
ABSTRACT
Macroautophagy/autophagy is vital for intracellular quality control and homeostasis. Therefore, careful regulation of autophagy is very important. In the past 10 years, a number of studies have reported that estrogenic effectors affect autophagy. However, some results, especially those regarding the modulatory effect of 17β-estradiol (E2) on autophagy seem inconsistent. Moreover, several clinical trials are already in place combining both autophagy inducers and autophagy inhibitors with endocrine therapies for breast cancer. Not all patients experience benefit, which further confuses and complicates our understanding of the main effects of autophagy in estrogen-related cancer. In view of the importance of the crosstalk between estrogen signaling and autophagy, this review summarizes the estrogenic effectors reported to affect autophagy, subcellular distribution and translocation of estrogen receptors, autophagy-targeted transcription factors (TFs), miRNAs, and histone modifications regulated by E2. Upon stimulation with estrogen, there will always be opposing functional actions, which might occur between different receptors, receptors on TFs, TFs on autophagy genes, or even histone modifications on transcription. The huge signaling network downstream of estrogen can promote autophagy and reduce overstimulated autophagy at the same time, which allows autophagy to be regulated by estrogen in a restricted range. To help understand how the estrogenic regulation of autophagy affects cell fate, a hypothetical model is presented here. Finally, we discuss some exciting new directions in the field. We hope this might help to better understand the multiple associations between estrogen and autophagy, the pathogenic mechanisms of many estrogen-related diseases, and to design novel and efficacious therapeutics.
Abbreviations: AP-1, activator protein-1; HATs, histone acetyltransferases; HDAC, histone deacetylases; HOTAIR, HOX transcript antisense RNA
Introduction
Macroautophagy/autophagy is a biochemical process necessary for the maintenance of intracellular homeostasis, and its failure can lead to pathological problems such as metabolic diseases, tumors, and developmental defects. Considering that interfering with autophagy is a potential therapy for several diseases, autophagy has become a promising and attractive target for the pharmaceutical industry [Citation1,Citation2]. However, the regulatory mechanisms and pathophysiological states of autophagy have not been fully elucidated, especially for tumors [Citation2,Citation3]. Estrogen is a sex hormone that plays an important role not only in the development of female secondary sexual characteristics, but also in bone development, cardiovascular system protection, and homeostasis [Citation4]. In addition, the occurrence and development of many tumors have been shown to be estrogen dependent [Citation4]. Increasingly, ESR (estrogen receptor) ligands are being designed, synthesized, and used to treat related diseases, and some have already been used clinically [Citation5]. As early as a few decades ago, it was reported that estrogen affects the autophagy of renal tubules [Citation6,Citation7]. In the past 10 years, a number of studies have reported that ESR ligands affect autophagy-related proteins and morphologies in different tissues [Citation8–Citation40]. However, these reports are quite scattered (, ). Some results, especially those regarding the modulatory effect on autophagy of 17β-estradiol (E2), the major endogenous estrogen in mammals, seem inconsistent () [Citation8–Citation20]. In view of the importance of the crosstalk between estrogen signaling and autophagy, this review summarizes the ESR ligands reported to affect autophagy (characteristic autophagy proteins and key morphologies). In addition, tissue and subcellular distribution of the ESRs, autophagy-targeted TFs, miRNAs, and histone modifications are discussed as well. Finally, we examine the potential crosstalk between estrogen signaling and the autophagy pathway and summarize the molecular mechanisms involved in the regulation of autophagy by estrogen. We hope this will not only enrich our understanding of the known estrogenic regulation, but also promote more insight into the molecular mechanisms of estrogen’s action on autophagy.
Table 1. The regulation of E2 on autophagy.
Table 2. The regulation of autophagy by ESR ligands a.
Regulation of E2 on autophagy
As early as the 1970s, an association between castration and altered ultrastructural patterns of autophagosomes and lysosomes in different tissues was reported [Citation6,Citation7]. In early reports, castration leads to a higher rate of autophagy with an increased number of lysosomes and degrading organelles, which suggested for the first time that the systemic deprivation of steroid sex hormones could be linked to autophagy [Citation6,Citation7]. However, both the molecular mechanisms and signaling mediators of autophagy were poorly known then. In recent years, increasing research has shown that autophagy is involved in the effects of estrogen on various diseases ().
The process of autophagy requires the coordination of numerous proteins. To determine whether autophagy is inhibited or promoted not only depends on the single index of biomarker proteins such as MAP1LC3, but also on changes of autophagy flux, autophagy substrates, and key autophagy morphology [Citation41]. For example, chloroquine inhibits autophagy by blocking the fusion of lysosomes with autophagosomes, resulting in the accumulation of MAP1LC3-II and autophagosomes. Therefore, some of the actions on autophagy shown in need further confirmation. However, it is clear that E2 has multiple effects on autophagy. In many cases, E2 plays a role in promoting autophagy [Citation8–Citation16], but at other times, when cellular autophagy has been stimulated by hypoxia, lipopolysaccharides (LPS), spinal cord injury, or ovariectomy, E2 shows a restrictive effect on gene expression of some autophagy proteins [Citation17–Citation20]. Similar phenomena have also been observed for resveratrol, which can prevent the upregulation of autophagy induced by rapamycin ().
Both endogenous and exogenous ESR ligands perform biological functions with specific receptors, classical nuclear receptors (ESR1 and ESR2), and a membrane receptor, GPER1 (G protein-coupled estrogen receptor 1) [Citation5]. In the classical pathway, activated ESRs undergo conformational changes after binding with estrogen, release from the heat shock proteins (HSPs), and then transfer to the nucleus and form homologous or heterologous dimers. In the nucleus, ESRs interact with the estrogen responsive element (ERE) and regulate the expression of target genes, including some autophagy-related genes, directly or indirectly through TFs [Citation42]. Different from ESRs, GPER1 in the cytoplasm mainly takes part in the rapid response of cells to estrogen [Citation42].
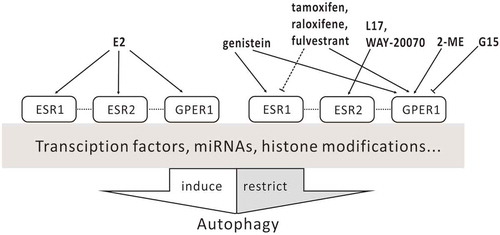
Based on the classical pathway, ESR antagonists such as tamoxifen, raloxifene, and fulvestrant have been designed, synthesized, and used in clinical treatment of ESR-positive breast cancer [Citation5]. However, these ESR antagonists are GPER1 agonists and are able to induce rapid activation of protein kinases, cAMP production, and gene transcription via GPER1 [Citation42]. Some specific regulators targeting GPER1 have been developed but have not yet been used in the clinic. Unexpectedly, there is a slight difference between their effects on autophagy ().
ESR distribution in organs and subcellular locations
As shown in , compared to ESRs, GPER1 is more widely expressed in human tissues. GPER1 is predominately expressed in the thyroid gland, skeletal muscle, adrenal gland, and kidney [Citation43]. In some organs, both ESR1 and ESR2 are expressed with GPER1 [Citation44], whereas in others, only one subtype of ESR is mainly expressed. It is worth mentioning that only ESR2 has ever been reported to be expressed in the stomach [Citation45].
Figure 1. Schematic view of ESR distribution in organs.
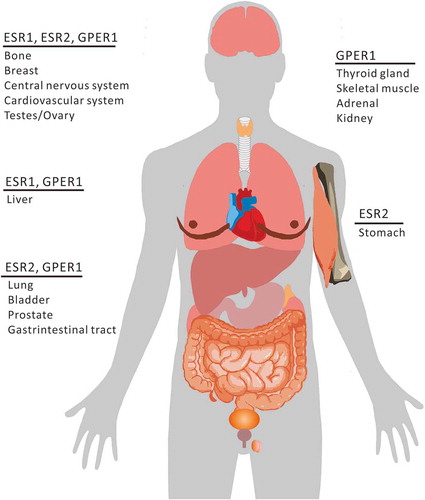
The expression of these receptors and their intracellular distribution are different in different cell types, and the effects of estrogen on their distribution are also different [Citation46–Citation49]. The subcellular localization of the receptors is closely related to their functions, and it changes in different cell types and physiological states. For example, upon binding with E2, ESRs located in endothelial cytosolic/nuclear and endothelial surface membranes are upregulated and activate NOS3 (nitric oxide synthase 3) via genomic and nongenomic pathways, respectively [Citation50]. The nitric oxide (NO) synthesized in this manner can suppress MTOR expression and induce autophagy [Citation51]. Mitochondrial ESRs (mtESRs) are expressed in many human cells including cardiac muscle, lens epithelium, and osteosarcoma, hepatoma, and breast cancer cells [Citation52–Citation55], and mediate the synthesis of mitochondrial respiratory chain proteins (MRCs) via induction with E2 [Citation55–Citation57]. The expression levels of mtESRs also increase with exposure to E2 [Citation55–Citation57]. However, the fate of mtESRs and their role during selective mitochondrial autophagy (mitophagy) is not yet clear. The subcellular localization of GPER1 might be related to different tumor characteristics. GPER1, being localized in the cytoplasm in breast cancer, is correlated with non-ductal carcinoma with better tissue differentiation and lower tumor grade, which is more common in luminal A and B breast cancers [Citation58]. In contrast, when GPER1 is localized in the nucleus, tumors are poorly differentiated or there is triple-negative breast cancer (TNBC), and the prognosis is even worse [Citation58]. Both ESR1 and GPER1 are distributed in lysosomes in an E2-dependent manner, revealing the existence of a lysosome-dependent degradation pathway for these receptors [Citation59,Citation60]. Whether ESR1 and GPER1 distributed in lysosomes participate in autophagy upon fusion with autophagosomes, and their corresponding pathophysiological effects, is unclear.
In general, the intracellular translocation of these receptors remains controversial and appears to be frequently determined by estrogen, though they are clearly involved in the association between estrogen and autophagy ().
Figure 2. Action of ESR ligands on autophagy. Dashed lines indicate tissue type-dependent activation or inhibition; dotted lines indicate crosstalk between receptors.
TFs involved
As shown in and , all of the receptors that might mediate the regulation of estrogen on core autophagy proteins via TFs. These TFs are involved in the regulation of all autophagic processes, including autophagy induction, vesicle nucleation and elongation, and retrieval.
Table 3. Autophagy proteins regulated by E2 via TFs.
Figure 3. Regulation of E2 on core autophagy proteins via TFs. JUN, subunit of AP-1; TP53, tumor protein p53.
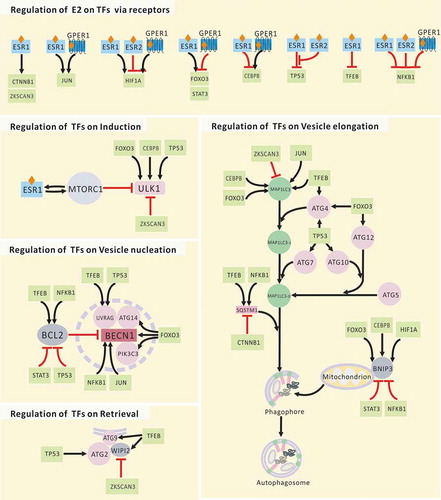
CEBPB
CEBPB links autophagy to the biological clock and maintains nutrient homeostasis by stimulating autophagic gene expression [Citation61]. The increase of nuclear CEBPB protein is dependent on MAPK14. The phosphorylation of MAPK14 is in turn dependent on PLA2G1B (phospholipase A2 group IB), PRKC/PKC activation, and oxidative stress [Citation62]. Expression levels of ESRs and PRKCA are inversely related [Citation63]. CREBBP/CREB (CREB binding protein) controls CEBPB expression by interacting with 2 sites near the TATA box [Citation64]. Recent research has shown that the breast tumor cell-activated phosphoinositide 3-kinase-AKT1 signaling pathway induces cytoplasmic GPER1 translocation of carcinoma-associated fibroblasts in an XPO1 (exportin 1)-dependent pattern, and leads to the activation of the estrogen-GPER1-cAMP-PRKA/PKA-CREB signaling axis that triggers the aerobic glycolysis switch in carcinoma-associated fibroblasts [Citation65].
FOXO3
The Atg14 gene is a direct target of FOXOs, and FOXOs positively regulate Atg14 gene expression [Citation66]. In skeletal muscle, both the ubiquitin-proteasomal and autophagic-lysosomal pathways, the 2 major systems of protein breakdown, are under control of FOXO3. BNIP3 appears to mediate the effect of FOXO3 on autophagy [Citation67]. The activation of FOXO3 by PRKAA2 (protein kinase AMP-associated catalytic subunit alpha 2) induces the expression of MAP1LC3-II, ATG8, BECN1, and BNIP3 in mouse skeletal muscle [Citation67,Citation68]. Constitutive activation of FOXO3 induces MAP1LC3-II and BNIP3 expression and leads to reversible heart atrophy in active Foxo3 transgene mice by constitutive tetracycline regulation [Citation69]. E2 strongly enhances the effects of FOXO3 via ESR1 [Citation70]. In addition, estrogen mediates the inactivation of FOXO3 by GPER1 [Citation71]. FOXO3 proteins in human and mouse have the same AKT1 phosphorylation sites and similar regulatory properties [Citation71].
TP53
TP53 is present in both the nucleus and cytoplasm, and its regulation in autophagy depends on its subcellular localization. The target autophagy genes of TP53 include ULK1, ULK2, ATG2B, ATG4A, ATG4C, ATG7, ATG10, VMP1 (vacuole membrane protein 1), and UVRAG [Citation72]. Initially, ESR1 was reported to be a suppressor of TP53 that can recruit nuclear receptor co-repressors (NCOR1 and NCOR2) and HDAC1 (histone deacetylase 1) [Citation73]. However, recent research has shown that ESR2 can attenuate the crosstalk between ESR1 and TP53 by reducing the recruitment of both NCOR1 and NCOR2 by ESR1. ESR2 antagonizes ESR1-TP53-mediated transcriptional regulation by reducing ESR1-TP53 binding via physical interaction [Citation74]. In addition, the MDM2 proto-oncogene, which is both a TP53 target gene and a negative feedback regulator of TP53, has bidirectional effects on ESR1 by direct interaction [Citation75].
ZKSCAN3
ZKSCAN3 and TFEB regulate lysosomal biogenesis and autophagy in an opposing manner [Citation76]. ZKSCAN3 is a repressor of several autophagy-related genes, including MAP1LC3 and WIPI2 (human homologs of yeast Atg18). WIPI2 has a positive role in the cycling of ATG9. ZKSCAN3 and TFEB are regulated in an opposing manner [Citation76]. For instance, PRKC leads to reduced phosphorylation, nuclear translocation, and activation of TFEB by inactivating GSK3B with a MTORC1-independent mechanism; however, PRKC activates MAPK8/JUN NH2-terminal kinase (mitogen-activated protein kinase 8) and MAPK14, which phosphorylate ZKSCAN3, leading to its translocation and inactivation. ZKSCAN3 phosphorylation at Thr153 by MAPK9 or MAPK14 is required for ZKSCAN3 cytoplasmic translocation and promotes lysosome biogenesis [Citation77]. The expression levels of ESRs and PRKC are inversely related [Citation63].
NFKB1
NFKB1 upregulates the expression of both BCL2 and BECN1 [Citation78,Citation79]. However, NFKB1 silences Bnip3 gene transcription by competing with E2F1 (E2F transcription factor 1) for binding to the Bnip3 promoter [Citation80]. The ubiquitin-binding protein SQSTM1 is required for RAS-induced NFKB1 activation in human tumors [Citation81]. ESRs mediate the inhibition of NFKB1 activity at several levels [Citation82]. Activation of GPER1 by the specific agonist G-1 leads to significant inhibition of the phosphorylation, nuclear localization, and transcriptional activities of NFKB1 [Citation83]. Both IL6 (interleukin 6) and VEGFA (vascular endothelial growth factor A) are inhibited accordingly [Citation83].
STAT3
Functional crosstalk between BCL2, RAC1, and activated STAT3 exists and promotes a permissive redox milieu for cell survival [Citation84]. STAT3 signaling events occur downstream of MMP14/MT1-MMP (matrix metallopeptidase 14). Gene silencing of MMP14 and STAT3 abrogates both STAT3 phosphorylation and BNIP3 expression [Citation85]. Activation of GPER1 inhibits HIF1A and STAT3 signals in TNBC cells [Citation83]. Upon LEP (leptin) treatment, ESR1 increases the transactivation and target gene expression of STAT3 by direct binding. In addition, the enhancement of LEP-mediated activation of STAT3 is independent of ESR1 ligands [Citation86].
TFEB
As a master regulator of lysosomal biogenesis, TFEB promotes the transcription of several lysosomal genes by directly binding to their promoters. Autophagy-related genes Map1lc3, Sqstm1, and Atg9 are also upregulated by TFEB [Citation76,Citation87]. Under well-nourished conditions, MTORC1 inhibits TFEB by its post-translational modification of phosphorylation. In contrast, under starvation conditions, lysosomal disruption and pharmacological inhibition of MTORC1 activates TFEB [Citation88,Citation89]. Upon estrogen stimulation, ESR1 binds to RPTOR (regulatory associated protein of MTOR complex 1) [Citation90]. The MTOR kinase can phosphorylate Ser104/106 of ESR1 and promote the transcription of ER target genes [Citation90,Citation91].
JUN
JUN is involved in the regulation of both BECN1 and MAP1LC3 transcription [Citation92,Citation93]. In ESR- or GPER1-positive GC-2 cells, E2 induces the MAPK1/3, JUN, and MAPK14-dependent mitochondrial apoptotic pathway [Citation94]. In ESR-negative GPER1 or CYP19A1/aromatase-positive SKBR3 cells, tamoxifen acts as a GPER1 agonist and recruits the FOS-JUN complex to AP-1-responsive elements that are located within the CYP19A1/aromatase promoter [Citation95].
HIF1A
The expression of BNIP3, BECN1, and ATG5 required for selective mitochondrial autophagy depends on HIF1A [Citation96]. ESR2 inhibits HIF1A activity by decreasing the binding of HIF1A to the promoter of the VEGFA gene, which attenuates VEGFA transcription induced by hypoxia [Citation97]. Activation of GPER1 by both E2 and G-1 increases VEGFA via upregulation of HIF1A via the GPER1-EGFR-MAPK1-FOS signaling pathway [Citation98]. The HIF1A gene itself contains an estrogen response element, and its expression is directly regulated by ESR1 [Citation99].
MiRNAs involved
In addition to the transcription factors listed above, E2 can regulate the expression of some autophagy-related proteins by suppressing or stimulating miRNA expression.
MIR214
Estrogen treatment downregulates the MIR214 family [Citation100]. MIR214 directly targets mitochondrial uncoupling proteins (UCPs) [Citation101]. UCPs are members of the larger family of mitochondrial anion carrier proteins (MACPs), and UCP2 is involved in uncoupling oxidative phosphorylation and facilitating energy dissipation as heat [Citation102]. UCP2 overexpression not only decreases nonspecific destructive intracellular reactive oxygen species (ROS), but also induces autophagy and endocrine resistance by the phosphorylation of AKT1 and MTOR [Citation21].
MIR21
Regulation of MIR21 transcription by E2 varies depending on experimental conditions, cell line, and control genes used in the analysis. In HepG2 human hepatoma cells, E2 inhibits MIR21 expression via ESR1 [Citation103]. In MCF-7 cells, MIR21 is upregulated by the ESR2 selective ligand diarylpropionitrile (DPN) [Citation104]. In systemic lupus erythematosus, estrogen-regulated STAT1 activates and promotes TLR8 expression via MIR21 [Citation105]. Both the expression and functional loss of STAT1 are related to the development of mammary adenocarcinomas [Citation106]. STAT1 suppresses ATG12 and BECN1 [Citation107]. MIR21 also targets BCL2 mRNA in rat and human beta cells [Citation108].
Long noncoding RNA HOTAIR
E2-GPER1 induces HOTAIR through the suppression of MIR148A [Citation109]. In hepatocellular carcinoma, HOTAIR activates autophagy by upregulating ATG3 and ATG7 [Citation110]. In endometrial cancer cells, HOTAIR regulates autophagy and contributes to cisplatin-induced resistance [Citation110]. In addition, BECN1, multidrug resistance (MDR), and ABCB1 (ATP binding cassette subfamily B member 1) are all regulated by HOTAIR [Citation111].
MIRLET7G
In MCF-7 cells, E2 suppresses the expression of MIRLET7G in a time- and MAP2K/MEK-MAPK-dependent manner [Citation104]. Mirlet7g regulates autophagy in mouse granulosa cells by targeting IGF1R (insulin like growth factor 1 receptor) and downregulates AKT-MTOR [Citation112]. Direct regulation of MIRLET7G by E2 via ESR1 has been reported [Citation104,Citation113].
In addition, MIR30D increases with hormone deprivation in MCF-7 cells. MIR30D regulates autophagy genes including BECN1, ATG5, ATG12, and ATG2 [Citation76]. However, dextran-coated charcoal stripping to treat serum in the original report could eliminate many hormones from the serum, which leaves the effect of estrogens on MIR30D unclear [Citation104].
Histone modifications involved
Histones are the chief protein components of chromatin. Various posttranslational modifications on histones are mainly produced by histone-modifying enzyme complexes in a dynamic manner [Citation76]. These post-translational modifications, including acetylation and methylation, can influence the overall chromatin structure and have clear functional consequences [Citation76]. Acetylation is a modulator of the ligand-dependent gene regulatory activity of ESRs [Citation114]. Estrogen can interfere with the nuclear regulation of autophagy by regulating proteins in histone modifier complexes. Such regulation of histone modification by estrogen is likely to play a role in estrogen-dependent signaling pathways in a variety of estrogen target tissues in both normal and pathological states.
HDACs
In skeletal muscle, HDAC1 and HDAC2 regulate autophagy by inducing autophagic gene expression and autophagosome formation. Loss of these HDACs leads to the accumulation of toxic autophagic intermediates in the myofibers of mice [Citation115]. In cardiomyocytes, HDAC inhibitors suppress autophagy. HDAC1 and HDAC2 are required for the autophagic response, and the overexpression of HDAC2 alone increases autophagy [Citation116]. E2 or dipropylnitrile and β-LGND2 (ESR2 agonists) comparably suppress HDAC2 production, phosphorylation, and the resulting prohypertrophic mRNA expression induced by angiotensin II [Citation117]. The expression of ESR1 can be reactivated by destabilization or reduction of the corepressor complex, formed by MTA1 (metastasis associated 1), IFI16 (interferon gamma inducible protein 16), and HDACs, on the ESR1 promoter [Citation118].
HATs
Several HATs have ubiquitination-linked enzymatic activity. As a HAT, KAT2B (lysine acetyltransferase 2B) acetylates both histone and non-histone proteins and promotes autophagy by inhibiting the AKT1-MTOR signaling pathway [Citation119]. DNA binding and transactivation activities of ESR1 are enhanced by acetylation at lysines 266 and 268 by EP300, which can be reversed by native deacetylases such as SIRT1 (sirtuin 1) [Citation114]. KAT7, another HAT, promotes the degradation of ESR1 through ubiquitination in a proteasome-dependent manner. KAT7 knockdown promotes ESR1 expression [Citation120]. In vitro, E2 inhibits the E3 ubiquitin ligase activity of KAT7 on ESR1 [Citation120].
Post-translational status of histone H3
ESR1 silences TP53-activated transcription by directing the assembly of SUV39H1/histone-lysine N-methyltransferase (suppressor of variegation 3–9 homolog 1) and histone H3 lys9 trimethylation (H3K9me3) at estrogen-repressed genes [Citation74]. ESR2 downregulates SUV39H1/H2 and releases the ESR1-induced transcriptional block by abrogating the repressive heterochromatin conformation of H3K9me3 [Citation74]. Furthermore, ESR2 stimulates the accumulation of trimethylated histone H3 lys4 (H3K4me3) and POLR2/RNA polymerase II on ESR1-repressed genes, which then induces the transcription of the repressed genes involved in TP53-based tumor suppression [Citation74].
Balancing acetylation at H4K16
Dynamic histone modifications play a pivotal role in cell-regulatory events [Citation121]. The acetylation of histone H4 at lysine 16 (H4K16) can influence higher-order chromatin structure, which plays an important role in transcription [Citation76]. Acetylated H4K16 is a primary histone substrate of native deacetylase SIRT1 [Citation76]. In addition, SIRT1 has several non-histone targets, including ATG5, ATG7, MAP1LC3, FOXOs, E2F1, TP73, PPARGC1A (PPARG coactivator 1 alpha), NFKB1, and TP53, all of which are involved in the regulation of autophagy [Citation76]. In oxygen-glucose deprived neurons, estrogen promotes PRKAA2 activation through ESR1 [Citation122]. Estrogen increases SIRT1 expression and activation [Citation123]. In sirt1-knockout neurons, estrogen-induced PRKAA2 activation disappears, which prevents the neuroprotection of estrogen [Citation122].
Concluding remarks and future perspectives
The modulation of estrogenic effects relating to autophagy could contribute to the development of potential strategies to treat numerous human diseases (, ). To clarify the estrogenic function of autophagy between normal physiological and pathological conditions, including tumors, there have been several studies on the estrogenic regulation of autophagy in ESR-positive breast cancers [Citation124–Citation128]. Arguably, much of what is known has come from this area, reflecting the fact that breast cancer is the most common cancer in women, and 70% are treated with drugs that target ESR action. The roles of autophagy in cancer have been confounded by numerous laboratory studies showing that both the enhancement and inhibition of autophagy seem to improve cancer treatment outcomes [Citation129,Citation130]. A similar situation is also seen in other diseases, such as clinically relevant murine models of ischemic stroke [Citation131]. Moreover, several clinical trials are already in place combining both autophagy inducers (rapamycin, everolimus) and autophagy inhibitors (chloroquine, 3-methyladenine) with endocrine therapies [Citation132,Citation133]. Not all patients experience benefit, which further confuses and complicates our understanding of the main effects of autophagy in breast cancer.
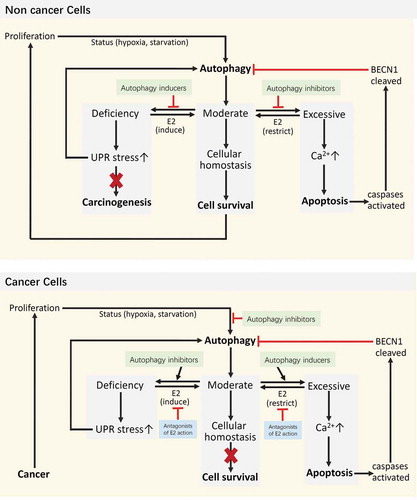
Based on previously reported models [Citation134,Citation135], a hypothetical model () is presented here to help understand how the estrogenic regulation of autophagy affects cell fate. Autophagy is vital for intracellular quality control and homeostasis. Therefore, careful regulation of autophagy is very important because either excessive or insufficient autophagy can be destructive to cells [Citation136]. It has been proven that estrogen can delay the apoptosis of breast cancer cells via ESRs [Citation137]. For the treatment of estrogen-related cancer cells, estrogen needs to be antagonized, which might be because of the protective effect of estrogen on cell balance via regulation of autophagy as shown here.
Figure 4. Model of how estrogenic regulation of autophagy affects cell fate. E2 helps maintain moderate autophagy and cellular homeostasis. Both deficient and excessive autophagy are abnormal. Deficient autophagy can lead to unfolded protein response (UPR) stress, which may reestablish homeostasis through the induction of autophagy. However, the UPR can further lead to carcinogenesis. The proliferation of cancer may induce a status of hypoxia and starvation, both of which can induce autophagy. Here, if a new balance is achieved, cells still have a chance to survive, which is bad for the treatment of cancer. Only when excessive autophagy releases enough calcium from the endoplasmic reticulum to the cytoplasm can apoptosis be triggered. Furthermore, activated caspases will cleave BECN1 and turn off autophagy. Autophagy inducers may prevent carcinogenesis when autophagy in non-cancer cells is deficient, or they may promote excessive autophagy in cancer cells and lead to apoptosis. Autophagy inhibitors seem to block the survival of cancer cells during starvation; however, the inhibition of autophagy cannot persist. The persistent stimulation of UPR stress also promotes autophagy.
As shown in this review, upon stimulation with estrogen, most autophagy-related genes are under the regulation of more than one TF, and most TFs are under the regulation of 2 or 3 receptors of estrogen. In addition, estrogen can interfere with autophagy via nuclear regulation by histone modifications. Thus, there will always be opposing functional actions, which might occur between different receptors, receptors on TFs, TFs on autophagy genes, or even histone modifications on transcription. In addition to those mentioned above, such as ESR1 and ESR2 [Citation74], TFEB and ZKSCAN3 [Citation76], and TP53 and MDM2 [Citation75], need to be further explored in the future. The huge signaling network downstream of estrogen can promote autophagy and reduce overstimulated autophagy at the same time, which allows autophagy to be regulated by estrogen in a restricted range. Estrogen’s mechanism of balancing autophagy is obviously more complicated than this. For instance, estrogen in the blood can cause negative feedback to reduce levels of hormones in the body. In addition to estrogen, other hormones, such as androgens, also play a role in autophagy [Citation138]. Some estrogen-regulated TFs and miRNAs can target ESRs, thereby forming another negative feedback loop to fine-tune estrogen-mediated cellular responses, including autophagy [Citation104,Citation139].
Because autophagy is tightly controlled by estrogen (), it would make sense to determine the fluctuation range of autophagy corresponding to estrogenic regulation under various physiological and pathological conditions. This leads to a major problem for autophagy research – autophagy is a dynamic process that is hard to assess in living organisms (especially humans) by existing methods in vitro [Citation129]. Considering the correlation between autophagy and cell substance balance, if we indirectly characterize autophagy by assessing the material and energy balance in cells instead of focusing solely on the process of autophagy itself, it will be easier to choose between autophagy inducers and inhibitors in the future. Of course, the optimal choice of markers remains to be determined. However, it is foreseeable that developing dynamic markers of cellular balance will be conducive to studying autophagy manipulation and clinical therapeutics.
Figure 5. Association between estrogen and autophagy. E2 balances the expression of core autophagy proteins through diverse transcription factors, miRNAs, and histone modifications via signaling pathways downstream of the receptors. The autophagic proteins controlled by E2 are involved in the entire process of autophagy. Lipids released by autophagy are the major source of cholesterol, the precursor of estrogen biosynthesis. E2 in the blood causes a negative feedback to reduce circulating levels of hormones. E2 activates NOS3 and initiates the synthesis of NO via membrane ESRs. NO induces autophagy by suppressing MTOR expression. In addition, some estrogen-regulated TFs and miRNAs can target ESRs. As the major mechanism for ESR degradation in eukaryotic cells, ESRs dissociate from complexes with HSPs upon binding of E2, are ubiquitinated by ubiquitin ligases (ULs), and are targeted for degradation. The fate of mtESRs and lysosomal ESR1 and GPER1 during autophagy is not yet clear. Ac, acetylation; GF, growth factor; HSPs, heat-shock proteins; HRAS, HRas proto-oncogene, GTPase; Me, methylation; RAF1, Raf-1 proto-oncogene, serine/threonine kinase; RTKs, receptor tyrosine kinases; SRC, SRC proto-oncogene, non-receptor tyrosine kinase; ULs, ubiquitin ligases.
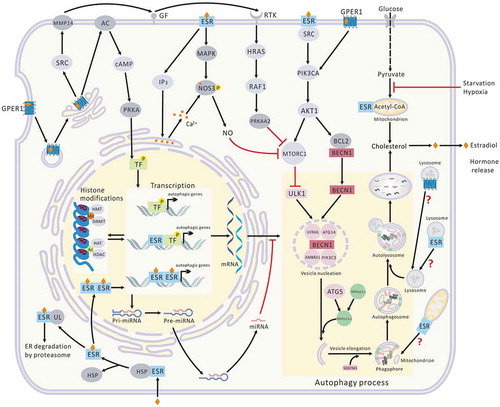
So far, studies of estrogen function on autophagy have mainly focused on breast tissue, however there are still many issues that need to be clarified. Take for example the TFs: although the effects of estrogen on TFs have mostly focused on breast cancer cells, the effects of TFs on autophagy are rarely seen in breast cancer cells (). In other tissues such as brain and bone, the role of autophagy has also been studied for its importance [Citation140,Citation141]. With regard to estrogenic regulation of autophagy in these tissues, there are also some related studies ( and ). Except for the issues mentioned above for breast cancer, more issues need to be solved for the diseases of these tissues. Take for example gliomas: there have been no clinical studies combining both autophagy inducers and autophagy inhibitors with ESR ligands, though they all have independent clinical trials [Citation142,Citation143]. The specificity of drug distribution in these tissues also need to be addressed [Citation143].
In addition, although GPER1 is more widely distributed in human tissues than ESRs and is expressed in TNBC cells and associated with drug resistance of ESR antagonists in endocrine therapy, GPER1 antagonists are very few and have not been developed to work in humans [Citation5]. Whether ESR1 and GPER1 distributed in lysosomes participate in autophagy upon fusion with autophagosomes, and the corresponding pathophysiological effects, are not clear. High levels of some autophagy biomarkers such as MAP1LC3, or autophagy morphologies such as autophagosomes triggered by the induction or accumulation caused by autophagy inhibition, need stricter distinction in future research. These directions might allow us to better understand the multiple associations between estrogen and autophagy, the pathogenic mechanisms of many estrogen-related diseases, and to design novel and efficacious therapeutics.
Acknowledgments
We thank Dr. Wei Liu and Dr. Eugene Chun, School of Molecular Sciences and Biodesign Institute, Arizona State University, USA, for critical reading of the manuscript. We thank Dr. Nian-Hong Chen, Laboratory of Signal Transduction, Department of Radiation Oncology, Memorial Sloan-Kettering Cancer Center, USA, for helpful discussion of the manuscript.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Cheng Y, Ren X, Hait WN, et al. Therapeutic targeting of autophagy in disease: biology and pharmacology. Pharmacol Rev. 2013;65:1162–1197.
- Levy JMM, Towers CG, Thorburn A. Targeting autophagy in cancer. Nat Rev Cancer. 2017;17:528–542.
- Mei Y, Thompson MD, Cohen RA, et al. Autophagy and oxidative stress in cardiovascular diseases. Biochim Biophys Acta. 2015;1852:243–251.
- Jia M, Dahlman-Wright K, Gustafsson J-Å. Estrogen receptor alpha and beta in health and disease. Best Pract Res Clin Endocrinol Metab. 2015;29:557–568.
- Meyer MR, Barton M. Estrogens and coronary artery disease: new clinical perspectives. Adv Pharmacol. 2016;77:307–360.
- Schiebler TH, Danner KG. The effect of sex hormones on the proximal tubules in the rat kidney. Cell Tissue Res. 1978;192:527–549.
- Park J, Shin H, Song H, et al. Autophagic regulation in steroid hormone-responsive systems. Steroids. 2016;115:177–181.
- Totta P, Busonero C, Leone S, et al. Dynamin II is required for 17β-estradiol signaling and autophagy-based ERα degradation. Sci Res. 2016;6:23727.
- Lahm T, Petrache I. LC3 as a potential therapeutic target in hypoxia-induced pulmonary hypertension. Autophagy. 2012;8:1146–1147.
- Lahm T, Albrecht M, Fisher AJ, et al. 17β-Estradiol attenuates hypoxic pulmonary hypertension via estrogen receptor-mediated effects. Am J Respir Crit Care Med. 2012;185:965–980.
- Kimura A, Ishida Y, Nosaka M, et al. Exaggerated arsenic nephrotoxicity in female mice through estrogen-dependent impairments in the autophagic flux. Toxicology. 2016;339:9–18.
- Yang YH, Chen K, Li B, et al. Estradiol inhibits osteoblast apoptosis via promotion of autophagy through the ER-ERK-mTOR pathway. Apoptosis. 2013;18:1363–1375.
- Brasseur K, Leblanc V, Fabi F, et al. ERα-targeted therapy in ovarian cancer cells by a novel estradiol-platinum(II) hybrid. Endocrinology. 2013;154:2281–2295.
- Li XZ, Sui C-Y, Chen Q, et al. The effects and mechanism of estrogen on rats with Parkinson’s disease in different age groups. Am J Transl Res. 2016;8:4134–4146.
- Chen KC, Lin C-M, Huang C-J, et al. Dual roles of 17-β estradiol in estrogen receptor-dependent growth inhibition in renal cell carcinoma. Cancer Genomics Proteomics. 2016;13:219–230.
- Guido C, Panza S, Santoro M, et al. Estrogen receptor beta (ERβ) produces autophagy and necroptosis in human seminoma cell line through the binding of the Sp1 on the phosphatase and tensin homolog deleted from chromosome 10 (PTEN) promoter gene. Cell Cycle. 2012;11:2911–2921.
- Hsieh DJ, Kuo -W-W, Lai Y-P, et al. 17β-Estradiol and/or estrogen receptor β attenuate the autophagic and apoptotic effects induced by prolonged hypoxia through HIF-1α-Mediated BNIP3 and IGFBP-3 signaling blockage. Cell Physiol Biochem. 2015;36:274–284.
- Wang F, Xiao J, Shen Y, et al. Estrogen protects cardiomyocytes against lipopolysaccharide by inhibiting autophagy. Mol Med Rep. 2014;10:1509–1512.
- Lin CW, Chen B, Huang K-L, et al. Inhibition of autophagy by estradiol promotes locomotor recovery after spinal cord injury in rats. Neurosci Bull. 2016;32:137–144.
- Yang Y, Zheng X, Li B, et al. Increased activity of osteocyte autophagy in ovariectomized rats and its correlation with oxidative stress status and bone loss. Biochem Biophys Res Commun. 2014;451:86–92.
- Yu, X., Luo A, Liu Y, et al. MiR-214 increases the sensitivity of breast cancer cells to tamoxifen and fulvestrant through inhibition of autophagy. Mol Cancer. 2015;14:208.
- Yu X, Li R, Shi W, et al. Silencing of MicroRNA-21 confers the sensitivity to tamoxifen and fulvestrant by enhancing autophagic cell death through inhibition of the PI3K-AKT-mTOR pathway in breast cancer cells. Biomed Pharmacother. 2016;77:37–44.
- Pawlik, A., Słomińska-Wojewódzka M, Herman-Antosiewicz A. Sensitization of estrogen receptor-positive breast cancer cell lines to 4-hydroxytamoxifen by isothiocyanates present in cruciferous plants. Eur J Nutr. 2016;55:1165–1180.
- Duan L, Danzer B, Levenson VV, et al. Critical roles for nitric oxide and ERK in the completion of prosurvival autophagy in 4OHTAM-treated estrogen receptor-positive breast cancer cells. Cancer Lett. 2014;353:290–300.
- Kim, H.S., Tian L, Jung M, et al. Downregulation of choline kinase-alpha enhances autophagy in tamoxifen-resistant breast cancer cells. PloS one. 2015;10:e0141110.
- Wu ST, Sun G-H, Cha T-L, et al. CSC-3436 switched tamoxifen-induced autophagy to apoptosis through the inhibition of AMPK/mTOR pathway. J Biomed Sci. 2016;23:60.
- Liu ZR, Song Y, Wan L-H, et al. Over-expression of miR-451a can enhance the sensitivity of breast cancer cells to tamoxifen by regulating 14-3-3ζ, estrogen receptor α, and autophagy. Life Sci. 2016;149:104–113.
- Nagelkerke A, Sieuwerts AM, Bussink J, et al. LAMP3 is involved in tamoxifen resistance in breast cancer cells through the modulation of autophagy. Endocr Relat Cancer. 2014;21:101–112.
- Kim DE, Kim Y, Cho D-H, et al. Raloxifene induces autophagy-dependent cell death in breast cancer cells via the activation of AMP-activated protein kinase. Mol Cells. 2015;38:138–144.
- Ruddy SC, Lau R, Cabrita MA, et al. Preferential estrogen receptor β ligands reduce Bcl-2 expression in hormone-resistant breast cancer cells to increase autophagy. Mol Cancer Ther. 2014;13:1882–1893.
- Prietsch RF, Monte LG, Da Silva FA, et al. Genistein induces apoptosis and autophagy in human breast MCF-7 cells by modulating the expression of proapoptotic factors and oxidative stress enzymes. Mol Cell Biochem. 2014;390:235–242.
- Alayev A, Berger SM, Kramer MY, et al. The combination of rapamycin and resveratrol blocks autophagy and induces apoptosis in breast cancer cells. J Cell Biochem. 2015;116:450–457.
- Turan B, Tuncay E, Vassort G. Resveratrol and diabetic cardiac function: focus on recent in vitro and in vivo studies. J Bioenerg Biomembr. 2012;44:281–296.
- Zhang X, Xu W, Su J, et al. The prosurvival role of autophagy in resveratrol-induced cytotoxicity in GH3 cells. Int J Mol Med. 2014;33:987–993.
- Graham CD, Kaza N, Klocke BJ, et al. Tamoxifen induces cytotoxic autophagy in glioblastoma. J Neuropathol Exp Neurol. 2016;75:946–954.
- Bai LY, Weng J-R, Hu J-L, et al. G15, a GPR30 antagonist, induces apoptosis and autophagy in human oral squamous carcinoma cells. Chem Biol Interact. 2013;206:375–384.
- Bravo D, Shogren KL, Zuo D, et al. 2-Methoxyestradiol-Mediated Induction of Frzb contributes to cell death and autophagy in MG63 osteosarcoma cells. J Cell Biochem. 2017;118:1497–1504.
- Li L, Li X, Han X, et al. An ovarian cancer model with positive ER: reversion of ER antagonist resistance by Src blockade. Oncol Rep. 2014;32:943–950.
- Simpkins F, Hevia-Paez P, Sun J, et al. Src Inhibition with saracatinib reverses fulvestrant resistance in ER-positive ovarian cancer models in vitro and in vivo. Clin Cancer Res. 2012;18:5911–5923.
- Dittmar, A.J., Drozda AA, Blader IJ. Drug repurposing screening identifies novel compounds that effectively inhibit toxoplasma gondii growth. mSphere. 2016;1:e00042–15.
- Klionsky, D.J., Abdelmohsen K, Abe A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy. 2016;12:1–222.
- Luo H, Liu M, Luo S, et al. Dynamic monitoring of GPER-mediated estrogenic effects in breast cancer associated fibroblasts: an alternative role of estrogen in mammary carcinoma development. Steroids. 2016;112:1–11.
- Carmeci C, Thompson DA, Ring HZ, et al. Identification of a gene (GPR30) with homology to the G-protein-coupled receptor superfamily associated with estrogen receptor expression in breast cancer. Genomics. 1997;45:607–617.
- Nilsson S, Gustafsson JA. Estrogen receptors: therapies targeted to receptor subtypes. Clin Pharmacol Ther. 2011;89:44–55.
- Matsuyama S, Ohkura Y, Eguchi H, et al. Estrogen receptor beta is expressed in human stomach adenocarcinoma. J Cancer Res Clin Oncol. 2002;128:319–324.
- Filardo E, Quinn J, Pang Y, et al. Activation of the novel estrogen receptor G protein-coupled receptor 30 (GPR30) at the plasma membrane. Endocrinology. 2007;148:3236–3245.
- Thomas P, Pang Y, Filardo EJ, et al. Identity of an estrogen membrane receptor coupled to a G protein in human breast cancer cells. Endocrinology. 2005;146:624–632.
- Razandi M, Alton G, Pedram A, et al. Identification of a structural determinant necessary for the localization and function of estrogen receptor alpha at the plasma membrane. Mol Cell Biol. 2003;23:1633–1646.
- Otto C, Rohde-Schulz B, Schwarz G, et al. G protein-coupled receptor 30 localizes to the endoplasmic reticulum and is not activated by estradiol. Endocrinology. 2008;149:4846–4856.
- Orshal JM, Khalil RA. Gender, sex hormones, and vascular tone. Am J Physiol Regul Integr Comp Physiol. 2004;286:R233–249.
- He H, Feng Y-S, Zang L-H, et al. Nitric oxide induces apoptosis and autophagy; autophagy down-regulates NO synthesis in physalin A-treated A375-S2 human melanoma cells. Food and Chem Toxicol. 2014;71:128–135.
- Solakidi S, Psarra A-MG, Sekeris CE. Differential subcellular distribution of estrogen receptor isoforms: localization of ERalpha in the nucleoli and ERbeta in the mitochondria of human osteosarcoma SaOS-2 and hepatocarcinoma HepG2 cell lines. Biochim Biophys Acta. 2005;1745:382–392.
- Solakidi S, Psarra A-MG, Sekeris CE. Differential distribution of glucocorticoid and estrogen receptor isoforms: localization of GRbeta and ERalpha in nucleoli and GRalpha and ERbeta in the mitochondria of human osteosarcoma SaOS-2 and hepatocarcinoma HepG2 cell lines. J Musculoskelet Neuronal Interact. 2007;7:240–245.
- Yang SH, Liu R, Perez EJ, et al. Mitochondrial localization of estrogen receptor beta. Proc Natl Acad Sci U S A. 2004;101:4130–4135.
- Chen JQ, Delannoy M, Cooke C, et al. Mitochondrial localization of ERalpha and ERbeta in human MCF7 cells. Am J Physiol Endocrinol Metab. 2004;286:E1011–1022.
- Chen JQ, Eshete M, Alworth WL, et al. Binding of MCF-7 cell mitochondrial proteins and recombinant human estrogen receptors alpha and beta to human mitochondrial DNA estrogen response elements. J Cell Biochem. 2004;93:358–373.
- Chen JQ, Russo PA, Cooke C, et al. ERbeta shifts from mitochondria to nucleus during estrogen-induced neoplastic transformation of human breast epithelial cells and is involved in estrogen-induced synthesis of mitochondrial respiratory chain proteins. Biochim Biophys Acta. 2007;1773:1732–1746.
- Samartzis EP, Noske A, Meisel A, et al. The G protein-coupled estrogen receptor (GPER) is expressed in two different subcellular localizations reflecting distinct tumor properties in breast cancer. PloS one. 2014;9:e83296.
- Totta P, Pesiri V, Marino M, et al. Lysosomal function is involved in 17β-estradiol-induced estrogen receptor α degradation and cell proliferation. PloS one. 2014;9:e94880.
- Cheng SB, Quinn JA, Graeber CT, et al. Down-modulation of the G-protein-coupled estrogen receptor, GPER, from the cell surface occurs via a trans-Golgi-proteasome pathway. J Biol Chem. 2011;286:22441–22455.
- Ma D, Panda S, Lin JD. Temporal orchestration of circadian autophagy rhythm by C/EBPβ. Embo J. 2011;30:4642–4651.
- Torres-Aviles, N.A., Albores-García D, Luna A, et al. Exposure to p,p’-DDE induces morphological changes and activation of the PKCalpha-p38-C/EBPbeta pathway in human promyelocytic HL-60 cells. Biomed Res Int. 2016;2016:1375606.
- Lahn M, Köhler G, Sundell K, et al. Protein kinase C alpha expression in breast and ovarian cancer. Oncology. 2004;67:1–10.
- Ramji DP, Foka P. CCAAT/enhancer-binding proteins: structure, function and regulation. Biochem J. 2002;365:561–575.
- Yu T, Yang G, Hou Y, et al. Cytoplasmic GPER translocation in cancer-associated fibroblasts mediates cAMP/PKA/CREB/glycolytic axis to confer tumor cells with multidrug resistance. Oncogene. 2017;36:2131–2145.
- Xiong X, Tao R, DePinho RA, et al. The autophagy-related gene 14 (Atg14) is regulated by forkhead box O transcription factors and circadian rhythms and plays a critical role in hepatic autophagy and lipid metabolism. J Biol Chem. 2012;287:39107–39114.
- Mammucari C, Milan G, Romanello V, et al. FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab. 2007;6:458–471.
- Sanchez AMJ, Csibi A, Raibon A, et al. AMPK promotes skeletal muscle autophagy through activation of forkhead FoxO3a and interaction with Ulk1. J Cell Biochem. 2012;113:695–710.
- Schips TG, Wietelmann A, Höhn K, et al. FoxO3 induces reversible cardiac atrophy and autophagy in a transgenic mouse model. Cardiovasc Res. 2011;91:587–597.
- Sisci D, Maris P, Cesario MG, et al. The estrogen receptor α is the key regulator of the bifunctional role of FoxO3a transcription factor in breast cancer motility and invasiveness. Cell Cycle. 2013;12:3405–3420.
- Zekas E, Prossnitz ER. Estrogen-mediated inactivation of FOXO3a by the G protein-coupled estrogen receptor GPER. BMC Cancer. 2015;15:702.
- Kenzelmann Broz D, Spano Mello S, Bieging KT, et al. Global genomic profiling reveals an extensive p53-regulated autophagy program contributing to key p53 responses. Genes Dev. 2013;27:1016–1031.
- Konduri SD, Medisetty R, Liu W, et al. Mechanisms of estrogen receptor antagonism toward p53 and its implications in breast cancer therapeutic response and stem cell regulation. Proc Natl Acad Sci U S A. 2010;107:15081–15086.
- Lu W, Katzenellenbogen BS. Estrogen Receptor-β modulation of the ERα-p53 loop regulating gene expression, proliferation, and apoptosis in breast cancer. Horm Cancer. 2017;8:230–242.
- Caldon CE. Estrogen signaling and the DNA damage response in hormone dependent breast cancers. Front Oncol. 2014;4:106.
- Fullgrabe J, Klionsky DJ, Joseph B. The return of the nucleus: transcriptional and epigenetic control of autophagy. Nat Rev Mol Cell Biol. 2014;15:65–74.
- Li Y, Xu M, Ding X, et al. Protein kinase C controls lysosome biogenesis independently of mTORC1. Nat Cell Biol. 2016;18:1065–1077.
- Tamatani M, Che YH, Matsuzaki H, et al. Tumor necrosis factor induces Bcl-2 and Bcl-x expression through NFkappaB activation in primary hippocampal neurons. J Biol Chem. 1999;274:8531–8538.
- Copetti T, Bertoli C, Dalla E, et al. p65/RelA modulates BECN1 transcription and autophagy. Mol Cell Biol. 2009;29:2594–2608.
- Shaw J, Yurkova N, Zhang T, et al. Antagonism of E2F-1 regulated Bnip3 transcription by NF-kappaB is essential for basal cell survival. Proc Natl Acad Sci U S A. 2008;105:20734–20739.
- Duran A, Linares JF, Galvez AS, et al. The signaling adaptor p62 is an important NF-kappaB mediator in tumorigenesis. Cancer Cell. 2008;13:343–354.
- Kalaitzidis D, Gilmore TD. Transcription factor cross-talk: the estrogen receptor and NF-kappaB. Trends Endocrinol Metab. 2005;16:46–52.
- Liang S, Chen Z, Jiang G, et al. Activation of GPER suppresses migration and angiogenesis of triple negative breast cancer via inhibition of NF-κB/IL-6 signals. Cancer Lett. 2017;386:12–23.
- Kang J, Chong SJF, Ooi VZQ, et al. Overexpression of Bcl-2 induces STAT-3 activation via an increase in mitochondrial superoxide. Oncotarget. 2015;6:34191–34205.
- Pratt J, Annabi B. Induction of autophagy biomarker BNIP3 requires a JAK2/STAT3 and MT1-MMP signaling interplay in Concanavalin-A-activated U87 glioblastoma cells. Cell Signal. 2014;26:917–924.
- Binai, N.A., Damert A, Carra G, et al. Expression of estrogen receptor alpha increases leptin-induced STAT3 activity in breast cancer cells. Int J Cancer. 2010;127:55–66.
- Settembre C, De Cegli R, Mansueto G, et al. TFEB controls cellular lipid metabolism through a starvation-induced autoregulatory loop. Nat Cell Biol. 2013;15:647–658.
- Napolitano G, Ballabio A. TFEB at a glance. J Cell Sci. 2016;129:2475–2481.
- Pena-Llopis S, Vega-Rubin-de-Celis S, Schwartz JC, et al. Regulation of TFEB and V-ATPases by mTORC1. Embo J. 2011;30:3242–3258.
- Alayev A, Salamon RS, Berger SM, et al. mTORC1 directly phosphorylates and activates ERα upon estrogen stimulation. Oncogene. 2016;35:3535–3543.
- Jerjees DA, Negm OH, Alabdullah ML, et al. The mammalian target of rapamycin complex 1 (mTORC1) in breast cancer: the impact of oestrogen receptor and HER2 pathways. Breast Cancer Res Treat. 2015;150:91–103.
- Li DD, Wang -L-L, Deng R, et al. The pivotal role of c-Jun NH2-terminal kinase-mediated Beclin 1 expression during anticancer agents-induced autophagy in cancer cells. Oncogene. 2009;28:886–898.
- Sun T, Li D, Wang L, et al. c-Jun NH2-terminal kinase activation is essential for up-regulation of LC3 during ceramide-induced autophagy in human nasopharyngeal carcinoma cells. J Transl Med. 2011;9:161.
- Chimento A, Sirianni R, Casaburi I, et al. 17β-Estradiol activates GPER- and ESR1-dependent pathways inducing apoptosis in GC-2 cells, a mouse spermatocyte-derived cell line. Mol Cell Endocrinol. 2012;355:49–59.
- Catalano, S, Giordano C, Panza S, et al. Tamoxifen through GPER upregulates aromatase expression: a novel mechanism sustaining tamoxifen-resistant breast cancer cell growth. Breast Cancer Res Treat. 2014;146:273–285.
- Zhang H, Bosch-Marce M, Shimoda LA, et al. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J Biol Chem. 2008;283:10892–10903.
- Lim, W, Park Y, Cho J, et al. Estrogen receptor beta inhibits transcriptional activity of hypoxia inducible factor-1 through the downregulation of arylhydrocarbon receptor nuclear translocator. Breast Cancer Res. 2011;13:R32.
- De Francesco EM, Pellegrino M, Santolla MF, et al. GPER mediates activation of HIF1α/VEGF signaling by estrogens. Cancer Res. 2014;74:4053–4064.
- Yang J, AlTahan A, Jones DT, et al. Estrogen receptor-α directly regulates the hypoxia-inducible factor 1 pathway associated with antiestrogen response in breast cancer. Proc Natl Acad Sci U S A. 2015;112:15172–15177.
- Williams KC, Renthal NE, Gerard RD, et al. The microRNA (miR)-199a/214 cluster mediates opposing effects of progesterone and estrogen on uterine contractility during pregnancy and labor. Mol Endocrinol. 2012;26:1857–1867.
- Yu G, Wang J, Xu K, et al. Dynamic regulation of uncoupling protein 2 expression by microRNA-214 in hepatocellular carcinoma. Biosci Rep. 2016;36:e00335.
- Diano S, Horvath TL. Mitochondrial uncoupling protein 2 (UCP2) in glucose and lipid metabolism. Trends Mol Med. 2012;18:52–58.
- Teng Y, Litchfield LM, Ivanova MM, et al. Dehydroepiandrosterone-induces miR-21 transcription in HepG2 cells through estrogen receptor β and androgen receptor. Mol Cell Endocrinol. 2014;392:23–36.
- Klinge CM. miRNAs and estrogen action. Trends Endocrinol Metab. 2012;23:223–233.
- Young NA, Valiente GR, Hampton JM, et al. Estrogen-regulated STAT1 activation promotes TLR8 expression to facilitate signaling via microRNA-21 in systemic lupus erythematosus. Clin Immunology. 2017;176:12–22.
- Chan SR, Vermi W, Luo J, et al. STAT1-deficient mice spontaneously develop estrogen receptor alpha-positive luminal mammary carcinomas. Breast Cancer Res. 2012;14:R16.
- McCormick J, Suleman N, Scarabelli TM, et al. STAT1 deficiency in the heart protects against myocardial infarction by enhancing autophagy. J Cell Mol Med. 2012;16:386–393.
- Sims EK, Lakhter AJ, Anderson-Baucum E, et al. MicroRNA 21 targets BCL2 mRNA to increase apoptosis in rat and human beta cells. Diabetologia. 2017;60:1057–1065.
- Tao S, He H, Chen Q. Estradiol induces HOTAIR levels via GPER-mediated miR-148a inhibition in breast cancer. J Transl Med. 2015;13:131.
- Yang L, Zhang X, Li H, et al. The long noncoding RNA HOTAIR activates autophagy by upregulating ATG3 and ATG7 in hepatocellular carcinoma. Mol Biosyst. 2016;12:2605–2612.
- Sun MY, Zhu J-Y, Zhang C-Y, et al. Autophagy regulated by lncRNA HOTAIR contributes to the cisplatin-induced resistance in endometrial cancer cells. Biotechnol Lett. 2017;39:1477–1484.
- Zhou J, Yao W, Liu K, et al. MicroRNA let-7g regulates mouse granulosa cell autophagy by targeting insulin-like growth factor 1 receptor. Int J Biochem Cell Biol. 2016;78:130–140.
- Qian P, Zuo Z, Wu Z, et al. Pivotal role of reduced let-7g expression in breast cancer invasion and metastasis. Cancer Res. 2011;71:6463–6474.
- Kim MY, Woo EM, Chong YTE, et al. Acetylation of estrogen receptor alpha by p300 at lysines 266 and 268 enhances the deoxyribonucleic acid binding and transactivation activities of the receptor. Mol Endocrinol. 2006;20:1479–1493.
- Moresi V, Carrer M, Grueter CE, et al. Histone deacetylases 1 and 2 regulate autophagy flux and skeletal muscle homeostasis in mice. Proc Natl Acad Sci U S A. 2012;109:1649–1654.
- Gammoh N, Lam D, Puente C, et al. Role of autophagy in histone deacetylase inhibitor-induced apoptotic and nonapoptotic cell death. Proc Natl Acad Sci U S A. 2012;109:6561–6565.
- Pedram A, Razandi M, Narayanan R, et al. Estrogen regulates histone deacetylases to prevent cardiac hypertrophy. Mol Biol Cell. 2013;24:3805–3818.
- Ma H, Li L, Dou G, et al. Z-ligustilide restores tamoxifen sensitivity of ERa negative breast cancer cells by reversing MTA1/IFI16/HDACs complex mediated epigenetic repression of ERa. Oncotarget. 2017;8:29328–29345.
- Jia YL, Xu M, Dou C-W, et al. P300/CBP-associated factor (PCAF) inhibits the growth of hepatocellular carcinoma by promoting cell autophagy. Cell Death Dis. 2016;7:e2400.
- Iizuka M, Susa T, Tamamori-Adachi M, et al. Intrinsic ubiquitin E3 ligase activity of histone acetyltransferase Hbo1 for estrogen receptor α. Proc Jpn Acad Ser B Phys Biol Sci. 2017;93:498–510.
- Fullgrabe J, Lynch-Day MA, Heldring N, et al. The histone H4 lysine 16 acetyltransferase hMOF regulates the outcome of autophagy. Nature. 2013;500:468–471.
- Guo JM, Shu H, Wang L, et al. SIRT1-dependent AMPK pathway in the protection of estrogen against ischemic brain injury. CNS Neurosci Ther. 2017;23:360–369.
- Elangovan S, Ramachandran S, Venkatesan N, et al. SIRT1 is essential for oncogenic signaling by estrogen/estrogen receptor α in breast cancer. Cancer Res. 2011;71:6654–6664.
- Schoenlein PV, Periyasamy-Thandavan S, Samaddar JS, et al. Autophagy facilitates the progression of ERalpha-positive breast cancer cells to antiestrogen resistance. Autophagy. 2009;5:400–403.
- Periyasamy-Thandavan S, Jackson WH, Samaddar JS, et al. Bortezomib blocks the catabolic process of autophagy via a cathepsin-dependent mechanism, affects endoplasmic reticulum stress and induces caspase-dependent cell death in antiestrogen-sensitive and resistant ER+ breast cancer cells. Autophagy. 2010;6:19–35.
- Karunakaran S, Ramachandran S, Coothankandaswamy V, et al. SLC6A14 (ATB0,+) protein, a highly concentrative and broad specific amino acid transporter, is a novel and effective drug target for treatment of estrogen receptor-positive breast cancer. J Biol Chem. 2011;286:31830–31838.
- Cook KL, Clarke R. Estrogen receptor-alpha signaling and localization regulates autophagy and unfolded protein response activation in ER+ breast cancer. Receptors Clin Investig. 2014;1:e316.
- Tran AT, Ramalinga M, Kedir H, et al. Autophagy inhibitor 3-methyladenine potentiates apoptosis induced by dietary tocotrienols in breast cancer cells. Eur J Nutr. 2015;54:265–272.
- Thorburn A, Thamm DH, Gustafson DL. Autophagy and cancer therapy. Mol Pharmacol. 2014;85:830–838.
- Lin S-R, Fu Y-S, Tsai M-J, et al. Natural compounds from herbs that can potentially execute as autophagy inducers for cancer therapy. Int J Mol Sci. 2017;18:1412.
- Buckley KM, Hess DL, Sazonova IY, et al. Rapamycin up-regulation of autophagy reduces infarct size and improves outcomes in both permanent MCAL, and embolic MCAO, murine models of stroke. Exp Transl Stroke Med. 2014;6:8.
- Martinez-Outschoorn UE, Pavlides S, Whitaker-Menezes D, et al. Tumor cells induce the cancer associated fibroblast phenotype via caveolin-1 degradation: implications for breast cancer and DCIS therapy with autophagy inhibitors. Cell Cycle. 2010;9:2423–2433.
- Massarweh S, Romond E, Black EP, et al. A phase II study of combined fulvestrant and everolimus in patients with metastatic estrogen receptor (ER)-positive breast cancer after aromatase inhibitor (AI) failure. Breast Cancer Res Treat. 2014;143:325–332.
- Periyasamy-Thandavan S, Jiang M, Schoenlein P, et al. Autophagy: molecular machinery, regulation, and implications for renal pathophysiology. Am J Physiol Renal Physiol. 2009;297:F244–256.
- Tyson JJ, Baumann WT, Chen C, et al. Dynamic modelling of oestrogen signalling and cell fate in breast cancer cells. Nat Rev Cancer. 2011;11:523–532.
- Petherick KJ, Williams AC, Lane JD, et al. Autolysosomal β-catenin degradation regulates Wnt-autophagy-p62 crosstalk. Embo J. 2013;32:1903–1916.
- Petrovski G, Zahuczky G, Katona K, et al. Clearance of dying autophagic cells of different origin by professional and non-professional phagocytes. Cell Death Differ. 2007;14:1117–1128.
- Blessing AM, Rajapakshe K, Reddy Bollu L, et al. Transcriptional regulation of core autophagy and lysosomal genes by the androgen receptor promotes prostate cancer progression. Autophagy. 2017;13:506–521.
- Dai R, Ahmed SA. Sexual dimorphism of miRNA expression: a new perspective in understanding the sex bias of autoimmune diseases. Ther Clin Risk Manag. 2014;10:151–163.
- Nikoletopoulou V, Papandreou M-E, Tavernarakis N. Autophagy in the physiology and pathology of the central nervous system. Cell Death Differ. 2015;22:398–407.
- Valenti MT, Dalle Carbonare L, Mottes M. Role of autophagy in bone and muscle biology. World J Stem Cells. 2016;8:396–398.
- Hainsworth, JD, Shih, KC, Shepard, GC, et al. Phase II study of concurrent radiation therapy, temozolomide, and bevacizumab followed by bevacizumab/everolimus as first-line treatment for patients with glioblastoma. Clin Adv Hematol Oncol. 2012;10:240–246.
- Odia Y, Kreisl TN, Aregawi D, et al. A phase II trial of tamoxifen and bortezomib in patients with recurrent malignant gliomas. J Neurooncol. 2015;125:191–195.
- Niehof M, Manns MP, Trautwein C. CREB controls LAP/C/EBP beta transcription. Mol Cell Biol. 1997;17:3600–3613.
- Chauhan S, Goodwin JG, Chauhan S, et al. ZKSCAN3 is a master transcriptional repressor of autophagy. Molecular Cell. 2013;50:16–28.
- Scherz-Shouval R, Weidberg H, Gonen C, et al. p53-dependent regulation of autophagy protein LC3 supports cancer cell survival under prolonged starvation. Proc Natl Acad Sci U S A. 2010;107:18511–18516.
- Webb AE, Brunet A. FOXO transcription factors: key regulators of cellular quality control. Trends Biochem Sci. 2014;39:159–169.
- Kouzmenko AP, Takeyama K-I, Ito S, et al. Wnt/beta-catenin and estrogen signaling converge in vivo. J Biol Chem. 2004;279:40255–40258.