
ABSTRACT
CTSD (cathepsin D) is one of the major lysosomal proteases indispensable for the maintenance of cellular proteostasis by turning over substrates of endocytosis, phagocytosis and autophagy. Consequently, CTSD deficiency leads to a strong impairment of the lysosomal-autophagy machinery. In mice and humans CTSD dysfunction underlies the congenital variant (CLN10) of neuronal ceroid lipofuscinosis (NCL). NCLs are distinct lysosomal storage disorders (LSDs) sharing various hallmarks, namely accumulation of protein aggregates and ceroid lipofuscin leading to neurodegeneration and blindness. The most established and clinically approved approach to treat LSDs is enzyme replacement therapy (ERT) aiming to replace the defective hydrolase with an exogenously applied recombinant protein. Here we reveal that recombinant human pro-CTSD produced in a mammalian expression system can be efficiently taken up by a variety of cell models, is correctly targeted to lysosomes and processed to the active mature form of the protease. In proof-of-principle experiments we provide evidence that recombinant human CTSD (rhCTSD) can improve the biochemical phenotype of CTSD-deficient hippocampal slice cultures in vitro and retinal cells in vivo. Furthermore, we demonstrate that dosing of rhCTSD in the murine CLN10 model leads to a correction of lysosomal hypertrophy, storage accumulation and impaired autophagic flux in the viscera and central nervous system (CNS). We establish that direct delivery of the recombinant protease to the CNS is required for improvement of neuropathology and lifespan extension. Together these data support the continuation of the pre-clinical studies for the application of rhCTSD in the treatment of NCL.
Abbreviations: AIF1/IBA1: allograft inflammatory factor 1; BBB: blood brain barrier; CNS: central nervous system; CTSB: cathepsin B; CTSD: cathepsin D; CTSL: cathepsin L; ERT: enzyme replacement therapy; GFAP: glial fibrillary acidic protein; INL: inner nuclear layer; LAMP1: lysosomal-associated membrane protein 1; LAMP2: lysosomal-associated membrane protein 2; MAP1LC3/LC3: microtubule-associated protein 1 light chain 3; LDL: low-density lipoprotein; LRP1: low density lipoprotein receptor-related protein 1; LSD: lysosomal storage disorder; MEFs: mouse embryonic fibroblasts; M6P: mannose 6-phosphate; mCTSD: mature CTSD; NCL: neuronal ceroid lipofuscinosis; ONL: outer nuclear layer; PB: phosphate buffer; proCTSD: pro-cathepsin D; LRPAP1: low density lipoprotein receptor-related protein associated protein 1; rhCTSD: human recombinant CTSD; SAPC: saposin C; SAPD: saposin D; ATP5G1: ATP synthase, H+ transporting, mitochondrial F0 complex, subunit C1 (subunit 9); SQSTM1/p62: sequestosome 1; TPP1: tripeptidyl peptidase I.
Introduction
Cellular homeostasis requires a fine balance between synthesis and degradation of macromolecules. The turnover of intracellular constituents is finely regulated and involves their routing to hydrolase rich lysosomes in a process named autophagy. Different mechanisms exist for lysosomal delivery of components destined for degradation. The ‘bulk’ degradation of macromolecules and organelles occurs via macroautophagy, hereafter referred to as ‘autophagy’. This process is initiated with the de novo formation of double-membrane–bound vesicles that elongate and sequester the cytosolic components, forming autophagosomes (). These vesicles are deprived of proteolytic enzymes and therefore need to fuse with hydrolase-rich lysosomes. Degradation of proteins within these newly formed autolysosomes relies on a family of lysosomal proteases called cathepsins, namely the cysteine proteinases CTSB (cathepsin B), CTSF and CTSL, and the aspartyl protease CTSD (cathepsin D). In mice CTSD deficiency or CTSB and CTSL double deficiency result in a severe blockage of the autophagic flux [Citation1] (), proving the crucial importance of these proteases for lysosomal proteolysis.
Figure 1. Therapeutic approach and purification of rhCTSD. A Scheme showing that CTSD activity is essential for the degradation of autophagic cargo under normal conditions (top panel) and its deficiency leads to impairment of autophagy (middle panel). We hypothesize that this blockage in autophagy can be corrected by providing exogenous recombinant human pro-CTSD (rhCTSD, lower panel). B Scheme of the production and purification procedure for rhCTSD: (1) rhCTSD secreted into the cell culture media by high producing clone of HEK-EBNA cells stably overexpressing proCTSD; (2) collection of the media supernatant followed by filtration and concentration of the protein; (3) nickel-affinity chromatography of the His-tagged proCTSD; (4) size-exclusion chromatography. C Top: elution profile of nickel-affinity purification of rhCTSD (protein absorbance at 280 nm [a.u.]). His-tagged rhCTSD is eluted by washing with 250 mM imidazole in PBS (red line, right axis). Bottom: Coomassie brilliant blue protein staining of the affinity chromatography fractions. D Top: Elution profile of the size-exclusion chromatography purification of rhCTSD (protein absorbance at 280 nm [a.u.]). Bottom: Coomassie protein staining and CTSD immunoblot of size-exclusion chromatography fractions. Only fractions containing monomeric rhCTSD were pooled and used for further studies.
![Figure 1. Therapeutic approach and purification of rhCTSD. A Scheme showing that CTSD activity is essential for the degradation of autophagic cargo under normal conditions (top panel) and its deficiency leads to impairment of autophagy (middle panel). We hypothesize that this blockage in autophagy can be corrected by providing exogenous recombinant human pro-CTSD (rhCTSD, lower panel). B Scheme of the production and purification procedure for rhCTSD: (1) rhCTSD secreted into the cell culture media by high producing clone of HEK-EBNA cells stably overexpressing proCTSD; (2) collection of the media supernatant followed by filtration and concentration of the protein; (3) nickel-affinity chromatography of the His-tagged proCTSD; (4) size-exclusion chromatography. C Top: elution profile of nickel-affinity purification of rhCTSD (protein absorbance at 280 nm [a.u.]). His-tagged rhCTSD is eluted by washing with 250 mM imidazole in PBS (red line, right axis). Bottom: Coomassie brilliant blue protein staining of the affinity chromatography fractions. D Top: Elution profile of the size-exclusion chromatography purification of rhCTSD (protein absorbance at 280 nm [a.u.]). Bottom: Coomassie protein staining and CTSD immunoblot of size-exclusion chromatography fractions. Only fractions containing monomeric rhCTSD were pooled and used for further studies.](/cms/asset/f4a79c8d-12c5-47a9-ac26-cd7642643218/kaup_a_1637200_f0001_oc.jpg)
CTSD is a lysosomal hydrolase ubiquitous to all cell types but particularly abundant in the central nervous system (CNS) [Citation2]. This enzyme is synthesized in the rough endoplasmic reticulum as a pre-pro-enzyme (zymogen) with the 20 amino-acid signal peptide being directly removed during translation. Once in the Golgi apparatus the 52-kDa pro-CTSD is tagged with 2 mannose 6-phosphate (M6P) residues, targeting it to the lysosomes. The routing of CTSD to these acidic organelles may also occur in a M6P-independent manner, depending on the cell type [Citation3]. The processing of the 44 amino-acid pro-peptide occurs when the enzyme reaches late endosomes in a reaction catalyzed by yet unidentified proteases and yielding a 48-kDa single-chain active intermediate. The final processing step consists of the cleavage of this intermediate form by CTSB and CTSL in the lysosome, producing the final mature form of the enzyme: one N-terminal 14-kDa light chain and one C-terminal 34-kDa heavy chain bound together by non-covalent interactions [Citation4].
Neuronal ceroid lipofuscinosis (NCLs), also known as Batten disease, are a group of rare recessive lysosomal storage disorders (LSDs) caused by mutations in 14 different genes and characterized by early blindness and a severe progressive neurodegeneration. The age of onset of the disease varies from infancy to late adulthood. CTSD deficiency in humans underlies the congenital form (CLN10, MIM610127) of NCL [Citation5], the earliest and most severe variant with onset before or around birth [Citation6]. Despite this variability, all NCL forms share unifying pathomorphological features, including selective damage and loss of neurons in the retina and the brain, neuroinflammation and accumulation of autofluorescent storage material – ceroid-lipofuscin [Citation7]. Autophagy deregulation has recently been recognized as a NCL hallmark [Citation8,Citation9]. Autophagic perturbations, such as accumulated autophagosomes and autophagic substrates, have been identified in several NCL mouse models, pointing to a possible blockage in the autophagic flux [Citation1,Citation10–Citation15]. These perturbations may thus play a central role in the etiology of NCLs and tackling them may be an efficient therapeutic avenue.
Collectively NCLs constitute the most common type of inherited neurodegenerative diseases in childhood, with an estimated worldwide incidence of 1:100,000 [Citation7]. To date the only Food and Drug Administration (FDA) approved specific therapy for NCL is enzyme replacement therapy (ERT) by intraventricular infusion of human recombinant TPP1 (tripeptidyl-peptidase 1; cerliponase alfa) [Citation16], the protease affected in late-infantile CLN2. Other LSDs have also been successfully treated by ERT, including Gaucher disease and mucopolysaccharidoses (I, II and VI) [Citation17]. ERT aims to replace the defective hydrolase by a recombinantly produced enzyme, typically administered by intravenous injection and delivered to the lysosomes via receptor-mediated endocytosis. We have previously developed an ERT strategy to treat α-mannosidosis, a LSD caused by the deficiency of the lysosomal acid hydrolase MAN/α-mannosidase [Citation18–Citation20]. The treatment of patients with the engineered human MAN/α-mannosidase (velmanase alfa) has been recently approved in Europe [Citation21]. A major hurdle for the treatment of neuronopathic LSDs such as NCLs by ERT is the delivery of therapeutic enzyme across the blood-brain barrier (BBB). The administration of cerliponase alfa by intraventricular infusion is unprecedented by allowing the direct targeting of neurons which results in attenuation of disease progression [Citation16].
Supplementation with an enzyme capable of ameliorating the blockage in autophagy may be a particularly promising strategy to treat various forms of NCL. CTSD is a good candidate for such an approach, considering that malfunction in this protease has been suggested as a potential pathogenic link between NCL variants [Citation22]. Here we evaluate the therapeutic potential of ERT with recombinant human pro-CTSD in a murine pre-clinical model of CLN10 [Citation23] ().
Results
Production of recombinant human pro-CTSD (rhCTSD)
Firstly, we devised a strategy to overexpress recombinant human pro-CTSD (rhCTSD) in human HEK 293-EBNA cells (). A signal-peptide was included upstream of pro-CTSD to drive the secretion of the protease. After establishment of a stable cell line overexpressing rhCTSD, one high producing clone was selected for enzyme production. Secreted rhCTSD was purified from the cell supernatant by nickel affinity chromatography through the His-tag added to the enzyme’s N terminus (). The purification involved an additional size exclusion chromatography step, yielding a highly purified, monomeric and stable enzyme solution (). The identity of the purified protease as rhCTSD was confirmed by mass spectrometry analysis (data not shown).
Cellular uptake of rhCTSD
We then examined whether the purified rhCTSD could be taken up and processed by mouse embryonic fibroblasts (MEFs) deficient in CTSD. Culturing of MEFs in the presence of 20 µg/mL of rhCTSD in the media resulted in a gradual time-dependent uptake of the enzyme (). The amount of rhCTSD used in these experiments was chosen based on preliminary studies (not shown). After 30 min exposure only the full length pro-CTSD was detected intracellularly. However, after 4 h the 48 kDa single-chain form and the 34-kDa heavy-chain of mature CTSD (mCTSD) could be detected (). This proteolytic processing reflects the transport of the protein to the lysosomes, where the final cleavage mediated by cysteine CTSB and CTSL takes place [Citation24]. In agreement, inhibition of cysteine cathepsins with leupeptin resulted in decreased rhCTSD processing and reduced mCTSD levels (Fig. S1A). The levels of mCTSD continued to increase until reaching a plateau after 24 h of incubation. Overall, our data demonstrate that rhCTSD is taken up by the murine cells and regularly processed to mCTSD. Immunohistochemistry analysis corroborated the delivery of rhCTSD to LAMP1 (lysosomal-associated membrane protein 1)-positive endolysosomes (). The levels of endocytosed rhCTSD varied among the cells probably due to differential endocytosis rates. CTSD activity levels assessed by a fluorometric assay increased in parallel with the levels of mCTSD protein, and were markedly increased after 4 h and reached a plateau after 24 h, suggesting that the mCTSD formed in endolysosomes is active ().
Figure 2. In vitro uptake of rhCTSD by mouse embryonic fibroblasts (MEFs). A Immunoblot of CTSD-deficient MEFs after incubation with 20 µg/mL rhCTSD for different periods (0.5–48 h). B Immunocytochemistry of CTSD and LAMP1 in CTSD-deficient MEFs after 24 h incubation with 20 µg/mL rhCTSD. Scale bar: 20 µm. C CTSD activity assayed with artificial fluorescent substrate in CTSD-deficient MEFs incubated with 20 µg/mL rhCTSD for different periods (0.5–48 h). D Immunoblot of CTSD-deficient MEFs after co-incubation with rhCTSD 20 µg/mL and 10 mM mannose-6-phosphate (M6P), 50 mM mannose (Mann) or 750 nM LRP1/LDLR agonist LRPAP1 for 6 h.
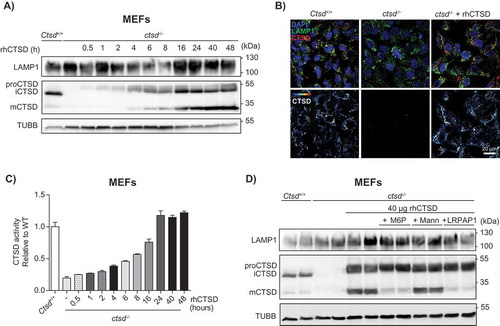
To clarify the nature of the receptor mediating the endocytosis of rhCTSD into MEF cells, a competition experiment was carried out with free mannose-6-phosphate (M6P), mannose and LRPAP1 (low density lipoprotein receptor-related associated protein 1), a chaperone for LRP1 (LDL receptor related protein 1) and other members of the low-density lipoprotein (LDL) receptor family [Citation25,Citation26]. Co-incubation of 40 µg rhCTSD with 10 mM M6P and 750 nM LRPAP1, but not with 50 mM mannose, reduced the delivery of rhCTSD to the lysosomal compartment, as evidenced by the reduced levels of mCTSD protein and enzymatic activity (, Fig. S1B). In line with these data, we observed a significant decrease in the uptake of isotope-labelled [125I]-rhCTSD by LRP1-deficient MEFs when compared to wild-type (WT) cells (Fig. S1C). Together these data demonstrate that the uptake of rhCTSD to the endolysosomal compartment is mediated by M6P and LRP1 receptors, as it also occurs with the endogenous enzyme [Citation3].
We also assessed the half-life of rhCTSD in MEFs by culturing the cells for 24 h in the presence of enzyme and then replacing the culture media. Three days after rhCTSD was removed from the culture media, more than 50% of mCTSD protein levels remained in the cells as compared to day zero, indicating a half-life of longer than 72 h in these cells (Fig. S1D).
Uptake of rhCTSD in the CLN10 mouse model
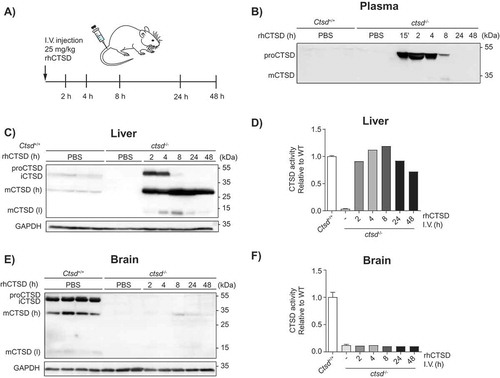
As a next step, we studied the uptake and processing of rhCTSD in CTSD-deficient mice (ctsd-/-). For this purpose, a range of enzyme concentrations was firstly tested taking into account previous studies with other recombinant lysosomal enzymes (not shown) [Citation18–Citation20]. Based on these preliminary tests, we decided to give post-natal day 20 (P20) ctsd-/- animals a dose of 25 mg/kg of rhCTSD by intravenous (i.v.) injection in the tail vein to follow the fate of rhCTSD at different time points (). The recombinant protease was rapidly taken up from the circulation, as indicated by the observation that 8 h after the rhCTSD injection only minor amounts of the enzyme were detectable in plasma by western blot (). The calculated half-life of rhCTSD in the circulation was approximately 4 h. In the liver, 2 h after injection high levels of proCTSD and mCTSD were observed by immunoblot analysis (). ProCTSD levels quickly decreased and after 4 h almost only mCTSD was present. This suggests that liver cells quickly take up and process rhCTSD to the active mCTSD form. Accordingly, CTSD activity in the liver of ctsd-/- mice injected with recombinant enzyme increased steadily until 8 h post-injection, after which it started to gradually decrease again (). These data suggest that the half-life of mCTSD in liver cells exceeds 48 h. In conclusion, rhCTSD is quickly taken up from the circulation and processed to the mature active form by hepatic cells. Uptake of recombinant enzyme also took place in other visceral tissues such as the spleen and the kidney (not shown). In contrast, uptake by the CNS was quite limited after the single i.v. injection of rhCTSD at P20. A faint signal of mCTSD could be detected at all time points, being highest 8 h post-injection (). No increase in protease activity could however be detected with the assay used (). This suggests that rhCTSD cannot easily cross the BBB of this murine model, even when higher doses of enzyme are applied (not shown).
Figure 3. In vivo uptake of rhCTSD by CTSD-deficient mice. A Scheme of the intravenous injection (i.v.) of 25 mg/kg rhCTSD in the tail vein of P20 ctsd-/- mice and sacrifice for organ collection after different time points (2–48 h). B Immunoblot of CTSD plasma levels at different time points after rhCTSD i.v. injection. C Immunoblot of CTSD protein levels and D CTSD activity (relative to WT) in the liver of ctsd-/- mice at different time points after rhCTSD i.v. injection. E Immunoblot of CTSD protein levels and F CTSD activity (relative to WT) in the brain of ctsd-/- mice at different time points after rhCTSD i.v. injection.
Dosing CLN10 mice with rhCTSD corrects visceral pathology
CTSD-deficient mice are a good phenocopy of CLN10, displaying the most dramatic NCL phenotype. Animals start losing weight early on and develop seizures, progressive retinal atrophy, gait and neurological abnormalities resulting in death around postnatal day 25 [Citation23,Citation27]. Pathologically the mice are characterized by lysosomal storage of autofluorescent ceroid lipofuscin, neuroinflammation and accumulation of autophagic vacuoles in neuronal and visceral tissues [Citation1,Citation27].
To test the ability of rhCTSD to correct the visceral phenotype of CTSD-deficient mice we performed repeated dosing of the therapeutic enzyme. Dosing started as early as postnatal day 3–5 (P3–5) with a bolus of 50 mg/kg rhCTSD administered via intraperitoneal (i.p.) injection. This was followed by four additional i.p. injections of 25 mg/kg during the next 2 weeks and one final i.v. injection of 25 mg/kg at day P19-21 (). After repeated dosing of rhCTSD, mCTSD was detectable in all the tissues analyzed – liver, spleen, kidney, intestine, thymus, skeletal muscle and, at minor levels, the brain ( and Fig. S2). In the liver, CTSD activity () and protein levels ( and ) were completely restored and lead to a correction of the various pathological markers analyzed (, and ). In particular, the levels of the LAMP1 protein, a marker for late-endosomes and lysosomes, elevated in the liver of untreated ctsd-/- mice were completely normalized in rhCTSD-treated mice (). In agreement, the increased activity of lysosomal hydrolase HEXB (hexosaminidase B) observed in the liver of untreated ctsd-/- mice was also corrected to WT levels in protease-treated animals (). Together these data indicate that the pathological hypertrophy of the lysosomal compartment caused by CTSD-deficiency is fully corrected. Liver damage in untreated ctsd-/- mice is visible upon toluidine blue staining revealing the presence of large empty spaces in the tissue (, Top panel). In rhCTSD-treated livers these structures were absent, indicating the prevention of liver damage (, Top panel). An additional established pathological hallmark of NCL is the lysosomal accumulation of small hydrophobic proteins called saposins as well as ATP5G1/subunit C of the mitochondrial ATP-synthase [Citation1,Citation27,Citation28]. Treatment with rhCTSD led to a significant reduction in the build-up of SAPC (saposin C) and SAPD in the liver, kidneys, intestine and skeletal muscle compared to untreated age-matched controls ( and ; Fig. S2). Immunoblot data show that ATP5G1 was significantly reduced after treatment in spleen () but not in liver (). In the liver CD68/macrosialin- and AIF1/IBA1 (allograft inflammatory factor 1)-positive macrophages, also known as Kupffer cells, were enlarged in untreated CTSD-deficient mice ( and Fig. S3A). By electron microscopy we observed the presence of polymorphic storage material in the enlarged vacuoles of these cells as well as ‘dying’ Kupffer cells within liver sinusoids (Fig. S3A). This vacuolization and the presence of storage material were fully reversed by the treatment with the human recombinant protease (, Fig. S3B).
Figure 4. ERT with rhCTSD in CLN10 mice. A Scheme of the rhCTSD injection schedule in ctsd-/- mice. Mice received an initial intraperitoneal (i.p.) injection of 50 mg/kg rhCTSD between P3-5, followed by a bi-weekly i.p. injection of 25 mg/kg and a final intravenous 25 mg/kg injection. Animals were sacrificed at P23. B CTSD (relative to WT) and HEXB (mU/mg) activity in lysates of liver of ctsd-/- mice treated with rhCTSD and age-matched controls. Data (n = 3-5, mean ± S.E.M.) were analyzed using an unpaired t-test. * P < 0.05, ** P < 0.01, *** P < 0.001, **** P < 0.0001. C Immunoblot of liver of P23 ctsd-/- mice treated with rhCTSD and age-matched PBS-injected controls. D Immunohistochemistry of liver of P23 ctsd-/- mice treated with rhCTSD and age-matched PBS-injected controls. Top panel – bright-field images of liver sections stained with toluidine blue (TB). Scale bar: 50 µm. White arrows show large empty spaces indicative of tissue damage. Middle panel – Immunohistochemistry of LAMP1 and CTSD. Nuclei are stained with DAPI. Scale bar: 40 µm. Bottom panels – immunostaining of SAPC and immune cell markers CD68 and AIF1/IBA1. Scale bar: 120 µm. White arrows indicate enlarged AIF1-positive Kupffer cells. E Immunoblot of spleen of P23 ctsd-/- mice treated with rhCTSD and age-matched PBS-injected controls. F Bright-field images of spleen sections of P23 ctsd-/- mice treated with rhCTSD and age-matched PBS-injected controls stained with TB. Scale bar: 20 µm. Black arrows indicate large abnormal vacuoles in macrophages of the red pulp.
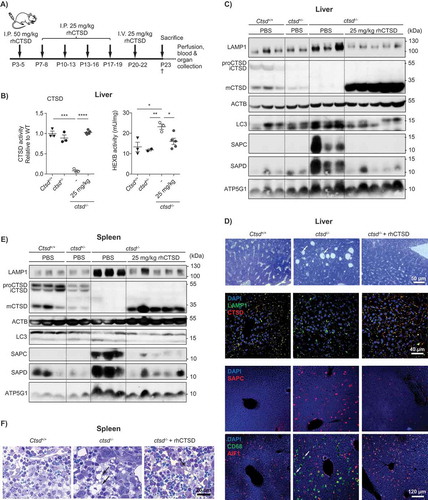
Similar results were found in the spleen, including a correction of mCTSD protein levels that was accompanied by a reduction of pathology markers LAMP1, ATP5G1, SAPC and SAPD (). The intense vacuolization of spleen cells (particularly sinusoidal cells) was also almost completely reversed in the treated ctsd-/- mice (, Fig. S3C,D). Additionally, a correction of autophagic flow was observed, as revealed by a reduction of the autophagy-associated protein lipidated MAP1LC3/LC3-II (microtubule-associated protein 1 light chain 3) in the spleen of rhCTSD-treated ctsd-/- mice when compared to untreated controls (). With this dosing regimen only minor levels of mCTSD were found in the CNS of treated ctsd-/- mice. The low levels of active enzyme were insufficient to delay the pathologic markers analyzed (i.e., lysosomal hypertrophy, autophagy deregulation, microgliosis, etc.; Fig. S2B).
Correction of the biochemical phenotype in neuronal and retinal cells
In order to understand why only minor levels of rhCTSD reach the CNS, we examined the ability of the recombinant enzyme to cross an in vitro BBB model. We analyzed [125I]-rhCTSD transcytosis across primary murine brain endothelial cells in the presence of the paracellular marker [14C]‑inulin for 90 minutes. Brain-to-blood transcytosis occurred at a slow rate and appeared to be, at least partially, dependent on the LRP1 receptor as demonstrated by the abrogation of transcytosis when LRP1-deficient endothelial cells were employed (Fig. S4). The levels of [125I]-rhCTSD transported in the blood-to-brain direction were below the detection level, indicating that the rate of transcytosis into the brain is negligible. These data are in line with our in vivo observations.
To overcome this obstacle and test the ability of the recombinant enzyme to reverse the biochemical phenotype in neurons we decided to test ERT with rhCTSD in organotypic brain slices. Hippocampal slices of ctsd-/- and control mice were prepared at P5 and maintained for 5 d in culture. 10 µg/mL rhCTSD was then added to the culture media for an additional culture period of 5 d. Examination of the brain slices by immunofluorescence (Fig. S5A) and immunoblot () revealed that, similarly to other cultured cells, rhCTSD reached the lysosomes of the hippocampal neurons where it was processed to mCTSD. In untreated ctsd-/- organotypic brain slices we found an increase of the autophagy marker membrane-bound LC3-II. The LC3-II levels (relative to GAPDH) presented a tendency of decrease ( and ) and the ratio of LC3-II to LC3-I (non-lipidated form) was significantly decreased ( and ) in ctsd-/- brain slices cultured in the presence of the recombinant zymogen. A decrease in LAMP1-positive endolysosomes was also observed by immunohistochemistry in treated ctsd-/- slices compared to untreated controls (Fig. S5A).
Figure 5. ERT with rhCTSD in murine hippocampal brain slices and retinas of CTSD-deficient mice. A Immunoblot of day in vitro (DIV) 10 hippocampal organotypic slices of ctsd-/- mice and controls treated with rhCTSD for 5 d. B Quantification of the LC3-II protein levels relative to GAPDH in organotypic brain slices treated with rhCTSD. C Quantification of the ratio LC3-II:LC3-I proteins in organotypic brain slices treated with rhCTSD. Data (n = 2-4, mean ± S.E.M.) were analyzed using an unpaired t-test. * P < 0.05, ** P < 0.01. D Immunohistochemistry showing CTSD and CD68 in the retina of PBS- (LA) and rhCTSD- (RA) injected eyes of P23 CTSD-deficient mice. Scale bar: 50 µm. E Quantification of the number of CD68-positive cells in the retina of PBS- and rhCTSD-injected CTSD-deficient eyes. Data (n = 5, mean ± S.E.M.) were analyzed using an unpaired t-test. * P < 0.05. F DAPI staining of WT and ctsd-/- retinas at P23 (i,ii). Indicated are the different cellular retinal layers: outer nuclear layer (onl), inner nuclear layer (inl), inner plexiform layer (ipl) and ganglion cell layer (gcl). Immunostaining of SAPD (iii, iv), ATP5G1 (v,vi), LAMP1 (vii,viii), LAMP2 (ix,x) and SQSTM1 (xi,xii) in the retina of PBS- (LA) or rhCTSD- (RA) injected eyes of P23 CTSD-deficient mice. Scale bar: 50 µm.
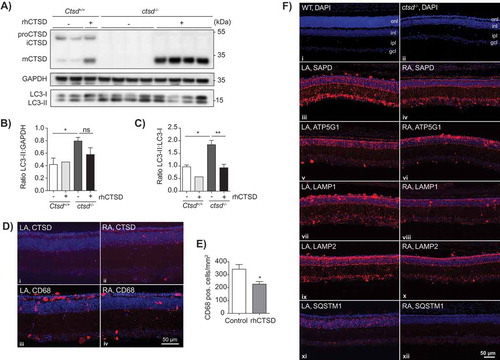
Retinal atrophy leading to blindness is a recognized hallmark of NCLs in patients and animal models [Citation29]. ctsd-/- mice present progressive retinal atrophy, visible from P12 onwards with the loss of photoreceptor cells in the outer nuclear layer (ONL) and later starting at P20 with the death of interneurons in the inner nuclear layer (INL) [Citation29]. We decided to test the ability of rhCTSD to clear storage material and rescue photoreceptor and other neuronal cell types in the retina of ctsd-/- mice. We anticipated that similarly to the BBB, the blood-retina barrier could have low permeability to rhCTSD. Therefore, we used previously established methodology [Citation30,Citation31] and injected 1 µL of 3.1 mg/mL (3.1 µg) rhCTSD into the vitreous cavity of CTSD-deficient animals at P7 and P14 and analyzed the retina at P23, when photoreceptor cells are almost completely lost. Immunohistochemical analysis revealed that rhCTSD was taken up by retinal glia cells, nerve cells and microglia/macrophages ( and Fig. S5B-F). The treatment was however unable to delay retinal degeneration and photoreceptor loss, at least in the current set up (). Nonetheless, we observed a significant decrease in the number of CD68-positive microglia/macrophages in retinas from rhCTSD-treated eyes when compared to retinas from the contralateral eyes of the same animals that had received injections of PBS ( and ), indicating attenuation of inflammation. Levels of the pathological markers LAMP1, LAMP2 (lysosomal-associated membrane protein 2), ATP5G1 and SAPD in retinas from rhCTSD-treated eyes were also significantly decreased when compared to retinas from the contralateral PBS-injected eyes (, Fig. S5G). Of note, we also found a complete abrogation of SQSTM1/p62 (sequestosome 1) puncta in rhCTSD-treated retinas as compared to control retinas, indicating that accumulated autophagy substrates were cleared by the protease ().
Intracranial dosing of rhCTSD
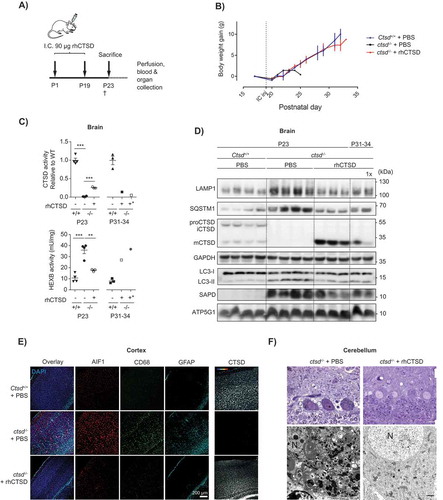
The positive results with the hippocampal slices and retinal cells led us to attempt the direct dosing of the recombinant protease in the CNS of ctsd-/- mice to overcome the hurdle posed by the BBB. For that, animals received an intracranial injection (i.c.) of rhCTSD (90 µg in 10 µL) at P1 (in the right hemisphere) followed by a similar injection (in the left hemisphere) at P19 (). The animals were sacrificed at P23, the standard age of sacrifice for untreated ctsd-/- mice. At this age ctsd-/- animals that had received rhCTSD intracranially appeared more active than PBS injected littermates and gained significantly more body weight than the controls (Fig. S6A). rhCTSD-injected ctsd-/- mice were also able to hold onto the grid of the cage for longer periods than PBS-injected controls (Video S1 and S2), indicating superior motor coordination and muscle strength. Biochemically, we observed significantly increased levels of CTSD protein and activity in brain lysates of P23 treated mice compared to untreated controls ( and ), as well as broad CTSD protein distribution within the different brain regions analyzed ( and Fig. S6B,C). Markers of lysosomal hypertrophy such as HEXB activity () and LAMP1 protein levels ( and Fig. S6D) were restored to WT levels in the CNS of P23 rhCTSD-treated mice. Storage material (i.e., SAPD) was significantly reduced in the brain of these animals and the autophagic parameters SQSTM1 and LC3-II were restored to WT levels (), indicating the clearance of autophagic substrates and reduction in the number of autophagosomes. Importantly, light and electron microscopy analysis also revealed a greatly reduced number of electron-dense inclusions in neurons in the cerebellum (), cortex, hippocampus (not shown) and spinal cord (Fig. S6E) of intracranially-dosed rhCTSD mice. Microgliosis (CD68 and AIF1) and astrogliosis (GFAP [glial fibrillary acidic protein]) which are visible in the cortex and thalamus of untreated P23 ctsd-/- mice [Citation32], were significantly less pronounced in animals that received the recombinant protease (, Fig. S6B). To have a first impression of whether the i.c. injection regimen is able to prolong the lifespan of ctsd-/- mice we assessed the wellbeing of 3 mutant mice that had received 2 i.c. rhCTSD injections. Whereas untreated animals do not survive past the age of P27, these mice survived until P33-34 (). At this age the animals showed good motor coordination and muscle strength (Video S3). The brain of one of these animals still showed considerable levels of CTSD protein and activity ( and ) but levels of most biochemical pathological markers (e.g., LAMP1, SQSTM1 and SAPD) were slightly increased when compared to P23 treated mice, with the remarkable exception of LC3-II levels which remained similar to the younger treated animals (). Surprisingly, a ctsd-/- mouse treated with single i.c. injection at P1 lived almost as long as the mice injected twice (P31 vs P34) and presented a comparable improvement in the biochemical markers analyzed (). Also remarkable is the fact that the recombinant zymogen delivered to the brain appeared to ‘leak’ from the CNS and was found in the liver of i.c. injected animals. The levels of rhCTSD in the liver of these mice were sufficient to correct the pathological markers analyzed (i.e., LAMP1, SQSTM1, LC3-II, SAPD and HEXB activity) to a similar extent as the i.p/i.v. injection regimen (Fig. S7A and S7B). It is of note that lysosomal ATP5G1 accumulation is reduced in the liver of rhCTSD-treated mice (Fig. S7C) although this therapeutic effect was not observed when total levels of ATP5G1 were analyzed by immunoblot ( and ; Fig. S7A).
Figure 6. ERT by i.c. injection of rhCTSD in CTSD-deficient mice. A Scheme of the rhCTSD injection schedule in ctsd-/- mice. Mice received an initial intracranial (i.c.) injection of 90 µg rhCTSD (in 10 µL) at P1 (in the right hemisphere) followed by a similar second i.c. injection at P19 (in the left hemisphere). Animals were sacrificed at P23. B Body weight gain (grams) of ctsd-/- mice that received 2 i.c. injections of rhCTSD and PBS-injected WT and ctsd-/- controls. The time point of the second i.c. injection is indicated with a dotted line. C CTSD (relative to WT) and HEXB (mU/mg) activity in lysates of brains of ctsd-/- mice treated with rhCTSD by i.c. and age-matched PBS-injected controls. Data (n = 3–4, mean ± S.E.M.) were analyzed using an unpaired t-test. ** P < 0.01, *** P < 0.001. D Immunoblot of brains of P23 ctsd-/- mice treated with rhCTSD by i.c. injections and age-matched PBS-injected controls. E Immunohistochemistry of the cortex of P23 ctsd-/- mice treated with rhCTSD and age-matched PBS-injected controls. Shown are immune cell markers CD68 and AIF1, astrocyte marker GFAP and CTSD. Nuclei are stained with DAPI. Scale bar: 200 µm. F Top panels: semithin sections (toluidine blue staining) of the cerebellum (Purkinje cells) of PBS and rhCTSD i.c. injected ctsd-/- mice at P23. Scale bar: 10 µm. Bottom panels: electron microscopy pictures of Purkinje cells of PBS and rhCTSD i.c. injected ctsd-/- mice at P23. N, nucleus. Scale bar: 1000 nm.
The formation of neutralizing antibodies is a major hurdle to the clinical application of ERT. Therefore, we analyzed the formation of antibodies against rhCTSD in the mice treated with i.p. and i.v. injections. Animals treated with this injection regime showed considerable levels of anti-rhCTSD antibodies, allowing the detection of the recombinant protein by immunoblot analysis with the mouse sera (Fig. S8A). On the contrary, i.c. injected mice did not present significant levels of antibodies raised against the therapeutic enzyme (Fig. S8B). Furthermore, the antibodies present in the sera of i.p and i.v. injected mice did not inhibit CTSD maturation and activity in vitro (Fig. S8C,D).
Discussion
Despite not being curative, ERT is still the most successful approach to treat LSDs. This strategy has recently been expanded to peptidases and NCLs with the approval of Brineura (cerliponase alfa) for the treatment of CLN2 patients by intraventricular injection every 15 d [Citation16]. The patients treated with recombinant human TPP1 (tripeptidyl peptidase 1) present less motor and language decline than historical controls, demonstrating the potential of the therapy to tackle the severe neurological symptoms of the disease [Citation16]. A common hallmark of most NCL variants that is amenable to therapeutic interventions is the autophagy impairment. Accumulation of autophagosomes and impaired degradative capacity have been described in various NCL models, leading us to speculate that correcting this dysfunction may represent a promising therapeutic avenue. For playing a critical role in the intralysosomal digestion of autophagic material, CTSD may be a good candidate for such a broad range therapy. The mature double-chain form of CTSD is active at the very acidic pH of lysosomes (pH 3.5–4.5) where it participates in the bulk endo-proteolysis preceding the complete degradation of protein macro-complexes [Citation2,Citation33], showing a particular affinity for substrates enriched in hydrophobic (aromatic) amino acids [Citation2,Citation34]. The activity of this aspartic protease is essential to preserve the homeostasis of neuronal cells given its involvement in the degradation of oxidized or unfolded protein aggregates delivered to lysosomes via autophagy and endocytosis [Citation2]. This importance is highlighted by the early lethality in mice and humans suffering from CLN10 due to CTSD dysfunction. Impairment of CTSD maturation has also been suggested as an underlying contributor to the pathology of various other NCL forms [Citation22]. Furthermore, many other neurodegenerative diseases are characterized by the accumulation of toxic protein aggregates. For example, SNCA (synuclein alpha), APP (amyloid beta precursor protein) and HTT (huntingtin) are all physiological substrates of CTSD that accumulate during Parkinson, Alzheimer, and Huntington disease, respectively [Citation2].
Here we present evidence in proof-of-principle experiments that administration of rhCTSD can reverse the biochemical phenotype of a CLN10 mouse model. We show that recombinant human proCTSD (rhCTSD) can be delivered to the lysosome of mammalian cells, where it is rapidly processed to the mature active form of the enzyme. The uptake of rhCTSD appears, as the endogenous enzyme, to depend on the M6P and LRP1 receptors. The propensity for one receptor over another may depend on the cell type and it is possible that additional receptors can also mediate this uptake. We further confirm that the final maturation step of proCTSD to mCTSD is partially dependent on the activity of cysteine proteases. Nonetheless, processing still occurs when these hydrolases are inhibited, showing that the cleavage of the single chain intermediate can be catalyzed by other enzymes. Thus far we have observed uptake and processing of rhCTSD by all the mammalian cell models tested, including murine primary cortical neurons and CLN10 patient fibroblasts (not shown).
The recombinant proCTSD produced by us appears to be rather stable at 4°C and even at 37°C. The half-life of the mature form of the protease in MEFs surpasses 72 h, indicating a rather high stability in lysosomes. In ctsd-/- mice, intravenously administered enzyme is quickly taken up from the circulation within 8 h. Most of it seems to be captured by hepatic cells, where the zymogen is processed within the first couple of hours to the mature active enzyme. Mature CTSD also seems to have a rather long half-life in hepatic lysosomes, largely exceeding 48 h. Additionally, our in vivo and in vitro data indicate that the transport of rhCTSD by transcytosis across the BBB is very limited. Transcytosis from the brain to the blood seems to be slow and to occur in LRP1-dependent manner. These observations are in agreement with a previous report showing that CTSD can be secreted by CNS neurons and drained to the periphery via lymphatic routes [Citation35].
Already at birth, ctsd-/- mice present with accumulation of small hydrophobic proteins (e.g., saposins and ATP5G1) as well as disturbed autophagic flow in the viscera and CNS [Citation1,Citation27]. Here we show that an early treatment regimen with i.p. injections starting as early as P3-5 followed by an i.v. injection at weaning can reverse the main biochemical pathological markers in the viscera of the mutant mice, including the prevention of protein accumulation and restoration of the number of LC3-II positive autophagosomes. This provides evidence that the recombinant enzyme can carry out its proteolytic function within lysosomes and thereby assure the maintenance of the homeostasis of the lysosome-autophagy axis. In accordance to previous reports [Citation35,Citation36], we show that a correction of the visceral pathology is insufficient to delay the premature death of CTSD-deficient mice.
We also demonstrate that rhCTSD corrects the autophagic defect in CTSD-deficient hippocampal organotypic slices and decreases lysosomal hypertrophy and the number of microglia/macrophages in the retina of animals treated with intravitreal enzyme injections.
We further show, in a proof-of-principle experiment, that when rhCTSD is directly applied to the CNS, it can delay the major neuropathological symptoms of ctsd-/- mice. The regimen with i.c. injections might even be able to increase the lifespan of the CLN10 model, although more detailed studies are necessary to determine the exact extension. Nonetheless, the fact that animals having received therapeutic enzyme only once or twice can outlive untreated controls and show an improvement of disease markers is remarkable given the severity and progressive nature of the disease in this model. Finally, in accordance with previous studies [Citation35,Citation36], we show that rhCTSD delivered to the CNS corrects not only the pathological markers in the brain but also in the viscera and that CNS administration is a prerequisite for a correction of weight gain and lifespan extension. In accordance, a recent conditional knock-out model has demonstrated that correction of CTSD levels in microglia and pericytes delays death by 5 days, further highlighting the fact that expression of CTSD in CNS neurons is crucial for increasing lifespan [Citation36]. It is possible, even though we could not show it, that rhCTSD reaches the periphery via lymphatic routes, as previously suggested [Citation35].
Further studies should be carried out to compare the efficacy of different enzyme formulations as well as the various methods available for delivery of the therapeutic enzyme to the CNS. These studies will be essential for assessing the potential clinical application of recombinant proCTSD, given the severity and early onset of the disease. Furthermore, it would be of interest to evaluate whether rhCTSD can exert comparable beneficial effects in pre-clinical models of other NCL variants that present common pathological features such as disturbed autophagy. Such studies will reveal whether ERT with a bulk-protease, such as CTSD, has the potential to treat other neurological diseases characterized by the accumulation of neurotoxic protein aggregates.
Materials and methods
Cloning
Human pro-Cathepsin-D cDNA flanked by NheI (5ʹ) and NotI (3ʹ) restriction sites was synthesized by Life Technologies GmbH (Darmstadt, Germany) based on the cDNA sequence AAA51922.1 (European Nucleotide Archive), SEQ ID NO: 1. Using the mentioned restriction sites the pro-CTSD cDNA was cloned into the pCEP-Pu vector (Invitrogen, V04450), which includes upstream of the insert coding regions for the secretion signal peptide from SPARC (secreted protein acidic and cysteine rich) and for a hexa-histidinyl tag. The pCEP-Pu vector also contains an EBV origin of replication (oriP) for episomal maintenance of the plasmid [Citation37].
HEK-EBNA cells
Human embryonic kidney (HEK) 293 cells stably expressing the Epstein-Barr virus nuclear antigen 1 under the control of the CMV promoter (HEK 293-EBNA) were purchased from Invitrogen (R620-07). Cells were maintained in Dulbecco’s modified Eagle medium (DMEM; Life Technologies, 41965) containing 4 mM L-glutamine and 4.5 g/L glucose and supplemented with 10% (v:v) fetal bovine serum (Biochrom AG, S0115), 1% PenStrep (Sigma, P0781) and 0.25 mg/mL G-418 (Life Technologies, 11811–023) in a humidified 5% CO2 atmosphere at 37°C.
Stable cell line production
HEK 293-EBNA cells were transfected with pCEP-Pu containing pro-CTSD as follows: 2 × 106 were seeded in a 10-cm cell culture dish and transfected 24 h later using polyethylenimine (Polysciences, 24765) according to the manufacturer’s instructions. 48 h later, expressing cells were selected with 0.25 mg/mL G-418 and 1 µg/mL puromycin for 3 weeks. A high-producing stable clone was selected by serial dilution.
rhCTSD production and purification
Routinely, rhCTSD was produced by seeding 4 × 106 cells in 5 175-cm2 cell culture flasks with 35 mL DMEM (supplemented with 10% fetal bovine serum [FBS], 1% PenStrep, 0.25 mg/mL G-418, and 1 µg/mL puromycin). After the cells reached approximately 80% confluency the medium was replaced by 100 mL DMEM supplemented with 2.5% FBS and 1% PenStrep per flask. The medium was harvested after one week and the cell culture supernatant filtrated using a paper filter followed by vacuum filtration with Stericup (0.22 µm; Millipore, SCGPU10RE). The sample was then concentrated to a final volume of 50 mL via an Amicon system and an ultrafiltration disk with a 10 kDa cutoff (Millipore, PLGC07610). Recombinant protein was purified via its N-terminal His-tag using a His-Trap 1 mL column (GE Healthcare, 29–0510-21) on an Aekta Purifier System (GE Healthcare) and eluted with 250 mM imidazole (Roth, X998.4) in phosphate buffered saline, pH 7.4 (PBS: 137 mM NaCl, 2.7mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4). The protein was further purified via size exclusion chromatography on a Superdex 75 column (GE Healthcare, GE17-5174–01). Finally, monomeric rhCTSD was concentrated using a Vivaspin 20 tube with 10 kDa cutoff (Sartorious, VS2002). In vitro studies indicate that the purified rhproCTSD is stable when stored in PBS at 4°C (no signs of breakdown up to 48 h) and even at 37°C (only minor degradation after 48 h). For longer storage purified, monomeric rhCTSD was aliquoted, snap-frozen and kept at −80°C.
MEF isolation
To isolate mouse embryonic fibroblasts (MEFs) Ctsd+/- mice were mated. Pregnant female Ctsd+/- mice were sacrificed at day 13.5 post coitum. Both uterine horns were prepared and placed in a Petri dish containing sterile PBS. Each embryo was separated from its placenta and amniotic sac and transferred to a fresh Petri dish with PBS. Embryo heads were removed and used for genotype determination using the DirectPCR® Lysis Reagent Tail (Peqlab, 102-T) and proteinase K (Sigma, 1.24568) digestion. Furthermore, all red organs were dissected. In a 3.5 cm culture dish containing 2 mL of trypsin/EDTA (0.5 mg/mL/0.22 mg/mL in PBS) the remaining tissue was minced using a razor blade and incubated for 15 min at 37°C. Single cells were collected in culture medium and the suspension spun down at 300 x g and room temperature for 5 min. The pellets containing the fibroblasts were resuspended in 10 mL culture medium and added to one 10-cm culture dish per embryo. MEFs were immortalized by transfection of the SV40 large T antigen.
CTSD-deficient mice
CTSD-deficient ctsd-/- mice were bred from heterozygous founders and genotyped as previously described [Citation23]. All animals were housed in individually ventilated cages (IVC) to generate a specific pathogen-free environment. The room temperature was maintained at 19–22°C with a humidity of 45–60% and light conditions of 12 h lighting followed by 12 h darkness were applied in turns. Access to water and standard laboratory animal food (pellets by Sniff Spezialdiäten, V1534) was granted ad libitum. Animal handling and care were performed in agreement with the German animal welfare law according to the guidelines of the Christian-Albrechts-University of Kiel and the University Medical Center Hamburg-Eppendorf. Experiments involving animals were approved by the Ministry of Energy, Agriculture, the Environment and Rural Areas Schleswig-Holstein under the reference number V242–40536/2016 (81–6/16) and the Behörde for Gesundheit und Verbraucherschutz, Freie und Hansestadt Hamburg under reference number Ü2/17.
rhCTSD uptake experiments
Approximately 1 × 106 Ctsd+/+ or ctsd-/- MEF cells were seeded per well of 6-well plates. The cells were allowed to attach overnight. Starting the following day 20 µg/mL rhCTSD in PBS was added to the media of ctsd-/- MEFs at different time points (48, 40, 24, 16, 8, 6, 4, 2, 1 and 0.5 h). The cellular uptake of rhCTSD was then evaluated by immunoblot and CTSD activity assay. To test the processing and uptake route the following inhibitors were co-incubated with rhCTSD: leupeptin (25 µM; Enzo Life Sciences, ALX-260–009-M025), mannose-6-phosphate (10 mM; Sigma, M6876-10MG), mannose (50 mM; Serva, 28460) and LRPAP1 (750 nM; kindly provided by Prof. Thomas Braulke, University Hamburg, Germany).
Western blot
The cells were washed 3 times with PBS and lysed in 150 µL RIPA buffer (150 mM NaCl, 50 mM Tris-HCl, pH 7.4, 2 mM EDTA, 10 mM NaF, 1 mM Na3VO4, 1 mM PMSF [Sigma, P7626] 0.5% sodium deoxycholate [Applichem, A1531], 1% Triton X-100 [Sigma, X100], supplemented with 1x complete Protease Inhibitor Cocktail [Roche, 11836145001]) by shaking for 1 h at 4°C. Lysates were cleared by centrifugation at 4°C for 15 min at 12,000 x g and protein concentrations were determined using the BCA method (Thermo Fisher Scientific, 23225). The samples were denatured with 5× Laemmli buffer (50% [v:v] 1 M Tris-HCl, pH 6.8, 50% [v:v] 100% glycerol, 10% [w:v] DTT, 10% [w:v] SDS, 0.01% [w:v] bromophenol blue), boiled for 4 min at 100°C and separated by electrophoresis on 12.5% (w:v) SDS-PAGE gel or NuPage gradient gel (Thermo Fisher Scientific, NP0336BOX) running continuously at 80V. Proteins were transferred to a nitrocellulose membrane (Whatman, GE Healthcare, 10426994) using the semi-dry blotting. The membranes were blocked with 5% dry milk in TBS-T (20 mM Tris HCl, pH 7.0, 150 mM NaCl, 0.1% [v:v] Tween® 20 [Roth, 9127.2). The following primary antibodies were used: rat anti-LAMP1 (Developmental Studies Hybridoma Bank, 1D4B), rabbit anti-SQSTM1 (Enzo Life Science, BHL‐PW‐9860), goat anti-CTSD (Santa Cruz Biotechnology, sc-6486), mouse anti-TUBB/tubulin (Developmental Studies Hybridoma Bank, E7), rabbit anti-GAPDH (glyceraldehyde-3-phosphate dehydrogenase; Santa Cruz Biotechnology, sc‐25778), rabbit anti-ACTB/β-actin (Sigma‐Aldrich, A2066), rabbit anti-LC3 (MBL, PM036), rabbit anti-SAPC and SAPD (a kind gift from G. Grabowski, University of Cincinnatti, USA), rabbit anti-ATP5G1/SCMAS (a kind gift from E. F. Neufeld, UCLA, USA). The blots were washed with TBS-T and incubated 1 h at room temperature (RT) with secondary antibodies coupled to horseradish‐peroxidase (Dianova, goat anti-mouse 115–035-146, goat anti-rat 112–035-143, goat anti-rabbit 111–035-144, rabbit anti-goat 305–035-003) saturated in 5% milk. Following further washing horseradish peroxidase activity was determined using an ImageQuant™ LAS 680 (GE Healthcare). The Amersham ECL Advanced Western Blotting Detection kit (GE Healthcare, RPN2135) was used in combination with ImageJ software to detect and quantify antibody signals.
CTSD activity assay
Cells were lysed for 30 min at 4°C with shaking in 150 μL of 50 mM sodium acetate, pH 5.5, 0.1 M NaCl, 1 mM EDTA, and 0.2% Triton X-100. Lysates were clarified by centrifugation and immediately used for determination of proteolytic activity. For this, 2 μL of cell lysates were incubated at 37°C for 60 min in lysis buffer (100 μL) containing 10 µM of CTSD and E substrate (Enzo Life Sciences, P-145) and 25 µM leupeptin. The AMC released as a result of proteolytic activity was quantified with a Synergy™ HT Multi-Detection microplate reader (exc: 360 nm; em: 440 nm, band pass 40), and normalized to total protein content.
Immunocytochemistry of MEF cells
Approximately 0.5 × 106 Ctsd+/+ or ctsd-/- MEF cells were seeded per well of a 6-well plate (with 13 mm coverslips). The cells were allowed to attach overnight. The following day 20 µg/mL rhCTSD in PBS was added to the media of ctsd-/- MEFs. After 24 h, the cells were washed 3 times with PBS fixed with 99% ice-cold methanol for 20 min at room temperature. After permeabilization and blocking (PBS with 10% FBS and 0.2% saponin [Roth, 4185]) coverslips were incubated overnight with rabbit anti-CTSD (kindly provided by Prof. Andrej Hasilik, Münster, Germany) and rat anti-LAMP1 (Developmental Studies Hybridoma Bank, 1D4B) antibodies diluted 1:500 in blocking buffer. After incubation with fluorophore-conjugated secondary antibodies (Thermo Fisher Scientific, Alexa Fluor Goat anti-rat 488 A11006, Goat anti-rabbit 594 A11037) and washing, coverslips were embedded in mounting solution. Images were analyzed with an Olympus FV1000D Laser Scanning Confocal Microscope (model: FV10-292–115) with a 1003 lens (UPLSAPO 1003 numerical aperture [NA]: 1.40). Image acquisition was performed with the FV10-ASW 4.2 Viewer software (Olympus, Germany).
Repeated dosing of rhCTSD in CLN10 mice
Mice were genotyped at postnatal day P3. Starting between P3-5 mice received one i.p. injection of 50 mg/kg rhCTSD (in PBS). After that, animals received four i.p. injections of 25 mg/kg (2 per week) and one final intravenous injection of 25 mg/kg between P20-P21. At P23 mice were anaesthetized by intra-peritoneal injection of 10 mg/mL Ketamine (Bremer Pharma GmbH, 26706) and 6 mg/ml Rompun® (Bayer, KPOCCNU) in 0.9% (w/v) NaCl solution and transcardially perfused with phosphate buffer 0.1 M.
Intravitreal injections of rhCTSD and analyses of retinas
Intravitreal injections were performed as described previously [Citation30,Citation31]. In brief, animals were deeply anesthetized, a glass micropipette attached to a syringe was inserted into the vitreous cavity at the junction between sclera and cornea. 1 µL of vitreous fluid was removed, and rhCTSD was injected into one eye and 1 µL of PBS into the contralateral eye of the same animals. To analyze the uptake of rhCTSD into the retina, and the impact of the recombinant enzyme on the density of CD68-positive microglia/macrophages and on expression levels of SAPD, ATP5G1, LAMP1, LAMP2 and SQSTM1, we performed 2 injections at P7 and P14 and analyzed the retinas at P23. To identify the retinal cell types that have taken up the injected enzyme, injections were performed at P12 and analysis was done at P14.
Animals were sacrificed, eyes removed, fixed overnight in 4% paraformaldehyde (PFA), dehydrated in an ascending series of sucrose, and frozen. Cryostat sections (25-µm in thickness) from the central retina were blocked in PBS containing 0.1% bovine serum albumin (Sigma, A3608) and 0.3% Triton X-100, incubated with primary antibodies overnight, washed, incubated with Cy2- or Cy3-conjugated secondary antibodies (Jackson ImmunoResearch, donkey anti-rat Cy2 712–545-153, donkey anti-rabbit Cy2 711–545-152, donkey anti-goat Cy3 705–165-147, donkey anti-rabbit Cy3 711–165-152, donkey anti-rat Cy3 712–165-150), washed again, stained with DAPI and mounted onto slides. The following primary antibodies were used: anti-SAPD (kind gift of Konrad Sandhoff, University Bonn, Germany), anti-ATP5G1 (Abcam, ab181243), anti-LAMP1 (Santa Cruz Biotechnology, sc-19,992), anti-LAMP2 (Developmental Studies Hybridoma Bank, clone ABL-93), anti-SQSTM1 (Enzo Life Science, BML-PW9860), anti-CD68 (AbD Serotec, MCA 1957), anti-CTSD (Santa Cruz Biotechnology, sc-6486), anti-PRKCA/PKCα (protein kinase C, alpha; Santa Cruz Biotechnology, sc-208), anti-GFAP (Dako Cytomation GmbH, ZO334) and anti-CALB2/Calretinin (Fitzgerald Industries International, 7699/3H). CD68-positive cells with a clearly visible DAPI-stained nucleus were counted in central retina section of rhCTSD- and contralateral PBS-injected retinas from 5 animals, the retina area was determined and the density of positive cells calculated. Z-stacks were taken from retinal regions close to the optic disc with an AxioObserverZ.1 microscope equipped with an ApoTome2 (Zeiss, Oberkochen, Germany) and processed with Zen 2.3 pro software (Zeiss) and Fiji ImageJ software (National Institutes of Health, Bethesda, MD, USA).
Intracranial injections
Animals were genotyped at postnatal day 0 to be dosed at day P1 and P19. Intracranial dosing of the enzyme was performed on inhalation anaesthetized mice using isoflurane (2% in oxygenized air). Ten µL of enzyme solution containing 90 µg of rhCTSD in PBS, or PBS only for controls, was injected in the cauda putamen using a microsyringe (30 G) using a spacing devise with an injection depth of 1.15 mm over a period of 3 minutes.
Tissue collection and processing
The animals were anaesthetized by intra-peritoneal injection of 10 mg/mL Ketamine and 6 mg/mL Rompun® in 0.9% (w:v) NaCl solution and then transcardially perfused with 0.1 M phosphate buffer (PB), pH 6.8. Blood was collected directly from the right atrium of the heart prior to perfusion. Plasma was isolated by incubating the blood for 0.5 h at RT, followed by 1 h at 4°C and finally centrifugation for 30 min at 2000 x g to clear red blood cells and platelets. The supernatant (plasma) was collected and frozen at −80°C. All tissues harvested were divided in 2 parts. One part was snap frozen in liquid N2 and stored at −80°C for biochemical analyses and the other part was fixed in 4% (w:v) PFA in PB at RT for 4 h for immunohistochemical analyses. The fixed tissues were subsequently washed in PB at 4°C overnight, immersed in 30% sucrose in PBS, and stored at 4°C. Sections (35 μm) were cut with a Leica SM 2000 R sliding microtome (Leica Microsystems) with dry-ice cooling and stored in PBS containing 0.02% (w/v) sodium azide.
Preparation of tissue for biochemical analysis
Samples were stored at −80°C until homogenization in 1:10 ratio of tissue-weight to volume of PBS with 0.1% (v:v) Triton X-100 and protease inhibitor cocktail. The tissues were homogenized with 3 porcelain beads (PeqLab, 91-PCS-CK14B) in 2-mL screw-cap microcentrifuge tubes, samples were crushed with a Precellys® 24 homogenizer (Bertin) set at 6 m.s−1 for 20 s, twice, with samples kept on ice in between runs. Lysates were isolated from glass beads by pipetting into sterile 1.5-mL microcentrifuge tubes.
Immunohistochemistry tissues
After blocking nonspecific antibody binding sites of the slices with blocking solution (0.5% Triton X-100, 4% normal goat serum [Gibco, 16210–064] in PB), the sections were incubated with the primary antibodies in blocking solution overnight at 4°C. The following antibodies were used: rat anti-LAMP1 (1D4B, DSHB), rabbit anti-CTSD, rat anti-CD68 (FA-11, Biolegend), rabbit anti-AIF1/IBA1 (GTX100042, GeneTex), mouse anti-GFAP (G3893, Sigma) and rabbit anti-SAPC. After 3 washing steps in washing buffer (0.25% Triton X-100 in PB) sections were incubated with Alexa Fluor-conjugated secondary antibodies (Thermo Fisher Scientific, Alexa Fluor donkey anti-rat 488 A21208, donkey anti-rabbit 647 A31573, donkey anti-mouse 647 A31571, donkey anti-rabbit 594 A21207) for 2 h at room temperature, washed again 3 times in washing buffer, and finally coverslipped in Mowiol/DABCO. Images were analyzed with an Olympus FV1000D Laser Scanning Confocal Microscope (model: FV10-292–115) with a 1003 lens (UPLSAPO 1003 numerical aperture [NA]: 1.40). Image acquisition was performed with the FV10-ASW 4.2 Viewer software (Olympus, Germany).
Electron microscopy
For electron microscopy (EM) deeply anesthetized mice were perfused with 0.1 M phosphate buffer, pH 7.4, 1% procaine (Sigma, P9879) followed by 6% glutaraldehyde (Merck, 1.04239.1000) in PB supplemented with 1% Procain. Organs were stored in 3% glutaraldehyde in 0.1 M PB pH 7.4. Tissue samples were postfixed with 2% osmium tetroxide, dehydrated and embedded in Araldite (Araldite, 25165618). Semi-thin sections were stained with toluidine blue. Ultrathin sections were processed according to standard techniques, examined and photographed using an EM902 (Zeiss, Jena Germany) electron microscope equipped with a Megaview III digital camera (Albert Tröndle, Moorenweis, Germany).
Preparation of murine organotypic hippocampal slices
Organotypic hippocampal slice cultures were prepared from P5 ctsd-/- and Ctsd+/+ littermates as previously described [Citation38]. Briefly, P5 pups were decapitated, brains removed, placed in cold preparation medium (248 mM sucrose, 26 mM NaHCO3, 10 mM D-glucose, 4 mM KCl, 5 mM MgCl2, 1 m CaCl2, 2 mM kynurenic acid, 0.001% phenol red – oxygenated with 95% O2:5% CO2) and hippocampi dissected. Perpendicular slices of 400-μm thickness were cut using a McIlwain tissue chopper (Mickle Laboratory Engineering, Surrey, UK). After separating the slices in fresh preparation medium, only excellent quality slices were transferred onto millicell membranes (3 slices per membrane, Merck Millipore, PICM0RG50) placed in 6 well-plates with pre-warmed and CO2 equilibrated 1 mL OHSC medium (Minimum essential medium [Gibco, 21090–022] with 25% heat inactivated horse serum [Gibco 26050–088] in HBSS [Sigma, H9269]), supplemented with 1.25 M glucose, 2 mM glutamine (Gibco, 25030–024), 1 M HEPES (SIGMA, H0887), 1x SM1 (Stemcell, 5711), 1x antibiotics (Invitrogen, 15140122). Slices were cultured at 37°C, 5% CO2, 95% humidity. Feeding of slices was performed by exchanging 0.9 mL of medium at DIV2 and thereafter every 3 d. After 5 d recovery 10 µg/mL rhCTSD in PBS was added to the culture media. The slices were exposed to rhCTSD for 5 d. For western blotting 3 hippocampal slices were pooled together and lysed in RIPA buffer (see above).
Statistical analysis
Data are expressed as mean ± S.E.M. unless otherwise stated. For statistical analysis an unpaired student’s t-test was employed using GraphPad Prism 5 (Graph Pad Software, Inc., San Diego, USA): * P < 0.05; ** P < 0.01; *** P < 0.001; **** P<0.0001.
Supplemental Material
Download MS Word (27.7 KB)Supplemental Material
Download MP4 Video (13 MB)Supplemental Material
Download MP4 Video (88 MB)Supplemental Material
Download MP4 Video (36.4 MB)Supplemental Material
Download Zip (75.5 MB)Acknowledgments
We would like to thank Meryem Senkara, Marlies Rusch and Dagmar Niemeier for their crucial technical support, Elizabeth Neufeld for kindly providing the ATP5G1 antibody and Gregory Grabowski for the SAPC and SAPD antibodies, Thomas Braulke for the LRPAP1 peptide and Diego Yepes and Andreas Tholey for support in the mass spectrometry.
Disclosure statement
Authors of this publication receive research funding from OrfoNeuro ApS which is developing products related to research described in this publication. .
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
Related Research Data
References
- Koike M, Shibata M, Waguri S, et al. Participation of autophagy in storage of lysosomes in neurons from mouse models of neuronal ceroid-lipofuscinoses (Batten disease). Am J Pathol. 2005;167:1713–1728.
- Vidoni C, Follo C, Savino M, et al. The Role of Cathepsin D in the Pathogenesis of Human Neurodegenerative Disorders. Med Res Rev. 2016;36:845–870.
- Markmann S, Thelen M, Cornils K, et al. Lrp1/LDL Receptor Play Critical Roles in Mannose 6-Phosphate-Independent Lysosomal Enzyme Targeting. Traffic. 2015;16:743–759.
- Laurent-Matha V, Derocq D, Prébois C, et al. Processing of human cathepsin D is independent of its catalytic function and auto-activation: involvement of cathepsins L and B. J Biochem. 2006;139:363–371.
- Steinfeld R, Reinhardt K, Schreiber K, et al. Cathepsin D deficiency is associated with a human neurodegenerative disorder. Am J Hum Genet. 2006;78:988–998.
- Ketterer S, Gomez-Auli A, Hillebrand LE, et al. Inherited diseases caused by mutations in cathepsin protease genes. Febs J. 2017;284:1437–1454.
- Jalanko A, Braulke T. Neuronal ceroid lipofuscinoses. Biochim Biophys Acta. 2009;1793:697–709.
- Eskelinen E-L, Saftig P. Autophagy: A lysosomal degradation pathway with a central role in health and disease. Biochim Biophys Acta Mol Cell Res. 2009;1793:664–673.
- Seranova E, Connolly KJ, Zatyka M, et al. Dysregulation of autophagy as a common mechanism in lysosomal storage diseases. Essays Biochem. 2017;61:733–749.
- Brandenstein L, Schweizer M, Sedlacik J, et al. Lysosomal dysfunction and impaired autophagy in a novel mouse model deficient for the lysosomal membrane protein Cln7. Hum Mol Genet. 2016;25:777–791.
- Thelen M, Dae M, Schweizer M, et al. Disruption of the autophagy-lysosome pathway is involved in neuropathology of the nclf mouse model of neuronal ceroid lipofuscinosis. PLoS One. 2012;7:e35493.
- Leinonen H, Keksa-Goldsteine V, Ragauskas S, et al. Retinal Degeneration In A Mouse Model Of CLN5 Disease Is Associated With Compromised Autophagy. Sci Rep. 2017;7:1597.
- Cannelli N, Garavaglia B, Simonati A, et al. Variant late infantile ceroid lipofuscinoses associated with novel mutations in CLN6. Biochem Biophys Res Commun. 2009;379:892–897.
- Cao Y, Espinola JA, Fossale E, et al. Autophagy is disrupted in a knock-in mouse model of juvenile neuronal ceroid lipofuscinosis. J Biol Chem. 2006;281:20483–20493.
- Vidal-Donet JM, Cárcel-Trullols J, Casanova B, et al. Alterations in ROS activity and lysosomal pH account for distinct patterns of macroautophagy in LINCL and JNCL fibroblasts. PLoS One. 2013;8:e55526.
- Schulz A, Ajayi T, Specchio N, et al. Study of Intraventricular Cerliponase Alfa for CLN2 Disease. N Engl J Med. 2018;378:1898–1907.
- Parenti G, Andria G, Ballabio A. Lysosomal Storage Diseases: from Pathophysiology to Therapy. Annu Rev Med. 2015;66:471–486.
- Damme M, Stroobants S, Lüdemann M, et al. Chronic enzyme replacement therapy ameliorates neuropathology in alpha-mannosidosis mice. Ann Clin Transl Neurol. 2015;2:987–1001.
- Blanz J, Stroobants S, Lüllmann-Rauch R, et al. Reversal of peripheral and central neural storage and ataxia after recombinant enzyme replacement therapy in α-mannosidosis mice. Hum Mol Genet. 2008;17:3437–3445.
- Roces DP, Lüllmann-Rauch R, Peng J, et al. Efficacy of enzyme replacement therapy in α-mannosidosis mice: a preclinical animal study. Hum Mol Genet. 2004;13:1979–1988.
- Harmatz P, Cattaneo F, Ardigò D, et al. Enzyme replacement therapy with velmanase alfa (human recombinant alpha-mannosidase): novel global treatment response model and outcomes in patients with alpha-mannosidosis. Mol Genet Metab. 2018;124:152–160.
- Chandra G, Bagh MB, Peng S, et al. Cln1 gene disruption in mice reveals a common pathogenic link between two of the most lethal childhood neurodegenerative lysosomal storage disorders. Hum Mol Genet. 2015;24:5416–5432.
- Saftig P, Hetman M, Schmahl W, et al. Mice deficient for the lysosomal proteinase cathepsin D exhibit progressive atrophy of the intestinal mucosa and profound destruction of lymphoid cells. Embo J. 1995;14:3599–3608.
- Gieselmann V, Hasilik A, von Figura K. Processing of human cathepsin D in lysosomes in vitro. J Biol Chem. 1985;260:3215–3220.
- Mazella J, Zsürger N, Navarro V, et al. The 100-kDa neurotensin receptor is gp95/sortilin, a non-G-protein-coupled receptor. J Biol Chem. 1998;273:26273–26276.
- Willnow TE. Receptor-associated protein (RAP): a specialized chaperone for endocytic receptors. BiolChem. 1998;379:1025–1031.
- Koike M, Nakanishi H, Saftig P, et al. Cathepsin D deficiency induces lysosomal storage with ceroid lipofuscin in mouse CNS neurons. J Neurosci. 2000;20:6898–6906.
- Siintola E, Partanen S, Strömme P, et al. Cathepsin D deficiency underlies congenital human neuronal ceroid-lipofuscinosis. Brain. 2006;129:1438–1445.
- Koike M, Shibata M, Ohsawa Y, et al. Involvement of two different cell death pathways in retinal atrophy of cathepsin D-deficient mice. Mol Cell Neurosci. 2003;22:146–161.
- Bartsch U, Bandtlow CE, Schnell L, et al. Lack of evidence that myelin-associated glycoprotein is a major inhibitor of axonal regeneration in the CNS. Neuron. 1995;15:1375–1381.
- Jung G, Sun J, Petrowitz B, et al. Genetically modified neural stem cells for a local and sustained delivery of neuroprotective factors to the dystrophic mouse retina. Stem Cells Transl Med. 2013;2:1001–1010.
- Partanen S, Haapanen A, Kielar C, et al. Synaptic Changes in the Thalamocortical System of Cathepsin D-Deficient Mice. J Neuropathol Exp Neurol. 2008;67:16–29.
- Erickson AH, Isidoro C, Mach L, et al. Cathepsins: getting in Shape for Lysosomal Proteolysis [Internet]. In: Brix K, Stöcker W, editors. Proteases: structure and Function. Vienna: Springer Vienna; 2013. p. 127–173.
- Scarborough PE, Dunn BM. Redesign of the substrate specificity of human cathepsin D: the dominant role of position 287 in the S2 subsite. Protein Eng. 1994;7:495–502.
- Shevtsova Z, Garrido M, Weishaupt J, et al. CNS-expressed cathepsin D prevents lymphopenia in a murine model of congenital neuronal ceroid lipofuscinosis. Am J Pathol. 2010;177:271–279.
- Ketscher A, Ketterer S, Dollwet-Mack S, et al. Neuroectoderm-specific deletion of cathepsin D in mice models human inherited neuronal ceroid lipofuscinosis type 10. Biochimie. 2016;122:219–226.
- Koch M, May U, Kuhns S, et al. Interleukin 27 induces differentiation of neural C6-precursor cells into astrocytes. Biochem Biophys Res Commun. 2007;364:483–487.
- Mikhaylova M, Bär J, van Bommel B, et al. Caldendrin Directly Couples Postsynaptic Calcium Signals to Actin Remodeling in Dendritic Spines. Neuron. 2018;97:1110–1125.e14.