
ABSTRACT
The role protein aggregates play in the pathogenesis of neurodegenerative diseases has been a question since their initial observation. In this autophagic punctum, we discuss our recent findings of how the selectivity scaffold/adaptor WDFY3/Alfy is required for the turnover of aggregated mutant HTT (huntingtin; mHTT) in the adult brain, and how it confers resistance to Huntington disease (HD)-like symptoms. Depletion of WDFY3 in a mouse model of HD accelerates mHTT accumulation, and this is accompanied by an accelerated onset of motoric and neuropathological phenotypes, indicating that WDFY3 levels and the rate of aggregate accumulation can modify disease pathogenesis. Given that the accelerated accumulation is also recapitulated in medium spiny neurons created via direct conversion from human HD fibroblasts, we propose that WDFY3 is a genetic modifier of HD and suggest that it may also influence aging and the pathogenesis of other neurological disorders.
The accumulation of insoluble protein deposits is a shared feature across neurodegenerative diseases, but their role in pathogenesis has been a subject of debate. One of the difficulties of examining this closely has been our inability to separate the accumulation of the aggregated protein from the expression of the aggregation-prone protein itself. The large PtdIns3P-binding protein WDFY3 mediates the selective macroautophagic turnover of aggregated disease-relevant proteins in cellular systems while leaving the non-aggregated pool of protein untouched; but whether this observation extends to the mammalian brain was previously unexplored. In Fox et al. [Citation1] we sought first to determine whether WDFY3 plays a similar role in the adult brain, and then to examine its importance in a human disease-relevant system by genetically depleting WDFY3 in three Huntington disease models. We also employed WDFY3 as a tool to assess the potential toxicity of insoluble mutant HTT aggregates, and to determine how selective macroautophagy of disease-relevant aggregates may confer disease resistance.
The cause of HD is an expansion of a polymorphic CAG repeat within exon 1 of the HD gene that exceeds 40 repeats, causing the protein product to accumulate and aggregate in different neuronal subtypes within the adult brain. To determine WDFY3’s role in aggregate clearance in vivo, we created a conditional HD model that expresses mHTT under tet-regulation (HD103Q), and crossed this mouse with an inducible knockout (iKO) model of Wdfy3, that allowed us to eliminate WDFY3 in the adult brain, to determine acutely when WDFY3 is necessary during aggregate clearance. This mouse, called HDAlfy, provided a correlate in the mammalian brain of our previous findings in cellular systems, which demonstrated that WDFY3 is required for mHTT turnover, without affecting general macroautophagy.
Next, to test the hypothesis that aggregate turnover modifies disease pathogenesis, we created a heterozygous Wdfy3 knockout mouse and crossed it with an HD mouse model, BACHD. Depletion to 50% of normal WDFY3 levels in BACHDAlfy mice leads to an accelerated accumulation of detergent-insoluble mHTT aggregates. These mice also have an earlier onset of HD-like motor behaviors and more striatal GFAP reactivity, an indicator of neuronal stress. However, BACHDAlfy mice do not show accelerated loss of brain cells or volume. These data suggest that partial loss of WDFY3 affects protein aggregation and is sufficient to accelerate behavioral and neural pathogenesis, but not direct cytotoxicity.
We additionally worked with medium spiny neurons generated through direct neuronal conversion from HD patient fibroblasts. Application of a WDFY3 shRNA, triggering a 60% drop in WDFY3 levels, increases the appearance of mHTT aggregates in this model, again without affecting general autophagy or cytotoxicity. Therefore, across three disparate HD models, our findings indicate that WDFY3 is essential for the removal of mHTT deposits, and that reducing WDFY3-mediated turnover in a disease state leads to biochemical, pathological, and behavioral consequences. We propose that WDFY3 therefore has the potential to act as a genetic modifier of HD pathogenesis, and that this role for WDFY3 may extend to a variety of proteinopathies ().
Figure 1. Schematic model summarizing findings. In HD, translation of the trinucleotide repeat expansion that exceeds 40 repeats in the HD gene leads to expression of mutant HTT (mHTT), which leads to the symptoms and neuropathology associated with the disease. The expanded polyglutamine also causes mHTT to misfold, and the resulting aggregates can worsen symptoms. These aggregated structures are handled by WDFY3-mediated selective macroautophagy in the adult brain, and decreasing WDFY3 levels will augment aggregation, and drive pathology.
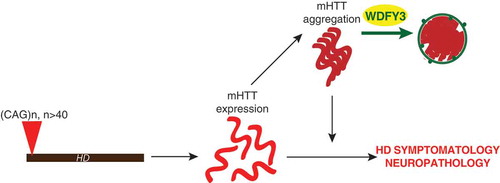
Given that multiple cellular systems exist to turn over aberrant proteins such as mHTT, including the ubiquitin-proteasome system, it is unlikely that neurons are completely reliant on WDFY3-mediated or selective macroautophagy to maintain homeostasis. Indeed, the impact of the depletion of WDFY3 by 50% is only observed upon the presence of an additional stressor – the expression of a protein that has a strong propensity to misfold. Instead, we suggest that WDFY3 may act as the cell’s final recourse to remove existing deposits when their presence becomes overwhelming. Our study focused on HD models, but WDFY3’s role as a macroautophagic scaffold/adaptor is more generalized, encompassing many disease-relevant deposits, ubiquitinated structures, and large protein complexes. Though our findings are suggestive of a toxic role for insoluble mHTT deposits, this toxicity may be indirect: if WDFY3 coordinates the turnover of essential biological complexes, then the distraction presented by accumulated misfolded proteins might be sufficient to wreak havoc on critical systems. As some studies suggest, the formation of the proteinaceous inclusions in many of these diseases may initially be a protective reflex; however, over time, the burden of eliminating these structures becomes overwhelming. This reasoning could contribute to an explanation of why age plus the toxicity of a disease-relevant protein ultimately converges upon symptomology during adulthood. Consequently, the rate of aggregated protein accumulation matters for symptom onset.
Our group and others have previously defined an essential role for WDFY3 in development from flies to humans, but given that the majority of neurodegenerative disorders occur later in life, it will be essential to define the basal role of WDFY3 in the normal aging brain. If aggrephagy is indeed a rate-limiting step in alleviating the cellular stress that disrupts neural circuitry during aging and disease, then driving selective turnover by upregulating WDFY3 or increasing its efficiency could have therapeutic value in HD and other disorders.
Disclosure Statement
The authors have no conflicts to declare
Additional information
Funding
Reference
- Fox LM, Kim K, Johnson CW, et al. Huntington’s disease pathogenesis is modified in vivo by Alfy/Wdfy3 and selective macroautophagy. Neuron. 2020 Mar 4;105(5):813–821.e6. doi:10.1016/j.neuron.2019.12.003. Epub 2019 Dec 30.