobiol. 1998;68:115–122.
- Martins WK, Gomide AB, Ét C, et al. Membrane damage by betulinic acid provides insights into cellular aging. Biochim Biophys Acta Gen Subj. 2017;1861:3129–3143.
- Kessel D, Reiners JJ. Effects of combined lysosomal and mitochondrial photodamage in a non-small-cell lung cancer cell line: the role of paraptosis. Photochem Photobiol. 2017;17:2045–2054.
- Spring BQ, Sears RB, Zheng LZ, et al. A photoactivable multi-inhibitor nanoliposome for tumour control and simultaneous inhibition of treatment escape pathways. Nat Nanotechnol. 2016;11:378–387.
- Pavani C, Francisco CML, Gobo NRS, et al. Improved photodynamic activity of a dual phthalocyanine–ALA photosensitiser. New J Chem. 2016;40:9666–9671.
- Garg AD, Maes H, Romano E, et al. Autophagy, a major adaptation pathway shaping cancer cell death and anticancer immunity responses following photodynamic therapy. Photochem Photobiol Sci. 2015;14:1410–1424.
- Martins RM, Amino R, Daghastanli KR, et al. A short proregion of trialysin, a pore-forming protein of Triatoma infestans salivary glands, controls activity by folding the N-terminal lytic motif. FEBS J. 2008;275:994–1002.
- Weinstein JN, Yoshikami S, Henkart P, et al. Liposome-cell interaction: transfer and intracellular release of a trapped fluorescent marker. Science. 1977;195:489–492.
- Stewart JC. Colorimetric determination of phospholipids with ammonium ferrothiocyanate. Anal Biochem. 1980;104:10–14.
- Wu EL, Cheng X, Jo S, et al. CHARMM-GUI Membrane Builder toward realistic biological membrane simulations. J Comput Chem. 2014;35:1997–2004.
- Jo S, Kim T, Iyer VG, et al. CHARMM-GUI: a web-based graphical user interface for CHARMM. J Comput Chem. 2008;29:1859–1865.
- Klauda JB, Brooks BR, MacKerell AD, et al. An ab initio study on the torsional surface of alkanes and its effect on molecular simulations of alkanes and a DPPC bilayer. J Phys Chem B. 2005;109:5300–5311.
- Hoover. Canonical dynamics: equilibrium phase-space distributions. Phys Rev A Gen Phys. 1985;31:1695–1697.
- Darden T, York D, Pedersen L. Particle mesh Ewald: an N ⋅log(N) method for Ewald sums in large systems. J Chem Phys. 1993;98:10089–10092.
- Phillips JC, Braun R, Wang W, et al. Scalable molecular dynamics with NAMD. J Comput Chem. 2005;26:1781–1802.
- Zoete V, Cuendet MA, Grosdidier A, et al. SwissParam: a fast force field generation tool for small organic molecules. J Comput Chem. 2011;32:2359–2368.
- Singh UC, Kollman PA. An approach to computing electrostatic charges for molecules. J Comput Chem. 1984;5:129–145.
- Besler BH, Merz KM, Kollman PA. Atomic charges derived from semiempirical methods. J Comput Chem. 1990;11:431–439.
- Frisch MJ, Trucks GW, Schlegel HB, et al., Gaussian 03, Revision A.1, Gaussian, Inc., Pittsburgh PA, 2003.
- Busch NA, Yarmush ML, Toner M, et al. Formalism for aggregation of peroxidized lipids and plasma membrane stability during photolysis. Biophys J. 1998;75:2956–2970.
- Braslavsky SE. Glossary of terms used in photochemistry, 3rd edition (IUPAC Recommendations 2006). Pure Appl Chem. 2007;79:293–465.
- Bolte S, Cordelières FP. A guided tour into subcellular colocalization analysis in light microscopy. J Microsc. 2006;224:213–232.
- Galluzzi L, Zamzami N, De la Motte Rouge T, et al. Methods for the assessment of mitochondrial membrane permeabilization in apoptosis. Apoptosis. 2007;12:803–813.
- Mukhopadhyay P, Rajesh M, Haskó G, et al. Simultaneous detection of apoptosis and mitochondrial superoxide production in live cells by flow cytometry and confocal microscopy. Nat Protoc. 2007;2:2295–2301.
- Kovalenko OA, Santos JH. Analysis of oxidative damage by gene-specific quantitative PCR. Curr Protoc Hum Genet. 2009;62:19.1.1–19.1.13.
- Ponti M, Forrow SM, Souhami RL, et al. Measurement of the sequence specificity of covalent DNA modification by antineoplastic agents using Taq DNA polymerase. Nucleic Acids Res. 1991;19:2929–2933.
- Santos JH, Mandavilli BS, Van Houten B. Measuring oxidative mtDNA damage and repair using quantitative PCR. Methods Mol Biol. 2002;197:159–176.