
ABSTRACT
Elucidation of the membranes contributing to autophagosomes has been a critical question in the field, and an area of active research. Recently, we showed that key events in autophagosome formation, from PtdIns3P formation/WIPI2 recruitment to LC3-GABARAP membrane conjugation, occur on the RAB11A-positive compartment (recycling endosomes). This observation raised the question of how the LC3-positive autophagosome precursors detach from the recycling endosome. We recently observed that DNM2 (dynamin 2) mediates this step, and described how the DNM2R465W mutation that causes centronuclear myopathy (CNM) leads to the accumulation of autophagic structures on recycling endosomes, thereby stalling macroautophagy/autophagy. This physiologically important step highlights the importance of understanding release of nascent autophagosomes from the recycling endosomes as part of the autophagy itinerary.
Autophagy is an important cellular process that mediates the sequestration and lysosomal degradation of cytoplasmic contents via double-membraned autophagosomes. While diverse cellular membranes appear to contribute to autophagosome formation, our data have argued that key events, from PtdIns3P formation through LC3 conjugation, occur on the RAB11A-positive recycling endosomes. Indeed, WIPI2 binding to RAB11A, a marker protein for recycling endosomes, is required for WIPI2 recruitment and subsequent steps in autophagosome biogenesis. The extent of compromise of autophagosome formation and autophagic substrate clearance resulting from interruption of the WIPI2-RAB11A interaction suggested that these events on recycling endosomes are relevant to most, if not all, mammalian autophagosomes. Furthermore, the tubular-vesicular nature of recycling endosomes can explain how autophagosomes have double membranes and also how extracellular domains of TFRC (transferrin receptor), which traffics through recycling endosomes, are found between the inner and outer membranes of autophagosomes or autophagosome precursors. A corollary of the proposition that recycling endosomes are the core platforms on which key events of autophagosome formation occur, is that impairing separation of the nascent autophagosomes from the RAB11A platform should cause accumulation of LC3-positive structures on recycling endosomes and impair autophagy.
We tested if DNM2 (dynamin 2) was a key mediator of nascent autophagosome release from recycling endosomes. DNM2, a GTPase that can mediate membrane fission, is implicated in many cellular processes, although it is most prominently associated with the regulation of clathrin-mediated receptor endocytosis. Some mutations in DNM2 cause Charcot–Marie–Tooth disease, a form of hereditary motor and sensory neuropathy, whereas other mutations cause centronuclear myopathy (CNM), a disease that manifests with muscle weakness and wasting. The DNM2R465W CNM mutation results in the accumulation of immature autophagic structures and centrally nucleated myofibers similar to those seen in autophagy-deficient mouse muscles. We found that DNM2 is required for autophagosome release from recycling endosomes, and scission of this compartment. DNM2 is recruited to sites of autophagosome formation via direct interactions of its LC3-interacting region (LIR) with LC3 family members. Cells with LIR mutations in DNM2 (DNM2W525L) have a defect in autophagy caused by an accumulation of LC3-positive structures on recycling endosomes. These data support our hypothesis that the recycling endosomes are indeed the platform where key steps of autophagosome formation (PtdIns3P formation through LC3 conjugation) occur.
The DNM2R465W mutation that causes CNM mimics the phenotype of DNM2 with a LIR mutation. EM of cells with the CNM R465W mutation revealed autophagosomes still attached to tubular structures (very likely recycling endosomes). However, R465W is not in the DNM2 LIR. Our data suggested that DNM2R465W is perfectly functional in autophagy in isolation, but the mutation increases its binding to another partner, ITSN1 (intersectin 1), on the plasma membrane. This decreases the amount of DNM2 on the recycling endosomes and impairs autophagy. Decreasing the level of ITSN1 restores DNM2R465W localization to LC3 on recycling endosomes, and rescues autophagy function. Similarly, overexpression of ITSN1 in normal cells mimics the effects of the DNM2 CNM or LIR mutations or DNM2 knockdown, supporting the competition mechanism we proposed (). These data suggest that DNM2 is important for the release of autophagosomes from the recycling endosome where they originate.
Figure 1. The figure shows DNM2 functions connected with two different binding partners. The DNM2R465W CNM mutation favors DNM2 binding with ITSN1, sequestering DNM2 away from its interaction with LC3 and its function in autophagy. Loss of DNM2 from the recycling endosomes impairs the release of autophagic structures from this compartment. The LIR domain of DNM2, responsible for its interaction with LC3 on recycling endosomes, is highly conserved in DNM1 and DNM3, as well as in other species.
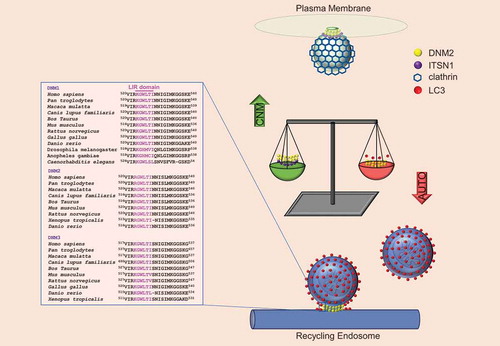
The DNM2R465W CNM mutation reinforces the fact that most proteins have multiple functions in cells. This mutation reveals an extraordinary disease mechanism where the mutant protein binds more strongly to one of its partners, thus sequestering it away from another partner (LC3), resulting in a compromise of one of its functions (autophagy). While we are not aware of other examples of this type of mutation, we suspect that this phenomenon may be common in disease and in physiological regulation. We have called this a “Location, location, location” mutation as the mislocalization of the potentially functional protein causes loss of function. This mutation and the DNM2 LIR mutant act as dominant-negative mutations, compatible with the oligomerization of DNM2 and previous observations that selective knockdown of the mutant allele in heterozygous mice with this DNM2R465W mutation ameliorates disease.
DNM2 is ubiquitously expressed and is part of a larger family of dynamin proteins including DNM1 and DNM3 that are mostly expressed in the brain. The presence of other DNM isoforms, such as DNM1 and DNM3 in neuronal tissues, may buffer/outcompete effects of DNM2 mutations in the central nervous system. Indeed, the LIR domain of DNM2 is perfectly conserved in DNM1 and DNM3, as well as in other species, raising the possibility that DNM1 and DNM3 may also interact with LC3 for nascent autophagosome release from recycling endosomes.
Acknowledgments
We are grateful to the UK Dementia Research Institute (funded by the MRC, Alzheimer’s Research UK and the Alzheimer’s Society) (DCR) and The Roger de Spoelberch Foundation (DCR) for funding.
Disclosure statement
No potential conflict of interest was reported by the authors.