
ABSTRACT
Massive expansions of the hexanucleotide in C9orf72 are the primary genetic origins of familial amyotrophic lateral sclerosis (ALS) and frontal temporal dementia (FTD). Current studies have found that this repeat sequence participates in the disease process by producing neurotoxic substances and reducing the level of C9orf72 protein; however, the progress in the functional study of C9orf72 is slow. Recently, a stable complex, consisting of C9orf72, SMCR8, and WDR41, has been implicated in regulating membrane trafficking and macroautophagy. We reported the cryo-electron microscopy (cryo-EM) structure of the C9orf72-SMCR8-WDR41 complex (CSW complex), unveiling that the CSW complex is a dimer of heterotrimers. Intriguingly, in the heterotrimer of the C9orf72-SMCR8-WDR41, C9orf72 interacts with SMCR8 in a manner similar to the FLCN-FNIP2 complex. Nevertheless, WDR41 is connected to the DENN domain of SMCR8 through its N-terminal β-strand and C-terminal helix but does not directly interact with C9orf72. Notably, the C9orf72-SMCR8 complex was demonstrated to act as a GAP for RAB8A and RAB11A in vitro.
Abnormal expansion of hexanucleotide (GGGGCC) in the intron of C9orf72 accounts for most cases of familial amyotrophic lateral sclerosis (ALS) and frontal temporal dementia (FTD), two of the most prevalent neurodegenerative diseases in adult. Two hypotheses, toxic “gain of function” and “loss of function”, have been proposed to explain the mechanism of disease onset. The neuron toxicity from gain of function attributes to the neurotoxic substances, including RNA G-quadruplexes and dipeptide repeat aggregates that are produced from the expansion of the hexanucleotide. Synergically, the reduction of the mRNA and protein of C9orf72 due to the mutation are named “loss of function”. Substantial evidence indicates that C9orf72, in a complex with SMCR8 (C9orf72-SMCR8 complex subunit) and WDR41 (WD40 repeat domain 41), functions as a guanine nucleotide exchange factor of RAB8A and RAB39B and regulates the autophagic flux by interacting with the ULK1 complex directly or indirectly. Nevertheless, the precise function of C9orf72 is still elusive, which hinders our understanding of the pathogenic mechanism of abnormal C9orf72.
In order to comprehensively understand the function of the CSW complex, multi-disciplinary methods, including, but not limited to, single-particle cryo-EM analysis, bioinformatics analysis, and biochemical assays, were applied [Citation1]. First of all, we obtained the recombinant protein of the CSW complex in a eukaryotic expression system, and solved the structure using cryo-EM at a resolution at 3.2 Å. Remarkably, we found that the ratio of the three proteins in the CSW complex is 2:2:2, rather than the previously supposed 1:1:1. Notably, the structure of the CSW complex shows a dimeric assembly of heterotrimers of C9orf72-SMCR8-WDR41 (). Intriguingly, we found that both the C-terminal region of C9orf72 and the DENN domain of SMCR8 are necessary for the dimerization of C9orf72-SMCR8-WDR41 using analytical ultracentrifugation experiments and analytical gel filtration. This observation shows a novel mechanism of interaction between the DENN domains.
Figure 1. Model of the CSW complex. (A) The overall structure of the CSW complex. The structure shows a two-fold symmetry. C9orf72, SMCR8, and WDR41 are colored in light blue, light green, and orange, respectively. The domains are denoted. The arginine finger is highlighted in red dashed lines. The dimer interface of the two protomers is denoted in blue dashed lines. (B) The proposed model of how the CSW complex promotes the fusion of endosomes: the CSW complex tethers two endosomes using WDR41, and promotes the fusion by activating RABs located on the surface of endosomes. C9orf72, SMCR8, and WDR41 are colored in blue, green, and yellow, respectively, while RABs are shown in dark blue. The arginine finger is highlighted as a dark pink line. The organization of domains is based on the structure in (A).
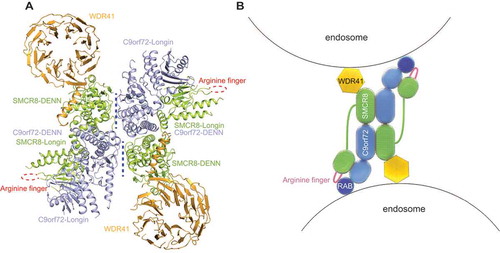
Besides, the relationship among the three subunits was defined. In the structure of the C9orf72-SMCR8-WDR41 protomer, C9orf72 and SMCR8 interact with each other in a way that resembles the FLCN-FNIP2 complex. Meanwhile, WDR41 is bound to the DENN domain of SMCR8 through its N-terminal β-strand and C-terminal helix without directly contacting with C9orf72 (). Mutagenesis analysis shows that the triad of L445, F446, and L449 from the C-terminal helix of WDR41 is essential to the interaction with SMCR8, suggesting that the hydrophobic interaction plays a critical role in the binding. However, a mutant of SMCR8, of which the eight residues on the DENN that interact with L445, F446, and L449 of WDR41 were mutated into alanine, shows a weak binding to WDR41, indicating that more residues of SMCR8 participate in this interaction.
Both C9orf72 and SMCR8 belong to the DENN family of proteins, and, generally, these proteins function as guanine nucleotide exchange factors of RAB GTPases. In order to assess the stimulatory effect of the CSW and the C9orf72-SMCR8 complex on the potential target RABs, we carried out a bioluminescence-based GTPase activity assay. Unexpectedly, among five reported RABs, only RAB8A and RAB11A can be stimulated by CSW or C9orf72-SMCR8. However, we did not observe that the CSW or C9orf72-SMCR8 complex can facilitate GDP dissociation from RAB8A and RAB11A. It is worth noting that the structure of the C9orf72-SMCR8 complex is similar to that of the FLCN-FNIP2 complex. Impressively, C9orf72 corresponds to FNIP2, whereas SMCR8 corresponds to FLCN. Intriguingly, the FLCN-FNIP2 complex functions as the GTPase activating protein (GAP) of RRAGC or RRAGD, which inspired us to think about whether the CSW and the C9orf72-SMCR8 complex may function as a GAP for RAB8A and RAB11A. Similarly, bioluminescence-based GAP activity titration experiments showed that with the stimulation of the CSW or the C9orf72-SMCR8 complex, the amount of GTP consumed by both RAB8A and RAB11A increases significantly. Moreover, Arg164 of FLCN is the arginine finger of the FLCN-FNIP2 complex. Sequence alignment shows that Arg147 of SMCR8 is highly conserved in animals and corresponds to Arg164 of FLCN. Excitingly, mutation of Arg147 to alanine abolishes the GAP activity of both the CSW complex and the C9orf72-SMCR8 complex. Remarkably, dimerization of C9orf72-SMCR8 and WDR41 are not required for the GAP activity. Of note, research from Hurley and Ferguson labs indicated that the CSW complex shows GAP activity for ARF1, and WDR41 is in charge of localizing the CSW complex to the lysosome by binding to SLC66A1. Together, the GAP function of the CSW complex in membrane trafficking and autophagy has been demonstrated. Based on these findings, a model for how the CSW complex functions on the membrane is proposed: the CSW binds with two different vesicles containing RAB8A or RAB11A using WDR41 and then activates the RABs on the membrane to facilitate the fusion of the two vesicles ().
The life expectancy of patients with ALS is approximately 2–4 years from onset. Although more than 60 academic institutions and companies have conducted more than 200 clinical trials for the treatment of ALS, no curative drug has been found for this disease. Unfortunately, the pathogenesis of ALS has not been fully understood. Thereby, the function of C9orf72, one of the most hopeful targets for ALS and FTD, has been placed under the spotlight. Hopefully, the RAB GAP activity of the CSW complex revealed in this study not only provides a basis for further research on the physiological function of the CSW complex, but also sheds light on the pathogenic mechanism of C9orf72 in ALS and FTD.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Reference
- Tang D, Sheng J, Xu L, et al. Cryo-EM structure of C9ORF72-SMCR8-WDR41 reveals the role as a GAP for Rab8a and Rab11a. Proc Natl Acad Sci U S A. 2020;117:9876–9883.