
ABSTRACT
The disruption of MTOR-regulated macroautophagy/autophagy was previously shown to cause autistic-like abnormalities; however, the underlying molecular defects remained largely unresolved. In a recent study, we demonstrated that autophagy deficiency induced by conditional Atg7 deletion in either forebrain GABAergic inhibitory or excitatory neurons leads to a similar set of autistic-like behavioral abnormalities even when induced following the peak period of synaptic pruning during postnatal neurodevelopment. Our proteomic analysis and molecular dissection further revealed a mechanism in which the GABAA receptor trafficking function of GABARAP (gamma-aminobutyric acid receptor associated protein) family proteins was compromised as they became sequestered by SQSTM1/p62-positive aggregates formed due to autophagy deficiency. Our discovery of autophagy as a link between MTOR and GABA signaling may have implications not limited to neurodevelopmental and neuropsychiatric disorders, but could potentially be involved in other human pathologies such as cancer and diabetes in which both pathways are implicated.
MTOR dysregulation is well-documented to be involved in the pathogenesis of autism spectrum disorder (ASD), and there is growing evidence pointing to a contribution to other neurodevelopmental and neuropsychiatric disorders as well. As a downstream pathway regulated by MTOR signaling, recent studies suggest that there is reduced autophagic flux in the brains of ASD patients compared to healthy controls. Moreover, autophagy deficiency in excitatory neurons results in neuronal hyperexcitability, increased epileptic activity, and autistic-like social behavioral deficits. Together, these findings indicate that MTOR regulation of autophagy is critically important to the proper functioning of the brain, and a disruption of which manifests as behavioral abnormalities relevant to ASD and other neurological disorders. Unfortunately, it has been unclear how autophagy deficiency affects neuronal function and behavior.
For many neuropsychiatric and neurodevelopmental disorders including ASD, a predominant hypothesis proposes that changes in what is commonly known as the excitatory-inhibitory balance or ratio (E-I balance/ratio), contributed by excitatory and inhibitory neurotransmission, affect how different parts of the brain communicate with each other and are thus thought to lead to behavioral abnormalities. We therefore examined how autophagy deficiency in mature GABAergic inhibitory neurons of the forebrain may affect their functions and disturb behavior in adolescent mice [Citation1]. To our surprise, both conditional deletion of Atg7 in GABAergic interneurons or excitatory neurons leads to a common set of ASD-like behavioral abnormalities including social deficits, increased distress and anxiety, suggesting that a similar molecular mechanism is responsible.
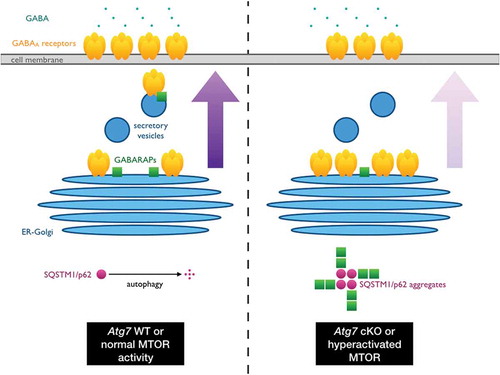
When autophagy is suppressed or disrupted, the cargo carrier SQSTM1/p62 accumulates and in turn forms aggregates in affected cells. We reasoned that aggregation of other proteins and/or co-aggregation of proteins with SQSTM1 may occur in the absence of autophagy, leading to a reduction of the functional pool of such proteins, thereby causing catastrophic disturbances in neuronal functions. We thus employed an unbiased, quantitative mass spectrometry-based method to identify differentially aggregated proteins in Atg7 conditional knockout (cKO) brains by combining semi-denaturing detergent agarose gel electrophoresis (SDD-AGE) and stable isotope labeling in mammals (SILAM), and found GABARAPL2 to have differentially formed high-molecular weight species in the absence of autophagy. Further analyses demonstrated that GABARAPL2, along with its paralogs GABARAP and GABARAPL1, have undergone conformational changes and are all sequestered into the SQSTM1-positive aggregates formed in autophagy-deficient neurons of both Dlx5-Cre and Camk2-Cre Atg7 cKO mice. Importantly, we found that the surface expression of GABAA receptors in Atg7 cKO neurons is reduced, consistent with the suggested function of GABARAPs in the trafficking of GABAA receptors. Because GABA inputs are typically inhibitory in nature, we also observed that autophagy-deficient GABAergic interneurons are firing more frequently because they are effectively receiving less inhibition, thus disturbing the E-I balance in Atg7 cKO mice.
We subsequently performed a series of experiments demonstrating the SQSTM1-dependent effects on GABARAPs in Atg7 cKO neurons. Specifically, we found that (1) the knockdown of Sqstm1 rescues the GABAA receptor trafficking deficits in Atg7 cKO neurons, and (2) the overexpression of SQSTM1 in wild-type neurons similarly leads to the formation of SQSTM1-positive aggregates, which sequester GABARAPs, and result in a reduction of surface GABAA receptors. To examine whether this molecular mechanism has broader implications for neurodevelopmental and neuropsychiatric disorders, we induced MTOR hyperactivation via Tsc2 knockdown and observed that the resulting suppression of autophagy leads to a similar sequestration of GABARAPs by SQSTM1-positive aggregates and ultimately disrupts GABAA receptor trafficking.
Finally, we also observed using post-mortem human brain samples from ASD patients and healthy controls that a subset of ASD patients had increased amounts of detergent-insoluble GABARAPs and SQSTM1 in their brains, possibly pointing to compromised GABAA receptor trafficking and E-I imbalance as a pathogenic mechanism for those ASD patients.
Altogether, our study illustrates a physiological link between autophagy and GABA signaling (). Interestingly, previous studies demonstrated hyperactivity for both Atg7 cKO and Tsc1 mutant neurons, and even suggested that the hyperactivity is due to a reduction of inhibitory inputs received by the affected cells. Our findings thus reconcile those observations and bridge the gap between MTOR hyperactivation, disturbance of E-I balance, and abnormal ASD-like behaviors. Furthermore, given that MTOR and GABA signaling are intertwined in other human pathologies like cancer and diabetes, it will be of interest to examine how autophagy links the two pathways under those conditions, and whether therapeutic modulation of autophagy may offer beneficial effects.
Figure 1. SQSTM1/p62 aggregates link MTOR and GABA signaling. Under normal physiological conditions, GABARAP family proteins mediate the trafficking of GABAA receptors from the ER-Golgi to the cell surface of neurons to receive inhibitory signals from GABAergic interneurons. However, in autophagy-deficient or MTOR-hyperactivated neurons, SQSTM1/p62 accumulates and forms protein aggregates, which in turn sequester GABARAPs. As the conformational status and subcellular localization of GABARAPs are altered by this co-aggregation event, their ability to mediate GABAA receptor trafficking is reduced and thus the affected neurons become hyperactive due to a loss of inhibitory inputs. The resulting shift in excitatory-inhibitory balance in turn contributes to the autistic-like behavioral abnormalities exhibited by autophagy-deficient mice and mouse models or ASD patients with hyperactivated MTOR signaling due to mutations in upstream regulatory molecules such as PTEN and TSC1/2.
Disclosure statement
No potential conflict of interest was reported by the authors.
Related Research Data
Reference
- Hui KK, Takashima N, Watanabe A, et al. GABARAPs dysfunction by autophagy deficiency in adolescent brain impairs GABAA receptor trafficking and social behavior. Sci Adv. 2019;5:eaau8237.