
ABSTRACT
MCOLN3/TRPML3 is a Ca2+-permeable cation channel that is expressed in multiple subcellular compartments with dynamic localization. Our previous studies suggest that upon macroautophagy/autophagy induction MCOLN3/TRPML3 is recruited and provides Ca2+ for the fusion process in autophagosome biogenesis. However, how intracellular trafficking and the Ca2+ channel function of MCOLN3/TRPML3 are related to autophagy are not known. Here we report that MCOLN3/TRPML3 undergoes palmitoylation at its C-terminal region, which is required for dynamic trafficking and cellular function of MCOLN3/TRPML3 in autophagy. Palmitoylation regulated MCOLN3/TRPML3 surface expression and trafficking, but not channel properties or localization and function of intracellular MCOLN3/TRPML3. Activation of intracellular MCOLN3/TRPML3 induced robust Ca2+ release, which solely increased autophagy in Ca2+- and palmitoylation-dependent manners. Palmitoylation regulated not only intracellular MCOLN3/TRPML3 trafficking to autophagic structures but also autophagic flux in induced autophagy. Importantly, nutrient starvation activated MCOLN3/TRPML3 to release Ca2+ and increased the level of MCOLN3/TRPML3 palmitoylation. Disruption of MCOLN3/TRPML3 palmitoylation, however, abolished the starvation-induced MCOLN3/TRPML3 activation without affecting channel activity. These results suggest that trafficking and channel function of MCOLN3/TRPML3 are regulated in the context of autophagy, and palmitoylation is a prerequisite for the function of MCOLN3/TRPML3 as a Ca2+ channel in autophagosome formation by controlling its trafficking between subcellular compartments.
Abbreviations: 17-ODYA, 17-octadecynoic acid; 2-BP, 2-bromopalmitate; BFA, brefeldin A; DN, dominant-negative; GPN, glycyl-L-phenylalanine-beta-naphthylamide; HN, hydroxylamine; KD, knockdown; MCOLN3/TRPML3, mucolipin 3; MS, mass spectrometry; PAT, palmitoyl acyltransferase; PM, plasma membrane; WT, wild type; ZDHHC, a zinc-finger motif and an Asp-His-His-Cys sequence
Introduction
MCOLN3/TRPML3 (mucolipin 3) is a Ca2+ permeable non-selective cation channel [Citation1,Citation2] that is expressed in the endocytic and autophagic pathways [Citation3,Citation4]. The cellular function of MCOLN3/TRPML3 involves the regulation of endocytosis, membrane trafficking, and autophagy [Citation3]. In a previous study, we showed that overexpression of MCOLN3/TRPML3 leads to reduced endocytosis and increased autophagy, whereas both knockdown (KD) and expression of a dominant-negative (DN) had a reciprocal effect. The subcellular localization of MCOLN3/TRPML3 during these cellular events is dynamic, in that MCOLN3/TRPML3 accumulated in the plasma membrane (PM) upon inhibition of endocytosis, and is massively recruited to autophagosomes upon induction of autophagy [Citation3]. In another study, we found that MCOLN3/TRPML3 interacts with GABARAPL2/GATE-16 which is implicated in autophagosome biogenesis, and the interaction is enhanced in induced autophagy [Citation5]. Thus, it is suggested that MCOLN3/TRPML3 dynamically moves to subcellular sites for autophagosome formation where it may provide Ca2+ for the fusion process [Citation3,Citation6]. However, the underlying mechanism by which MCOLN3/TRPML3 shuttles dynamically between intracellular compartments and whether vesicular Ca2+ efflux via MCOLN3/TRPML3 truly contributes to the autophagy process are largely unknown.
Palmitoylation is a post-translational modification consisting of the attachment of the 16-carbon fatty acid palmitate to cysteine residues via a hydroxylamine (HN)-sensitive thioester bond [Citation7–Citation9]. This lipid modification increases protein hydrophobicity and thus affects protein structure and the affinity of proteins for cellular membranes or membrane domains [Citation9,Citation10]. Among lipid modifications, palmitoylation is unique because it is reversible and dynamically regulated by specific extracellular signals [Citation7,Citation9,Citation11]. The reversibility of protein palmitoylation allows proteins to rapidly shuttle between intracellular membrane compartments [Citation8,Citation11]. For this reason, palmitoylation has been recognized as an important mechanism for subcellular protein trafficking [Citation12].
Although transmembrane proteins do not require palmitoylation for stable membrane binding, palmitoylation can also affect their structure, assembly, maturation, trafficking, and function [Citation10,Citation13]. In addition, evidence suggests that protein palmitoylation controls many stages in the life cycle and physiological function of ion channels [Citation14,Citation15]. Palmitoylation is thought to control the 2 fundamental determinants of ion channel function: (1) the number of channels resident in a membrane and (2) the activity of the channel at the membrane [Citation14]. It controls the former by regulating channel trafficking and the latter by controlling channel kinetics and modulation by other post-translational modifications. Currently, more than 50 distinct ion channel subunits have been experimentally demonstrated to be palmitoylated [Citation10,Citation14].
Here we describe that MCOLN3/TRPML3 is a palmitoylated protein, and palmitoylation controls MCOLN3/TRPML3 trafficking and cellular function in autophagy. Moreover, we provide evidence that Ca2+ channel activity and trafficking of intracellular MCOLN3/TRPML3 are regulated in the context of autophagy, and Ca2+ release via MCOLN3/TRPML3 truly contributes to the autophagy process in a palmitoylation-dependent manner.
Results
MCOLN3/TRPML3 is palmitoylated on the C-terminal tail
We used the CSS-Palm v2.0 palmitoylation prediction algorithm [Citation16] to investigate whether and where MCOLN3/TRPML3 is palmitoylated. As shown in ), MCOLN3/TRPML3 is strongly predicted to be palmitoylated at the N-terminal Cys11 and C-terminal Cys549, Cys550 and Cys551 amino acid residues. Sequence alignments of these N- and C-terminal cysteine residues across different species and among the TRPML subfamily members indicate that the C-terminal cysteine residues are evolutionarily conserved ()). The C-terminal cysteine residues of TRPML1, which align with those of MCOLN3/TRPML3, were already experimentally confirmed to be palmitoylated [Citation17,Citation18], increasing the possibility that the predicted C-terminal residues of MCOLN3/TRPML3 are palmitoylation sites as well.
Figure 1. Palmitoylation sites and PATs of MCOLN3/TRPML3. (a) Schematic of MCOLN3/TRPML3 showing palmitoylation sites (gray and red circles) predicted by CSS-Palm v2.0 and sequence alignments of the indicated cysteine residues across different species and among MCOLN/TRPML subfamily members. (b) MCOLN3/TRPML3 palmitoylation assay. HEK293T cells expressing GFP-tagged MCOLN3/TRPML3 (upper panel) or HEK293T and SW13 cells (lower panel) were either treated with DMSO (control) or metabolically labeled with 100 μM 17-ODYA. After 6 h, membrane fractions were prepared followed by reaction between biotin-azide and 17-ODYA using click chemistry. Biotinylated proteins were affinity isolated using streptavidin beads and eluates were analyzed by western blot using anti-GFP antibody or anti-MCOLN3/TRPML3 antibody to detect palmitoylated proteins. Tris or HN were added to the reaction mixture and incubated for 1 h at RT before adding the beads. Input samples were 1% of protein lysate used in the click chemistry reactions. (c) MCOLN3/TRPML3 and cysteine mutant palmitoylation assays. HEK293T cells expressing GFP-tagged MCOLN3/TRPML3 and the indicated single or triple cysteine mutants were assayed as in (b). (d) Quantified data of ratios of palmitoylated MCOLN3/TRPML3s to total MCOLN3/TRPML3 protein levels (n = 3, *p < 0.05, ***p < 0.005 compared to WT, Student’s t-test). Data are presented as the mean ± SEM. (e) HEK293T cells treated with the indicated siRNAs targeting human ZDHHCs for 72 h were transfected with pEGFPC1-MCOLN3/TRPML3 for 24 h to be assayed as in (b). NC indicates negative control siRNA.
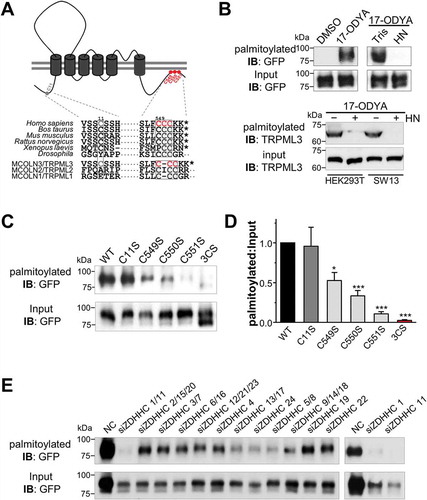
To directly assess whether MCOLN3/TRPML3 is a palmitoylated protein, we performed a recently developed palmitoylation assay [Citation19]. HEK293T cells expressing GFP-tagged wild-type (WT)-MCOLN3/TRPML3 were either treated with DMSO or metabolically labeled with 17-octadecynoic acid (17-ODYA), an alkyne-containing palmitate analog. The samples were then reacted with biotin-azide using click chemistry to biotinylate the 17-ODYA incorporated proteins. Biotinylated proteins were isolated with streptavidin beads and palmitoylation was detected by western blots using anti-GFP antibody. As shown in ), upper panel, MCOLN3/TRPML3 is robustly palmitoylated in 17-ODYA-labeled but not in DMSO-treated samples. To confirm that 17-ODYA is linked via a thioester bond and not via an amide linkage, 17-ODYA-incorporated and biotinylated proteins were treated with HN or with Tris as a control. Palmitoylation of MCOLN3/TRPML3 was completely abolished by HN treatment, indicating that it occurs exclusively on cysteine residues. Importantly, we also proved that the endogenous MCOLN3/TRPML3 is palmitoylated using anti-MCOLN3/TRPML3 antibody in HEK293T and SW13 cells (), lower panel). Antibody validation and expression of endogenous MCOLN3/TRPML3 in various cell lines are shown in Figures S1A-C.
Next, to determine MCOLN3/TRPML3 palmitoylation sites, we generated single (C11S, C549S, C550S and C551S) or C-terminal triple (C549, 550, 551S; designated as 3CS) site-directed cysteine-to-serine mutants and performed the same assay. The N-terminal MCOLN3C11S mutant showed similar palmitoylation levels to WT MCOLN3/TRPML3, suggesting that C11 is not a palmitoylation site in MCOLN3/TRPML3. Individual C-terminal single mutants showed reduced levels of palmitoylation but none of the single site mutants completely abolished MCOLN3/TRPML3 palmitoylation. Serine replacements at all C-terminal cysteine residues almost completely blocked palmitoylation of MCOLN3/TRPML3 ()). To confirm that these cysteine residues are indeed modified, we employed mass spectrometry (MS) analysis and identified C550 and C551 of MCOLN3/TRPML3 with high and low confidence, respectively, as palmitoylation sites (Figure S1E). We next performed an siRNA screen to identify the candidate enzymes among 23 palmitoyl acyltransferases (PATs) sharing a zinc-finger motif and an Asp-His-His-Cys sequence (ZDHHC). We verified KD efficacy by real-time RT-PCR (Fig. S1D), knocked down 12 sets of the 23 ZDHHC PATs as defined by phylogenetic tree analysis, and applied the same palmitoylation assay as in ). As a result, we found that KD of ZDHHC1/11 greatly reduced palmitoylation of MCOLN3/TRPML3 and individual KD of ZDHHC1 or ZDHHC11 also had a similar effect ()). Taken together, all these data demonstrate that MCOLN3/TRPML3 undergoes palmitoylation at its C-terminal end.
Palmitoylation controls cell surface expression of MCOLN3/TRPML3
In many ion channels, palmitoylation plays a significant role in controlling cell surface expression [Citation14,Citation20–Citation24]; therefore, we investigated if this is the case in MCOLN3/TRPML3. ) shows that MCOLN3/TRPML3(C11S) demonstrated normal surface expression, whereas MCOLN3/TRPML3(3CS) had markedly reduced surface expression compared to WT MCOLN3/TRPML3 in HEK293T and HeLa cells. We also observed a reduced surface level of MCOLN3/TRPML3 in cells treated with 2-bromopalmitate (2-BP), a potent palmitoylation inhibitor (), middle panel, and )). Because abolition of palmitoylation did not completely block the surface expression of MCOLN3/TRPML3, we next determined whole-cell current levels in cells expressing WT MCOLN3/TRPML3 and palmitoylation mutants. As was shown previously [Citation2,Citation25], alternative exposure of cells expressing MCOLN3/TRPML3 to Na+-free and Na+-containing solution resulted in a large inward rectifying current ()). Consistent with surface levels, MCOLN3/TRPML3(C11S) demonstrated no effect whereas MCOLN3/TRPML3(3CS) showed reduced current density at about 65% of WT MCOLN3/TRPML3 (). To fully activate MCOLN3/TRPML3 in another way, MCOLN3/TRPML3 current was elicited by SF-21, a small-molecule MCOLN/TRPML agonist [Citation26]. As shown in ), applying SF-21 to the cells induced a large inward current and consistently, the current density from MCOLN3/TRPML3(3CS) was only 66% of that from WT MCOLN3/TRPML3 ()). Importantly, the current-voltage (I/V) relationships in ) and () show that the 3CS mutation in MCOLN3/TRPML3 did not alter the channel properties. This indicates that the reduced current density from MCOLN3/TRPML3(3CS) reflects lower surface protein level and not changes in channel gating. We further measured PM Ca2+ influx in cells expressing WT MCOLN3/TRPML3 or palmitoylation mutants by increasing the Ca2+ concentration in the external solution ()). The amount of Ca2+ influx via MCOLN3/TRPML3(3CS) also decreased to about 65% of the WT MCOLN3/TRPML3 control. All these results indicate that C-terminal palmitoylation of MCOLN3/TRPML3 plays an important role in its surface expression.
Figure 2. Effect of palmitoylation on MCOLN3/TRPML3 surface expression. (a) Analysis of surface expression by biotinylation. HEK293T cells expressing WT MCOLN3/TRPML3, MCOLN3/TRPML3(C11S), or MCOLN3/TRPML3(3CS) treated with ethanol (control) or 2-BP, and HeLa cells expressing WT MCOLN3/TRPML3 or MCOLN3/TRPML3(3CS) were used for surface biotinylation assays. Total and surface proteins were analyzed by western blot using anti-GFP antibody. The data are representative of 3 independent experiments. (b) Densitometric analysis of surface MCOLN3/TRPML3s to total MCOLN3/TRPML3 protein levels (n = 3, *p < 0.05, ***p < 0.005 compared to WT, Student’s t-test) of western blots from HEK293T cells similar to those in (a). Data are presented as the mean ± SEM. (c) Whole cell current was measured in HEK293T cells expressing WT MCOLN3/TRPML3, MCOLN3/TRPML3(C11S), or MCOLN3/TRPML3(3CS). The MCOLN3/TRPML3 current was activated by sequentially exposing the cells to bath solutions containing 0 and 140 mM Na+. Current was measured by applying 100 ms ramps from −100 to + 100 mV and plotted at −100 mV. Dashed lines here and in (f) indicate the 0 current levels. (d) I/V relationships of the MCOLN3/TRPML3 currents recorded at the times shown in the filled circles in panel (c). (e) The current amplitudes of WT MCOLN3/TRPML3, MCOLN3/TRPML3(C11S), and MCOLN3/TRPML3(3CS) are plotted as the mean ± SEM of 10–13 cells (*p < 0.05, Student’s t-test). (f) The whole cell current was measured in HEK293T cells expressing WT MCOLN3/TRPML3 or MCOLN3/TRPML3(3CS) by applying 5 µM SF21 in a 140 mM Na+ bath solution. (g) I/V relationships of the MCOLN3/TRPML3 current recorded at the times shown in the filled circles in panel (f). (h) The current amplitudes of WT MCOLN3/TRPML3 and MCOLN3/TRPML3(3CS) are plotted as the mean ± SEM of 10 cells each (**p < 0.01, Student’s t-test).
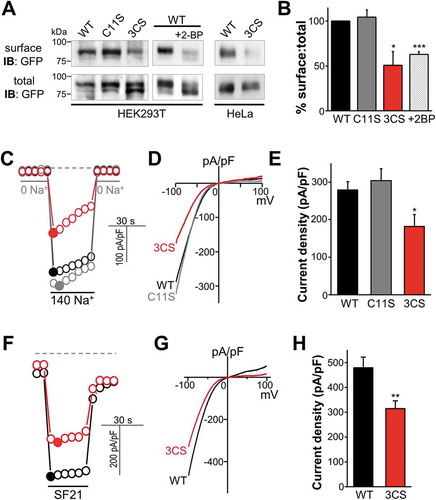
Figure 3. Effects of palmitoylation on MCOLN3/TRPML3 trafficking. (a) Schematic for MCOLN3/TRPML3 trafficking to and from the PM. (b and d) HEK293T cells expressing WT MCOLN3/TRPML3 or MCOLN3/TRPML3(3CS) were treated with either 20 µM BFA (b) for 4 h or 100 µM Dynasore (d) for 2 h at 37°C. MCOLN3/TRPML3 currents were measured by applying 5 µM SF21 in 140 mM Na+ bath solution. I/V relationships of the MCOLN3/TRPML3 current recorded from cells treated with the indicated compound (black and red traces) at the indicated time points are shown. (c and e) The current amplitudes and relative current density of WT MCOLN3/TRPML3 (black circle) and MCOLN3/TRPML3(3CS) (red circle) in experiments similar to those in panel (b and d) were plotted as the mean ± SEM of 10–21 cells (*p < 0.05, ***p < 0.005, Student’s t-test). (f) HeLa cells transfected with mCherry-MCOLN3/TRPML3 or mCherry-MCOLN3/TRPML3(3CS) were incubated with Alexa Fluor 488-EGF or Alexa Fluor 488-TF for 10 min, fixed, and imaged by confocal microscopy. Scale bars here and in all other confocal images: 20 µm. (g) ImageJ was used to count the number of puncta in 14–16 cells from 2–3 experiments and the number of particles is plotted as the mean ± SEM at the indicated conditions (***p < 0.005, Student’s t-test). Con indicates the empty vector control.
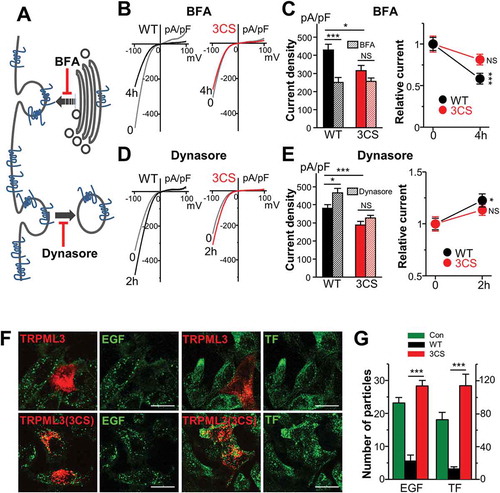
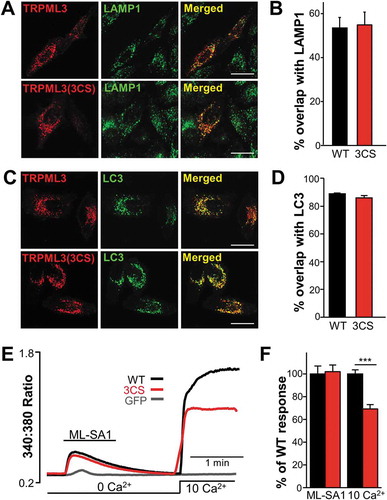
Figure 4. Organellar localization and Ca2+ release by WT MCOLN3/TRPML3 and MCOLN3/TRPML3(3CS). (a) HeLa cells transfected with plasmids encoding mCherry-MCOLN3/TRPML3 or mCherry-MCOLN3/TRPML3(3CS) were stained for LAMP1 (green) and imaged by confocal microscopy. (b) Images similar to those in panel (a) from 5–9 cells were used to determine the overlap with ImageJ. The results are given as the mean ± SEM. (c) HeLa cells transfected with a plasmid encoding mCherry-MCOLN3/TRPML3 or mCherry-MCOLN3/TRPML3(3CS) and GFP-LC3 were imaged by confocal microscopy. (d) Images similar to those in panel (c) from 7 cells were used to determine the overlap with ImageJ. The results are given as the mean ± SEM. (e) HEK293T cells expressing GFP (gray trace), WT MCOLN3/TRPML3 (black trace) or MCOLN3/TRPML3(3CS) (red trace) were preincubated in Ca2+-free medium to block PM Ca2+ influx. Organellar Ca2+ efflux was measured by applying 20 µM ML-SA1 in Ca2+-free medium and then, Ca2+ influx from the PM was measured by increasing Ca2+o to 10 mM. (f) Cellular responses to ML-SA1 and 10 mM Ca2+o were compared in cells transfected with WT MCOLN3/TRPML3 and MCOLN3/TRPML3(3CS). Results are plotted as the mean ± SEM of 28–29 cells (***p < 0.005, Student’s t-test).
Palmitoylation controls MCOLN3/TRPML3 trafficking between intracellular compartments, affecting MCOLN3/TRPML3 function in endocytosis
The surface level of a protein is determined by the balance between endocytosis from and delivery to the PM. To understand the underlying mechanism for lower MCOLN3/TRPML3 surface expression by palmitoylation inhibition, we investigated MCOLN3/TRPML3 trafficking at the PM. The rate of MCOLN3/TRPML3 endocytosis was measured using brefeldin A (BFA), which inhibits protein transport to the PM ()). HEK293T cells expressing either WT MCOLN3/TRPML3 or MCOLN3/TRPML3(3CS) were incubated with 20 μM BFA and current was recorded by applying SF-21 as in ). Surprisingly, current from WT MCOLN3/TRPML3, and not from MCOLN3/TRPML3(3CS), was reduced by 45% after BFA treatment compared to non-treated controls ()), indicating that endocytosis of MCOLN3/TRPML3 was altered by the palmitoylation-site mutation. Next, we examined MCOLN3/TRPML3 delivery to the PM using Dynasore, a cell-permeable inhibitor of DNM1 (dynamin 1) ()) [Citation27]. By blocking DNM1-dependent endocytosis, MCOLN3/TRPML3 current was significantly increased by 25% relative to a non-treated control in WT MCOLN3/TRPML3-expressing cells, but not in cells expressing MCOLN3/TRPML3(3CS) ()). This result is consistent with our previous findings showing accumulation of MCOLN3/TRPML3 in the PM upon inhibition of endocytosis by DN DNM1K44A [Citation3]. Together, these data indicate that MCOLN3/TRPML3 trafficking is regulated by palmitoylation.
We previously demonstrated that overexpression of MCOLN3/TRPML3 decreases, while KD increases, the rate of endocytosis [Citation3]. However, how MCOLN3/TRPML3 controls the rate of endocytosis is not known. Because palmitoylation has been implicated in many membrane trafficking processes [Citation8,Citation9], we examined whether the effect of MCOLN3/TRPML3 on endocytosis is also palmitoylation-dependent. HeLa cells expressing WT MCOLN3/TRPML3 or MCOLN3/TRPML3(3CS) were incubated with Alexa Fluor 488-tagged TF (transferrin) or EGF for 10 min and the uptake was quantified by particle counting. The images in ) and the summary in ) show that in both EGF and TF uptake, WT MCOLN3/TRPML3 reduced the rate of endocytosis as previously reported [Citation3,Citation4], whereas MCOLN3/TRPML3(3CS) reversed it to control levels or higher. These data combined with the results in suggest that the proper trafficking of MCOLN3/TRPML3 by palmitoylation is required for the function of MCOLN3/TRPML3 in membrane trafficking. Furthermore, these results imply that MCOLN3/TRPML3 palmitoylation may also play a role in autophagy, as the role of MCOLN3/TRPML3 in endocytosis is likely linked to its role in autophagy [Citation3]. Our previous study suggested that overexpression of MCOLN3/TRPML3 augments the autophagic response to result in accumulation of cellular materials in the autophagosomes and lysosomes, causing a feedback response to slow endocytosis [Citation3].
Localization and channel function of intracellular MCOLN3/TRPML3 are not affected by palmitoylation
The results in and show that localization and trafficking of MCOLN3/TRPML3 at the PM are controlled by palmitoylation. However, MCOLN3/TRPML3 is expressed not only in the PM but also in intracellular vesicular compartments, mainly in late endosomes and lysosomes and in LC3-positive autophagosomes [Citation3]. Thus, we investigated whether intracellular localization of MCOLN3/TRPML3 is also influenced by palmitoylation. To do this, HeLa cells expressing WT MCOLN3/TRPML3 or MCOLN3/TRPML3(3CS) were stained with LAMP1, a well-defined late endosome and lysosomal marker, and colocalization of MCOLN3/TRPML3 with LAMP1 was quantified from confocal images. As shown in ) and in previous studies [Citation3,Citation4], approximately 50% of WT MCOLN3/TRPML3 was colocalized with LAMP1, which was similar for MCOLN3/TRPML3(3CS) ()). In addition, overlap between MCOLN3/TRPML3 and LC3 that was nearly 90% was not altered by the 3CS mutation ()). Thus, localization of intracellular MCOLN3/TRPML3 appears not to be affected by palmitoylation.
To better characterize organelle-residing MCOLN3/TRPML3, we determined organellar Ca2+ efflux through MCOLN3/TRPML3 using a membrane-permeable TRPML agonist, ML-SA1 [Citation28]. HEK293T cells expressing WT MCOLN3/TRPML3 or MCOLN3/TRPML3(3CS) were incubated in Ca2+-free external solution to block Ca2+ influx from MCOLN3/TRPML3 localized in the PM. In this condition, ML-SA1 induced robust Ca2+ release from intracellular compartments via both WT and palmitoylation mutant MCOLN3/TRPML3 to a similar extent ()). This is in sharp contrast to results in the PM ( and )) showing reduced surface expression and the subsequent decreased activity of MCOLN3/TRPML3 by inhibition of palmitoylation. Thus, we conclude that the localization and the channel function of intracellular MCOLN3/TRPML3 are palmitoylation-independent.
Intracellular MCOLN3/TRPML3 activation solely can increase autophagy in Ca2+- and palmitoylation-dependent manners
Our previous studies [Citation3,Citation5] suggest that intracellular Ca2+ release through MCOLN3/TRPML3 may be involved in autophagosome formation. As we obtained robust Ca2+ efflux via MCOLN3/TRPML3 by agonist stimulation (), we examined the effect of intracellular MCOLN3/TRPML3 activation on autophagy and whether palmitoylation is involved in the process. To test this, HeLa cells transfected with a plasmid expressing LC3 only or co-transfected with WT MCOLN3/TRPML3 were treated with ML-SA1, and LC3 particles were counted. In this experiment, Ca2+ was removed from the medium to determine the effect of intracellular MCOLN3/TRPML3, not from the PM-localized MCOLN3/TRPML3. Because we observed ML-SA1-evoked small Ca2+ efflux in control cells (), gray line) most likely due to endogenous TRPML activation [Citation28], we first examined the effect of ML-SA1 on autophagy in control cells. As shown in and ), application of ML-SA1 to control cells induced a mild increase in autophagy, suggesting that organelle-residing endogenous TRPMLs participate in the autophagy process. Notably, expression of MCOLN3/TRPML3 resulted in marked autophagy and the addition of ML-SA1 induced a dramatic increase in the number of autophagosomes ( and )). This result indicates that autophagy can be induced solely by the activation of intracellular MCOLN3/TRPML3 regardless of autophagy signaling activation.
Figure 5. Differential effect of organellar Ca2+ efflux via WT MCOLN3/TRPML3 and MCOLN3/TRPML3(3CS) on autophagy. (a and b) HeLa cells transfected with a plasmid encoding GFP-LC3 only (a) or co-transfected with a plasmid encoding mCherry-MCOLN3/TRPML3 (b) were treated with vehicle or 20 µM ML-SA1 in Ca2+-free medium for 1 h at 37°C. (c) HEK293T cells transfected with a plasmid encoding WT MCOLN3/TRPML3 were preincubated with 10 µM BAPTA-AM for 30 min and intracellular Ca2+ was measured as in . (d) HeLa cells expressing GFP-LC3 and mCherry-MCOLN3/TRPML3 were preincubated with 10 µM BAPTA-AM in Ca2+-free medium for 30 min and treated as in panel (b). (e) Ca2+ was measured from HEK293T cells transfected with WT MCOLN3/TRPML3 by applying 500 µM GPN and then 20 µM ML-SA1. (f) HeLa cells expressing GFP-LC3 and mCherry-MCOLN3/TRPML3 were preincubated with 500 µM GPN and 10 µM CPA in Ca2+-free medium for 30 min and treated as in panel (b). (g) HeLa cells transfected with a plasmid encoding GFP-LC3 and mCherry-MCOLN3/TRPML3(3CS) were treated as in panel (b). (h) The normalized number of GFP-LC3 puncta in panel (a, b, d, f, g) was counted using ImageJ and given as the mean ± SEM of 23–33 cells for panel (a, b, g) and that of 4–14 cells for panel (D and F) (***p < 0.005, Student’s t-test).
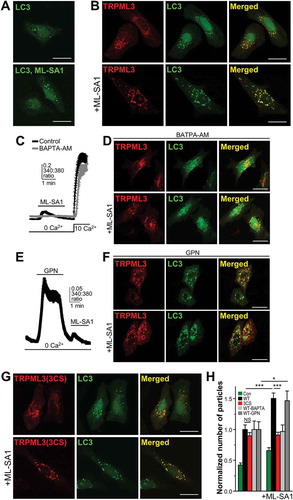
To determine whether the Ca2+ release through MCOLN3/TRPML3 activation in ) contributes to the increase of autophagy in ), we depleted the intracellular Ca2+ store using BAPTA-AM. As shown in ), pretreatment with BAPTA-AM for 30 min completely abolished ML-SA1-evoked Ca2+ release but did not affect PM Ca2+ influx. Importantly, autophagy increase by ML-SA1 was not observed in cells pretreated with BAPTA-AM, suggesting that Ca2+ release via intracellular MCOLN3/TRPML3 activation contributes to autophagy.
To further dissect the source of ML-SA1-induced Ca2+ release, we first used glycyl-L-phenylalanine-beta-naphthylamide (GPN) and then treated cells with ML-SA1. ) shows that ML-SA1-induced Ca2+ release remained even after depletion of the acidic Ca2+ store by GPN, suggesting that some MCOLN3/TRPML3 exists in non-acidic organelles. In this condition, ML-SA1 treatment still increased autophagy just as in ), increasing the possibility that MCOLN3/TRPML3-mediated Ca2+ release from a non-acidic Ca2+ store is indeed responsible for the increase of autophagy. ) shows that MCOLN3/TRPML3(3CS) elicited a similar level of increased autophagy compared to WT MCOLN3/TRPML3; however, ML-SA1 treatment led to no further increase of autophagy (), lower panel), despite similar amounts of Ca2+ release ()). These findings imply that a non-palmitoylable mutant may not traffic to sites for autophagosomes formation upon induction of autophagy by ML-SA1, thereby not exacerbating autophagy. Thus, the differential effect of WT MCOLN3/TRPML3 and a palmitoylation mutant on autophagy may be able to be observed only when massive trafficking of MCOLN3/TRPML3 is necessary, as reflected in induced autophagy by ML-SA1 ()).
Palmitoylation regulates autophagy-dependent MCOLN3/TRPML3 trafficking and function
To further investigate the effect of MCOLN3/TRPML3 palmitoylation on autophagy, we induced autophagy or inhibited MCOLN3/TRPML3 palmitoylation in different ways. First, starvation was employed to induce autophagy in cells expressing WT MCOLN3/TRPML3 or MCOLN3/TRPML3(3CS). As previously shown, overexpression of WT MCOLN3/TRPML3 caused a significant increase of autophagy in fed cells, and starvation of these cells resulted in a marked increase in the number of autophagosomes (), upper panels and ()). MCOLN3/TRPML3(3CS) increased autophagy to a similar extent with WT MCOLN3/TRPML3 in fed conditions (), lower panel); however, starvation of cells expressing MCOLN3/TRPML3(3CS) caused no further increase of autophagy (), lower panel and )). These results were once again confirmed in experiments using the ER stressor, CPA as an autophagy inducer ()). To inhibit MCOLN3/TRPML3 palmitoylation in another way, we made a C-terminal truncation mutant, MCOLN3/TRPML3(Δ5) that lacks the last 5 amino acids including the palmitoylation sites for MCOLN3/TRPML3 (), upper panel). As shown in ), the effect of the deletion on starvation-induced autophagy was also similarly observed, confirming that the above results are palmitoylation specific. Therefore, these findings together with previous results suggest that palmitoylation enables MCOLN3/TRPML3 translocation, which can further stimulate autophagy.
Figure 6. Effect of palmitoylation on MCOLN3/TRPML3 trafficking in the context of autophagy. (A-C) HeLa cells transfected with a plasmid encoding GFP-LC3 only (a) or co-transfected with a plasmid encoding mCherry-MCOLN3/TRPML3 or mCherry-MCOLN3/TRPML3(3CS) were kept in fed medium (b) or serum starved for 2 h (c). (d and e) The number of GFP-LC3 puncta in serum-starved cells (d) or in cells treated with 25 µM CPA for 3 h (e) was counted using ImageJ and given as the mean ± SEM of 12–28 cells (*p < 0.05, ***p < 0.005, Student’s t-test). (f) The upper panel shows the C-terminal amino acid sequence of WT MCOLN3/TRPML3 and MCOLN3/TRPML3(Δ5), where Δ5 indicates truncation of the last 5 amino acids of MCOLN3/TRPML3 including the palmitoylation sites. The number of GFP-LC3 puncta in cells co-transfected with WT MCOLN3/TRPML3 or MCOLN3/TRPML3(Δ5) was counted and given as the mean ± SEM of 11–14 cells (*p < 0.05, Student’s t-test). (g) HEK293T cells expressing GFP-tagged WT, 3CS or D458K MCOLN3/TRPML3 with FLAG-GABARAPL2 were kept in full medium or serum-starved for 2 h and immunoprecipitated samples with anti-GFP antibody were subjected to western blotting with anti-FLAG antibody. The inputs, corresponding to 5% of the amount used for each immunoprecipitation, were blotted with anti-FLAG antibody (middle panel) and anti-GFP antibody (lower panel). (h) Cell lysates in panel (g) were used for western blot analysis to assay endogenous LC3 levels. ACTB/β-actin was used as a loading control.
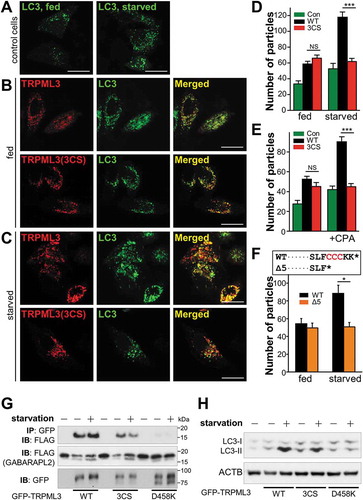
To examine whether MCOLN3/TRPML3 is indeed translocated upon induction of autophagy, we took advantage of the previously reported MCOLN3/TRPML3-GABARAPL2 relationship. Our previous study showed that MCOLN3/TRPML3-GABARAPL2 overlap was low in fed conditions but increased in starved conditions in the confocal images [Citation5]. Moreover, their interaction was enhanced upon starvation, indicating that MCOLN3/TRPML3 dynamically moves to GABARAPL2-residing compartments upon induction of autophagy [Citation5]. Thus, we investigated the effects of palmitoylation and Ca2+ channel function of MCOLN3/TRPML3 on MCOLN3/TRPML3-GABARAPL2 interaction and autophagy using palmitoylation-disabled and channel-dead mutants. ) clearly shows that the enhanced MCOLN3/TRPML3-GABARAPL2 interaction induced by starvation in WT MCOLN3/TRPML3 expressing cells disappeared in MCOLN3/TRPML3(3CS)-expressing cells, suggesting that the palmitoylation mutant was unable to traffic to autophagic structures. In addition, the interaction was completely abolished in DN channel-dead MCOLN3D458K transfected cells, indicating that MCOLN3/TRPML3 channel activity is essential for the interaction and autophagy. LC3-II levels in the same set of experiments also proved that only WT MCOLN3/TRPML3 dramatically increased autophagy upon starvation, but MCOLN3/TRPML3(3CS) and MCOLN3D458K had a less or minimal effect ()). Therefore, these data suggest that palmitoylation regulates MCOLN3/TRPML3 trafficking on demand upon autophagy stimulation, and channel activity of MCOLN3/TRPML3 is crucial to autophagy.
Both channel activity and palmitoylation of MCOLN3/TRPML3 have an impact on autophagy and vice versa
Next, to determine the effect of MCOLN3/TRPML3 channel function and trafficking on autophagy, we performed an autophagic flux assay using the vacuolar-type H+-ATPase inhibitor bafilomycin A1, or a combination of lysosomal protease inhibitors E64d and pepstatin A. First, MCOLN3/TRPML3 expression or channel activity was inhibited by siRNA ()), a cell-permeable antagonist, ML-SI1 ()), or a DN MCOLN3D458K, ()) and autophagic flux was observed. Inhibition of MCOLN3/TRPML3 only slightly increased the LC3-II level but significantly decreased autophagic flux, indicating that MCOLN3/TRPML3 mainly contributes to autophagosome formation rather than degradation.
Figure 7. Effect of channel function and palmitoylation of MCOLN3/TRPML3 on autophagy and vice versa. (a) HeLa cells transfected with either negative control siRNA (-) or siRNA targeting MCOLN3/TRPML3 (+) for 72 h were treated with 100 nM bafilomycin A1 in HBSS for 4 h or with 10 µg/ml E64d and 10 µg/ml pepstatin A in serum-free medium for 11 h and processed for western blot analysis to assay endogenous LC3 levels. (b) HeLa cells transfected with negative control siRNA in panel (a) were treated with 100 nM bafilomycin A1 in HBSS for 4 h in the presence and absence of 10 µM ML-SI1 and assayed as in panel (a). (c) HEK293T cells expressing control vector (-) or MCOLN3D458K (+) were treated with 100 nM bafilomycin A1 in HBSS for 4 h and assayed as in panel (a). (d) HeLa cells transfected with a plasmid encoding mCherry-tagged WT MCOLN3/TRPML3, MCOLN3/TRPML3(3CS), or MCOLN3/TRPML3(Δ5) were treated with DMSO or 100 nM bafilomycin A1 in PBS for 3 h and processed for western blot analysis to assay endogenous LC3 levels. The result is representative of 3 independent experiments. (e) Autophagic flux was calculated from the change in normalized LC3-II levels to ACTB/β-actin upon bafilomycin A1 treatment in (d). Values are relative to WT MCOLN3/TRPML3 and are plotted as the mean ± SEM from 3 independent experiments (***p < 0.005, Student’s t-test). (f) Intracellular Ca2+ was measured in HEK293T cells expressing WT MCOLN3/TRPML3, MCOLN3/TRPML3(3CS), or MCOLN3D458K while the cells were kept in fed medium and then exposed to HBSS. Organellar Ca2+ efflux was measured by applying 20 µM ML-SA1. (g) The increase of 340:380 ratio in panel (f) was presented as the mean ± SEM of 6–34 cells (***p < 0.005, Student’s t-test). (h) HEK293T cells expressing GFP-tagged WT MCOLN3/TRPML3 or MCOLN3/TRPML3(3CS) were kept in fed medium or serum-starved during treatment with 100 μM 17-ODYA for 6–8 h and assayed as in ). (i) Quantified data of ratios of palmitoylated MCOLN3/TRPML3 to total MCOLN3/TRPML3 protein levels in fed or starved condition are presented as the mean ± SEM (n = 3, *p < 0.05, Student’s t-test). (j) HEK293T cells were treated as in panel (h) and analyzed by western blot using anti-MCOLN3/TRPML3 antibody to detect endogenous palmitoylated MCOLN3/TRPML3.
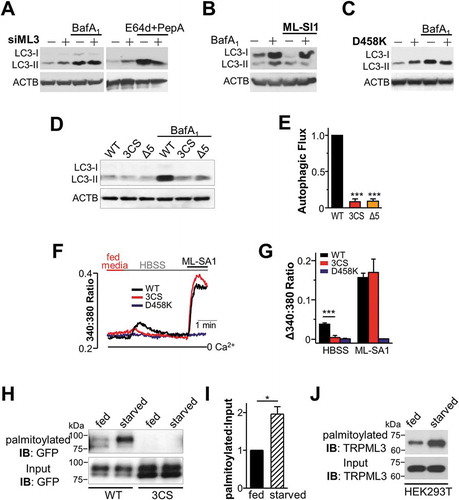
Second, to inhibit MCOLN3/TRPML3 trafficking, 2 forementioned palmitoylation mutants were used. Autophagic flux was dramatically decreased in MCOLN3/TRPML3(3CS)- or MCOLN3/TRPML3(Δ5)-expressing cells compared to WT MCOLN3/TRPML3-expressing cells (), suggesting that MCOLN3/TRPML3 palmitoylation is also required for proper autophagy. These findings together prove that both MCOLN3/TRPML3 channel activity and MCOLN3/TRPML3 palmitoylation are essential for MCOLN3/TRPML3 function in autophagy, that is to say autophagosome biogenesis. This corresponds to our previous study showing that the function of MCOLN3/TRPML3 in autophagy is to facilitate autophagosome formation by interaction with GABARAPL2 [Citation5].
As MCOLN3/TRPML3 activation solely increased autophagy without autophagy signaling activation (), we asked whether autophagy induction can activate MCOLN3/TRPML3 to release Ca2+. ) show a significant Ca2+ increase only in WT MCOLN3/TRPML3-expressing cells, but not in MCOLN3/TRPML3(3CS) nor DN MCOLN3D458K transfected cells when the media was changed to HBSS to starve the cells. However, ML-SA1-induced Ca2+ release was similarly observed between WT and palmitoylation mutant-expressing cells, as shown in . This result suggests that palmitoylation allows MCOLN3/TRPML3 to be translocated and activated by autophagy induction, leading to Ca2+ release and autophagosome formation. If MCOLN3/TRPML3 is dynamically translocated to autophagosomes upon induction of autophagy, its trafficking and thus palmitoylation would be increased in an autophagy-dependent manner. To test this possibility, we performed the same palmitoylation assay as in ) except that the cells were kept in serum-starved medium during metabolic labeling. The level of MCOLN3/TRPML3 palmitoylation in starved conditions was almost twice as much as that in fed conditions, but this was not observed with the palmitoylation mutant (). Moreover, this was the case with endogenous MCOLN3/TRPML3 of HEK293T cells ()). Together, all these data indicate that palmitoylation regulates MCOLN3/TRPML3 trafficking and subsequent activation in the context of autophagy upon cellular demand.
Discussion
In the present study, we propose palmitoylation as the underlying mechanism for the regulation of MCOLN3/TRPML3 trafficking and cellular function in autophagy. shows that MCOLN3/TRPML3 is a palmitoylated protein and the palmitoylation occurs at C-terminal cysteine residues of MCOLN3/TRPML3. Inhibition of MCOLN3/TRPML3 palmitoylation by amino acid substitutions or a palmitoylation inhibitor reduced the cell surface expression of MCOLN3/TRPML3 (). It turned out that the reduced surface expression of the palmitoylation mutant resulted from impaired MCOLN3/TRPML3 trafficking to the PM. When endocytosis or exocytosis was inhibited, the surface level of WT MCOLN3/TRPML3 was significantly changed, while that of the palmitoylation mutant was not (). However, MCOLN3/TRPML3 channel properties and activity remained intact in palmitoylation mutants, indicating that palmitoylation primarily regulates MCOLN3/TRPML3 trafficking between subcellular compartments, and does not play a role in channel kinetics.
Because MCOLN3/TRPML3 is expressed mainly in the intracellular vesicular compartments, an evaluation of palmitoylation on intracellular MCOLN3/TRPML3 was also necessary. As shown in , the localization and function of intracellular MCOLN3/TRPML3 appears not to be affected by palmitoylation. Therefore, we investigated the effect of palmitoylation on intracellular MCOLN3/TRPML3 trafficking between subcellular compartments. The data in clearly show that palmitoylation controls intracellular MCOLN3/TRPML3 trafficking when MCOLN3/TRPML3 is forced to move to other compartments as happens during autophagy activation. Inhibition of MCOLN3/TRPML3 palmitoylation did not increase MCOLN3/TRPML3-GABARAPL2 interaction and thus autophagy, suggesting that palmitoylation regulates the recruitment of MCOLN3/TRPML3 into newly forming autophagosomes upon induction of autophagy.
As Atg8-family proteins are implicated in autophagosome biogenesis [Citation29], we suggested that MCOLN3/TRPML3 might play a role in autophagosome formation through the interaction with GABARAPL2 by providing Ca2+ in the fusion process. Indeed, we observed robust Ca2+ efflux from intracellular compartments using the cell-permeable MCOLN/TRPML agonist ML-SA1 in cells overexpressing MCOLN3/TRPML3 (). Activation of MCOLN3/TRPML3 with ML-SA1 solely induced autophagy, and the elevated autophagy was completely prevented by pretreatment with BAPTA-AM, but not by GPN. These results suggest that Ca2+ efflux not from acidic Ca2+ stores via MCOLN3/TRPML3 activation truly contributes to autophagosome biogenesis. Induction of autophagy by MCOLN3/TRPML3 overexpression itself was not affected by palmitoylation in that both WT and palmitoylation mutant MCOLN3/TRPML3 increased autophagy to a similar extent. However, further increase of autophagy by ML-SA1 was completely abolished by inhibition of palmitoylation, despite the same amount of Ca2+ efflux. This suggests that proper trafficking of MCOLN3/TRPML3 by palmitoylation is required for the cellular function of MCOLN3/TRPML3 in autophagy ().
In , we provide the evidence that both MCOLN3/TRPML3 channel activity and palmitoylation are indispensable for autophagosome formation. Importantly, here we first show that MCOLN3/TRPML3 can be activated upon nutrient starvation resulting in Ca2+ release, which is also regulated by palmitoylation. We also show that MCOLN3/TRPML3 palmitoylation is dynamically regulated in an autophagy-dependent manner, in that the level of MCOLN3/TRPML3 palmitoylation was doubled in starved conditions. Therefore, together with previous results, these findings suggest that both trafficking and Ca2+ channel function of MCOLN3/TRPML3 are regulated by autophagy, and vice versa. When autophagy is induced, we think that MCOLN3/TRPML3 is recruited to autophagosome formation sites by palmitoylation and then activated to provide Ca2+ for the fusion-fission process. How the translocated MCOLN3/TRPML3 can be activated afterwards needs to be further investigated. Solving this problem is of particular interest as identification of the Ca2+ channel supplying Ca2+ in the autophagy process has been a long-standing question in the autophagy field.
Because autophagy is implicated in many human diseases, understanding how autophagy is regulated is becoming increasingly important. In the present study, we propose 2 mechanisms for autophagy regulation, MCOLN3/TRPML3 palmitoylation and MCOLN3/TRPML3 activation upon autophagy induction. In addition, our findings suggest that palmitoylation may serve as a general regulatory mechanism for other autophagy-related proteins with dynamic subcellular localization.
Materials and methods
Plasmid construction and mutagenesis
Human MCOLN3/TRPML3 in pEGFPC1 (Clontech, 6084-1), p3XFlag-CMV-7.1 (Sigma, E7533) and pCMV-HA (Clontech, 635690) was prepared as detailed previously [Citation3]. To generate mCherry-MCOLN3/TRPML3, mCherry was amplified by PCR and cloned ahead of MCOLN3/TRPML3 into the pCMV-HA-MCOLN3/TRPML3. The constructs were used to prepare single or triple cysteine mutants, a dominant-negative mutant, and a deletion mutant using the QuikChange mutagenesis kit (Stratagene, 200523). All mutations were confirmed by sequencing the entire DNA insert to verify the presence of the desired mutation and the absence of extraneous mutations. pEGFPC1‑LC3 was kindly provided by Dr. Tamotsu Yoshimori (Osaka University; Osaka, Japan). p3XFlag-CMV-7.1-GABARAPL2 was prepared as detailed previously [Citation5].
Antibodies and reagents
The following antibodies were used: anti-LAMP1 (BD biosciences, 555798), anti-GFP (Invitrogen, A11122), anti-LC3 (Thermo Fisher Scientific, PA1-16930), and anti-MCOLN3/TRPML3 (Alomone Labs, ACC-083). Other reagents used were Alexa Fluor 488-EGF (Invitrogen, E13345), Alexa Fluor 488-TF (Invitrogen, T13342), biotin-azide (Invitrogen, B10184), Fura-2 AM (Invitrogen, F1225), ML-SA1 (Merck Millipore, 648493), GPN (Cayman Chemical, 14634), 17-ODYA (Cayman Chemical, 90270), hydroxylamine hydrochloride (Sigma-Aldrich, 159417), brefeldin A (Toronto Research Chemicals, B677240), dynasore (Sigma-Aldrich, D7693), BAPTA-AM (Invitrogen, B1204), bafilomycin A1 (Merck Millipore, 196000), HBSS (Sigma-Aldrich, H8264), CPA (Sigma-Aldrich, C1530), E64d (Merck Millipore, 330005), pepstatin A (Sigma-Aldrich, P5318), and ML-SI1 (GW405833 hydrochloride; Sigma-Aldrich, G1421). The siRNA sequences for MCOLN3/TRPML3 were: siRNA 1: 5ʹ-CUCCUUCAGUGUUAAAGUA-3ʹ; siRNA 2: 5ʹ-GUUAAUCUUCGCAGUAAAC-3ʹ.
Cell culture and transfection
HEK293T, HeLa, SK-MEL-28, and SW13 cells were obtained from Korean Cell Line Bank (KCLB®, Seoul, South Korea). Cells were maintained in DMEM (Sigma-Aldrich, D5796) supplemented with 10% fetal bovine serum (Gibco, 16000-044) in a humidified incubator at 37°C and 5% CO2. Cells were plated 1 day before transfection with Lipofectamine 2000 (Invitrogen, 11668019) according to the manufacturer’s instructions.
Palmitoylation assay
HEK293T, SW13 or HEK293T cells transfected with the indicated plasmids or siRNAs were incubated in medium supplemented with 100 µM 17-ODYA (Cayman Chemical, 90270) for 6–8 h. Cells were then washed with phosphate-buffered saline (PBS; pH 7.4, 137 mM NaCl, 1.5 mM KH2PO4, 7.8 mM Na2HPO4, 2.7 mM KCl) and briefly sonicated in detergent-free lysis buffer (PBS containing 1 × EDTA-free protease inhibitor cocktail [Roche Diagnostics, 04693159001], 1 mM sodium vanadate [Sigma-Aldrich, 590088] and 10 mM sodium fluoride [Sigma-Aldrich, S7920]). Membrane fractions were made by ultracentrifugation (163,348 g for 30 min at 4°C) and then pellets were lysed in lysis buffer with 1% (v:v) Triton X-100 (Sigma-Aldrich, X100). An aliquot (94 µl) of each cleared lysate was reacted with 6 μl of freshly premixed click chemistry reagent (final 100 μM biotin-azide [1 µl of 10 mM stock in DMSO], 1 mM Tris [2-carboxyethyl]phosphine [Sigma-Aldrich, 646547; 2 µl of 50 mM stock in H2O], 100 μM Tris-[benzyltriazolylmethyl]amine [Sigma-Aldrich, 678937; 1 µl of 10 mM stock in DMSO], and 1 mM CuSO4 [Sigma-Aldrich, 469130; 2 µl of 50 mM stock in H2O]) [Citation19]. Reaction mixtures were incubated for 1 h at room temperature (RT) with intermittent mixing, and then 2 μl of 0.5 M EDTA was added to terminate the reactions. Unreacted biotin was quenched by the addition of an excess of 1 M Tris, pH 8.0. Biotinylated (palmitoylated) proteins were then affinity isolated using streptavidin beads (Thermo Fisher Scientific, 29202) by incubation at 4°C for 2 h. The beads were washed 3 times with PBS containing 0.5% Triton X-100 and treated with sample buffer for western blot analysis.
Reverse transcription and real-time PCR
Total RNA from the siRNA-transfected HEK293T cells were extracted using the RNeasy mini kit (QIAGEN, 74104) according to the manufacturer’s protocol. Total RNA (0.5 µg) was reverse-transcribed into complementary DNA (cDNA) using SuperScript™ III Reverse Transcriptase (Invitrogen, 18080-044). Quantitative real-time PCR was performed to compare ZDHHC mRNA expression levels to the negative controls. The sequences of the primer used are in . A 2-µl aliquot of cDNA was amplified in a 20-µl reaction volume, including 10 µl 2 × SYBR Premix Ex Taq (Takara Biotechnology, RR420) and 1 μl of each 10 μM primer (Bioneer) using the Thermal Cycler Dice Real Time System TP800 (Takara Biotechnology, Kyoto, Japan). The crossing threshold value for each PCR-amplified product was calculated by the Thermal Cycler Dice Real Time System software. Data for each ZDHHC amplification were normalized to the housekeeping gene. Each experiment was performed in triplicate and values were averaged.
Table 1. Real-time PCR primers for ZDHHCs.
LC-MS/MS analysis
LC-MS/MS analysis was performed at Medicinal Bioconvergence Research Center. Peptide sample was analyzed on a LTQ-Orbitrap Velos (Thermo Fisher Scientific, Bremen, Germany) connected to an Easy-nano LC II system (Thermo Fisher Scientific, Odense, Denmark) incorporating an autosampler. The dried peptide sample was resuspended in 70 μl of 0.1% formic acid, and an aliquot (7 μl) was injected to a reversed-phase peptide trap EASY-Column (L 2 cm, ID 100 μm, 5 μm, 120 Å, ReproSil-Pur C18-AQ, Thermo Fisher Scientific) and a reversed-phase analytical EASY-Column (L 10 cm, ID 75 μm, 3 μm, 120 Å, ReproSil-Pur C18-AQ, Thermo Fisher Scientific), and electrospray ionization was subsequently performed using a 30-μm (i.d.) nano-bore stainless steel online emitter (Thermo Fisher Scientific). The duration of the total LC gradient was 60 min. The peptides were eluted in a linear gradient of 10 ~ 40% Buffer B (0.1% formic acid in acetonitrile) over 40 min. The temperature and voltage applied to the capillary was 275°C and 1.9 V. All data were acquired with the mass spectrometer operating in automatic data-dependent switching mode. The MS survey was scanned from 350 to 2000 m/z with resolution set to 100,000.
Surface biotinylation assay
Cells were washed with PBS and incubated in 1 mg/ml Sulfo-NHS-LC-Biotin (Thermo Fisher Scientific, 21335) in PBS for 30 min on ice. Free biotin was quenched by the addition of 100 mM glycine in PBS. Lysates were prepared in lysis buffer by sonication, which were then centrifuged at 20,817 g for 20 min at 4°C, and protein concentration in the supernatants was determined. Volume and protein content were equally adjusted in all samples and 12.5% avidin beads (Thermo Fisher Scientific, 29202) were added to each sample. After incubation for 2 h at 4°C, the beads were collected by centrifugation, washed 3 times with 0.5% Triton X-100 in PBS, and the proteins were extracted in sample buffer. Collected proteins were analyzed by western blot.
Confocal microscopy and immunocytochemistry
HeLa cells expressing WT MCOLN3/TRPML3 or a cysteine mutant were grown on glass coverslips, fixed with 4% paraformaldehyde (Biosolution, BP031), and permeabilized by incubation with 0.1% Triton X-100 at RT for 20 min. After fixation, nonspecific sites were blocked with 5% goat serum (Sigma-Aldrich, A6003). Cells were stained with primary antibody overnight at 4°C and followed by incubation with fluorescent secondary antibody for 1 h at RT. Cover slips were mounted on glass slides and analyzed using a Zeiss LSM 710 confocal microscope. The images were analyzed off-line using NIH ImageJTM software.
Uptake of fluorescent EGF and TF
HeLa cells were grown on glass coverslips and transfected with a plasmid encoding WT MCOLN3/TRPML3 or a cysteine mutant. The cells were washed twice with PBS and incubated with 80 ng/ml labeled EGF (Invitrogen, E13345) for 10 min at 37°C. After a PBS wash, cells were incubated with unlabeled EGF for 1–2 min at 4°C and fixed with cold MeOH. For TF uptake, 1 µg/ml labeled TF (Invitrogen, T13342) was used. Cells were mounted on glass slides, fixed, and analyzed using confocal microscopy.
Current recordings and solutions
A tight seal, whole-cell configuration was used to record the whole cell current at RT as previously described [Citation2]. Currents were measured in response to voltage ramps (−100 to + 100 mV, 100 ms) applied every 5 sec from a holding potential of 0 mV. The pipette solution contained 150 mM KCl, 10 mM HEPES, 1.13 mM MgCl2, 5 mM ATP (Sigma-Aldrich, A9187), 10 mM BAPTA adjusted to pH 7.3 with KOH. The bath solution consisted of 140 mM NaCl, 5 mM KCl, 1 mM MgCl2, 1 mM CaCl2, 10 mM glucose, 10 mM HEPES, adjusted to pH 7.4 with NaOH. Na+-free solution contained 150 mM NMDG-Cl (Sigma-Aldrich, M2004) and 10 mM HEPES, adjusted to pH 7.4.
Measurement of [Ca2+]i
Transfected HEK293T cells on glass coverslips were loaded with Fura-2 by incubation with 2 μM Fura-2 AM for 30 min at 37°C and transferred to a perfusion chamber. The cells were continuously perfused with the indicated solutions. Fura-2 fluorescence was measured at excitation wavelengths of 340 and 380 nm and the results are given as the 340:380 fluorescence ratio.
Statistical analysis
Origin 7.0 (OriginLab Corporation) software was used for all analyses. All presented numeric values represent mean ± standard error of the mean. Statistical significance was analyzed using Student’s t-test and P-values <0.05 were regarded as significant.
Supplemental Material
Download TIFF Image (771.5 KB)Acknowledgments
This work was supported by the National Research Foundation of Korea (NRF) grants funded by the Korean Government (NRF-2014R1A1A2057830 to H.J.K. and 2018R1A4A1023822 to I.S.).
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplemental Material
Supplemental data for this article can be accessed here.
Additional information
Funding
Related Research Data
References
- Nilius B, Owsianik G. The transient receptor potential family of ion channels. Genome Biol. 2011;12(3):218.
- Kim HJ, Li Q, Tjon-Kon-Sang S, et al. A novel mode of MCOLN3/TRPML3 regulation by extracytosolic pH absent in the varitint-waddler phenotype. EMBO J. 2008 Apr 23;27(8):1197–1205.
- Kim HJ, Soyombo AA, Tjon-Kon-Sang S, et al. The Ca(2+) channel MCOLN3/TRPML3 regulates membrane trafficking and autophagy. Traffic. 2009 Aug;10(8):1157–1167.
- Martina JA, Lelouvier B, Puertollano R. The calcium channel mucolipin-3 is a novel regulator of trafficking along the endosomal pathway. Traffic. 2009 Aug;10(8):1143–1156.
- Choi S, Kim HJ. The Ca2+ channel MCOLN3/TRPML3 specifically interacts with the mammalian ATG8 homologue GATE16 to regulate autophagy. Biochem Biophys Res Commun. 2014 Jan 3;443(1):56–61.
- Kondratskyi A, Yassine M, Kondratska K, et al. Calcium-permeable ion channels in control of autophagy and cancer. Front Physiol. 2013;4:272.
- Fukata Y, Fukata M. Protein palmitoylation in neuronal development and synaptic plasticity. Nat Rev Neurosci. 2010 Mar;11(3):161–175.
- Aicart-Ramos C, Valero RA, Rodriguez-Crespo I. Protein palmitoylation and subcellular trafficking. Biochim Biophys Acta. 2011 Dec;1808(12):2981–2994.
- Linder ME, Deschenes RJ. Palmitoylation: policing protein stability and traffic. Nat Rev Mol Cell Biol. 2007 Jan;8(1):74–84.
- Chamberlain LH, Shipston MJ. The physiology of protein S-acylation. Physiol Rev. 2015 Apr;95(2):341–376.
- Salaun C, Greaves J, Chamberlain LH. The intracellular dynamic of protein palmitoylation. J Cell Biol. 2010 Dec 27;191(7):1229–1238.
- Guan X, Fierke CA. Understanding protein palmitoylation: biological significance and enzymology. Sci China Chem. 2011 Dec;54(12):1888–1897.
- Blaskovic S, Blanc M, van der Goot FG. What does S-palmitoylation do to membrane proteins? FEBS J. 2013 Jun;280(12):2766–2774.
- Shipston MJ. Ion channel regulation by protein S-acylation. J Gen Physiol. 2014 Jun;143(6):659–678.
- Shipston MJ. Ion channel regulation by protein palmitoylation. J Biol Chem. 2011 Mar 18;286(11):8709–8716.
- Ren J, Wen L, Gao X, et al. CSS-Palm 2.0: an updated software for palmitoylation sites prediction. Protein Eng Des Sel. 2008 Nov;21(11):639–644.
- Vergarajauregui S, Puertollano R. Two di-leucine motifs regulate trafficking of mucolipin-1 to lysosomes. Traffic. 2006 Mar;7(3):337–353.
- Chandra M, Zhou H, Li Q, et al. A role for the Ca2+ channel TRPML1 in gastric acid secretion, based on analysis of knockout mice. Gastroenterology. 2011 Mar;140(3):857–867.
- Martin BR, Cravatt BF. Large-scale profiling of protein palmitoylation in mammalian cells. Nat Methods. 2009 Feb;6(2):135–138.
- Thomas GM, Hayashi T, Chiu SL, et al. Palmitoylation by DHHC5/8 targets GRIP1 to dendritic endosomes to regulate AMPA-R trafficking. Neuron. 2012 Feb 9;73(3):482–496.
- Hayashi T, Thomas GM, Huganir RL. Dual palmitoylation of NR2 subunits regulates NMDA receptor trafficking. Neuron. 2009 Oct 29;64(2):213–226.
- Mattison HA, Hayashi T, Barria A. Palmitoylation at two cysteine clusters on the C-terminus of GluN2A and GluN2B differentially control synaptic targeting of NMDA receptors. PLoS One. 2012;7(11):e49089.
- Jeffries O, Geiger N, Rowe IC, et al. Palmitoylation of the S0-S1 linker regulates cell surface expression of voltage- and calcium-activated potassium (BK) channels. J Biol Chem. 2010 Oct 22;285(43):33307–33314.
- Gonnord P, Delarasse C, Auger R, et al. Palmitoylation of the P2X7 receptor, an ATP-gated channel, controls its expression and association with lipid rafts. FASEB J. 2009 Mar;23(3):795–805.
- Kim HJ, Li Q, Tjon-Kon-Sang S, et al. Gain-of-function mutation in MCOLN3/TRPML3 causes the mouse Varitint-Waddler phenotype. J Biol Chem. 2007 Dec 14;282(50):36138–36142.
- Grimm C, Jors S, Saldanha SA, et al. Small molecule activators of MCOLN3/TRPML3. Chem Biol. 2010 Feb 26;17(2):135–148.
- Kirchhausen T, Macia E, Pelish HE. Use of dynasore, the small molecule inhibitor of dynamin, in the regulation of endocytosis. Methods Enzymol. 2008;438:77–93.
- Shen D, Wang X, Li X, et al. Lipid storage disorders block lysosomal trafficking by inhibiting a TRP channel and lysosomal calcium release. Nat Commun. 2012;3:731.
- Weidberg H, Shvets E, Shpilka T, et al. LC3 and GATE-16/GABARAP subfamilies are both essential yet act differently in autophagosome biogenesis. EMBO J. 2010 Jun 2;29(11):1792–1802.