
Macroautophagy (afterward named autophagy) is a central process participating in coordinating energy metabolism, recycling of damaged organelles, and in degrading protein aggregates and pathogens. One of the important regulators of autophagy is the AMP-activated protein kinase (AMPK), a heterotrimeric complex with a catalytic PRKAA/α subunit and regulatory PRKAB/β and PRKAG/γ subunits. AMPK is a key sensor of the cellular energy status recognizing increasing ADP/AMP levels through its PRKAG/γ subunit in cells. Activation also requires phosphorylation of the PRKAA/α subunit at threonine 172 (T172) by STK11/LKB1 (serine/threonine kinase 11). Downstream effectors are components of the ULK1/Atg1 complex and the PIK3C3/VPS34-containing class III phosphatidylinositol 3-kinase (PtdIns3 K) complex, both of which are necessary for the induction of autophagy. Substrates of AMPK are ULK1 and two PtdIns3 K complex components, BECN1 (beclin 1) and PIK3C3/VPS34. These findings suggest that both complexes are directly controlled by the energy sensor AMPK. However, the precise mechanisms that are triggered by the AMPK-dependent phosphorylation are not well understood. Moreover, it is unclear whether the substrates noted above are the only downstream effectors of AMPK. With this information in mind, the question we wanted to address is whether additional substrates exist that connect AMPK to the induction of autophagy. In this effort we identified CCNY (cyclin Y)-CDK16 as a downstream, positive effector of AMPK-induced autophagy.
We first applied recombinant AMPK to a protein microarray in the presence of 33P-γ-ATP [Citation1]. This approach has advantages and disadvantages compared to other methods that are used to define substrates. Advantages include the fact that direct phosphorylation reactions can be observed. The proteins on the array, more than 9000, were expressed in insect cells using a baculovirus system. Thus, the proteins carry at least some post-translational modifications that might be relevant for being recognized by AMPK or any other kinase analyzed in such an experimental system. Moreover, the proteins are spotted in comparable concentrations, which might help to define low-abundance substrates. But of course it is an in vitro experiment, which is a major limitation. Of note is that these arrays are no longer manufactured, most likely a response to the ever more powerful mass spectrometry approaches that allow the analysis of entire phospho-proteomes of a cell.
Our in vitro and cellular analyses supported the mass spectrometry data that implicated serine 326 (S326) as the phospho-acceptor site on CCNY by AMPK. CCNY associates with different cyclin-dependent kinases (CDKs), including CDK14, CDK15, and CDK16. These different kinase complexes have been poorly studied. Unlike the situation for other CDKs, none of the three kinases seems to have a major impact on the cell cycle. Rather, they are associated with cellular differentiation and with intracellular transport processes. Because of the strong link between AMPK and autophagy, the three CCNY-CDK complexes were tested for effects on autophagy. Importantly, only CCNY in complex with CDK16, but neither with CDK14 nor CDK15, affects autophagy. The more detailed analysis revealed that overexpression of CCNY-CDK16 stimulates autophagy dependent on the presence of ULK1 and BECN1, suggesting that CCNY-CDK16 regulates autophagy upstream of the ULK1 and PtdIns3 K complexes (). In contrast, reducing or eliminating the expression of CCNY-CDK16 interferes with AMPK-induced autophagy, indicating that AMPK is indeed upstream of CCNY-CDK16. Mechanistically the S326 phosphorylation by AMPK promotes the interaction of CCNY with CDK16, which in turn autophosphorylates S336, which serves as a marker for active CCNY-CDK16.
Figure 1. Model of the interaction of AMPK with CCNY-CDK16 and downstream implications for the induction of autophagy. Under low energy status, AMPK induces autophagy by negatively regulating the MTOR complex, and thus reducing protein biosynthesis, and activating the ULK1/Atg1 and PIK3C3/VPS34-containing PtdIns3 K complexes that promote phagophore formation and cargo loading. Additionally, AMPK has to phosphorylate CCNY at S326 to activate the CCNY-CDK16 complex for the induction of autophagy. The phosphorylation of CCNY at S326 allows the interaction with CDK16 and promotes the kinase activity of the complex. This in turn stimulates auto-phosphorylation of CCNY at S336, a modification that indicates an active kinase complex. We suggest CCNY-CK16 phosphorylates factors associated with the autophagic machinery (red arrows). These factors might function early in the process, for example, as part of the ULK1/Atg1 and PIK3C3/VPS34 PtdIns3 K complexes, or further downstream, as indicated by the question marks.
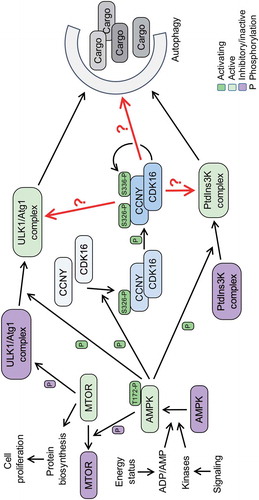
What needs to be addressed now? The key question is to identify the substrates of CCNY-CDK16 that mediate its stimulatory role on autophagy. Little is known about substrates of this kinase and even less about its specificity. Thus, efforts have to be developed to determine mechanistic links between CCNY-CDK16 and substrates relevant for regulation and/or execution of autophagy. One might assume that components of the ULK1 and PtdIns3 K complexes are substrates; however, the dependence of CCNY-CDK16-induced autophagy on ULK1 and BECN1 might simply suggest that some essential process is no longer functional and that CCNY-CDK16 is not sufficient to overcome the defect. Thus, critical substrates may exist in either of the two complexes, but further downstream targets are also possible.
Can one make predictions where in the autophagy process a CCNY-CDK16 substrate might be located? As discussed in our paper, CDK16 functions in vesicular transport and in actin cytoskeleton organization, both being relevant for autophagy. Thus, CCNY-CDK16 might have substrates that are involved in providing membrane components for the initial phagophore formation or that are important late in autophagy, for example, by bringing autophagosomes in close proximity to lysosomes. In this context the interaction between SEC23 of the COPII complex and CDK16 has to be mentioned. Besides directly regulating the induction of autophagy, CCNY-CDK16 could also be involved in balancing the cellular decisions between autophagy and apoptosis. Pointing to this function is the cyclin-dependent kinase inhibitor CDKN1B/p27KIP1, which promotes autophagy and inhibits apoptosis, and has been described as a CCNY-CDK16 substrate. We expect that screening for substrates and their functional characterization will provide answers to these questions.
Finally, the association of autophagy with disease, including inflammatory, tumorigenic and neurodegenerative processes, is worth pointing out. If CCNY-CDK16 is an important regulator of these diseases, alterations in the function of this kinase might be expected. Indeed, links to inflammatory bowel disease for CCNY and to cancer for CDK16 have been established. Moreover, the high expression in neuronal cells, with a functional and direct interaction with CDK5, suggests a link of CCNY-CDK16 to neurodegeneration. However, little direct evidence supports the suggestion that CCNY-CDK16 kinase activity is involved in these processes at present. Again, defining CCNY-CDK16 substrates may provide links toward one or the other disease that can be addressed experimentally. Thus, CCNY-CDK16 may emerge as a drug target.
Acknowledgements
The work in our laboratories was supported by the START and IZKF programs of the Medical School of the RWTH Aachen University, a VIDI-Innovational Research Grant from the Netherlands Organization of Scientific Research, and the Deutsche Forschungsgemeinschaft.
Disclosure statement
No potential conflict of interest was reported by the authors.
Reference
- Dohmen M, Krieg S, Agalaridis G, et al. AMPK-dependent activation of the cyclin Y/CDK16 complex controls autophagy. Nat Commun. 2020;11:1032.