
ABSTRACT
Hepatic lipid homeostasis is controlled by a coordinated regulation of various metabolic pathways involved in de novo synthesis, uptake, storage, and catabolism of lipids. Disruption of this balance could lead to hepatic steatosis. Peroxisomes play an essential role in lipid metabolism, yet their importance is often overlooked. In a recent study, we demonstrated a role for hepatic peroxisomal β-oxidation in autophagic degradation of lipid droplets. ACOX1 (acyl-Coenzyme A oxidase 1, palmitoyl), the rate-limiting enzyme of peroxisomal β-oxidation, increases with fasting or high-fat diet (HFD). Liver-specific acox1 knockout (acox1-LKO) protects mice from hepatic steatosis induced by starvation or HFD via induction of lipophagy. Mechanistically, we showed that hepatic ACOX1 deficiency decreases the total cytosolic acetyl-CoA levels, which leads to reduced acetylation of RPTOR/RAPTOR, a component of MTORC1, which is a key regulator of macroautophagy/autophagy. These results identify peroxisome-derived acetyl-CoA as a critical metabolic regulator of autophagy that controls hepatic lipid homeostasis.
Nonalcoholic fatty liver disease (NAFLD), the most common liver disease worldwide, is caused by dysregulation of lipid homeostasis, which is controlled by coordinated regulation of various metabolic pathways. Peroxisomes are involved in many aspects of lipid metabolism, including ether lipid synthesis, bile acid synthesis, α-oxidation of branched chain fatty acids and β-oxidation of very long chain fatty acids (VLCFA). The first step of peroxisomal β-oxidation is carried out by ACOX1, which is enriched in the liver, and further increases with fasting or high-fat feeding. However, the precise role of ACOX1-mediated fatty acid oxidation in the liver has remained unclear.
To understand the physiological significance of peroxisomal β-oxidation in hepatocytes, we generated mice with liver-specific knockout of Acox1 (acox1-LKO) [Citation1]. Surprisingly, these mice are protected against starvation- or high-fat diet-induced fatty liver, in contrast to liver-specific inhibition of mitochondrial fatty acid oxidation, which has been previously shown to exacerbate hepatic steatosis. Although acox1-LKO mice exhibit a compensatory increase in mitochondrial β-oxidation activity, it is unlikely that this alone accounts for the protection against hepatic steatosis, because previous studies demonstrate that increasing the liver mitochondrial β-oxidation capacity through hepatic overexpression of Cpt1 is not sufficient to prevent fatty liver. This is presumably because fatty acids must first be liberated from triglycerides through lipolysis before their oxidation in mitochondria. However, neither the gene expression of lipolytic enzymes (PNPLA2/ATGL and LIPE/HSL) nor their lipid droplet localization is altered in acox1-LKO liver [Citation1].
Besides cytosolic lipolysis, triglycerides can alternatively be hydrolyzed by lipophagy, a subtype of autophagy that involves encapsulation of lipid droplets within an autophagosome, which fuses with a lysosome, followed by hydrolysis of triglycerides by LIPA/LAL (lysosomal acid lipase A). Interestingly, the expression of the Lipa gene is increased in the liver of acox1-LKO mice, which prompted us to determine if inhibition of peroxisomal β-oxidation promotes autophagy/lipophagy. In support of this possibility, the protein levels of the autophagy markers SQSTM1/p62 and LC3 are significantly lower in the livers of acox1-LKO mice. Treatment of mice with leupeptin, an inhibitor of lysosomal proteases, results in SQSTM1 and LC3 accumulation, suggesting that acox1 knockout increases autophagic flux. Using hydrodynamic gene delivery to express a plasmid encoding mCherry-GFP dual tandem-tagged LC3 in livers of acox1-LKO and control mice, we confirmed that Acox1 inactivation promotes autophagy activation. Moreover, we demonstrated using immunofluorescence analysis that the colocalization between lipid droplets and lysosomes is increased with acox1 knockout, suggesting lipophagy activation [Citation1]. Together, these results indicate that inactivation of peroxisomal β-oxidation protects against hepatic steatosis by promoting hydrolysis of lipid droplets through lipophagy, followed by oxidation of the released fatty acids in mitochondria ().
Figure 1. A model depicting the proposed molecular mechanism through which peroxisomal β-oxidation regulates MTORC1 activation to inhibit lipophagy. The expression of the peroxisomal fatty acid oxidation enzyme ACOX1 increases with fasting or HFD, resulting in increased production of acetyl-CoA, which is channeled to lysosomes for acetylation of the MTORC1 component RPTOR; this leads to MTOR activation. Phosphorylation of ULK1 at Ser757 by MTOR suppresses the induction of autophagy and lipophagy. Fatty acids liberated through lipophagy may be oxidized in mitochondria.
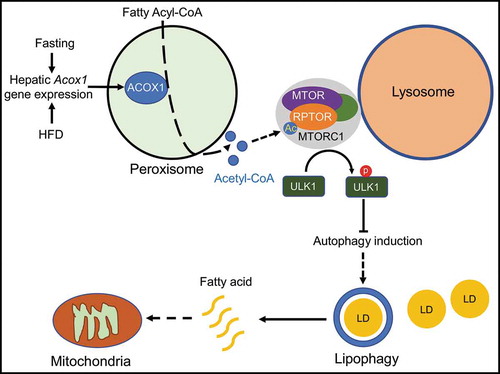
We next sought to understand how disruption of peroxisomal β-oxidation promotes autophagic degradation of lipid droplets in the liver. One of the products of fatty acid oxidation, in general, is acetyl-CoA, a key metabolic regulator of autophagy that has been previously shown to inhibit autophagy through an unclear mechanism. Remarkably, the total hepatic cytosolic acetyl-CoA pool is decreased 50% in fasted acox1-LKO mice as compared to control animals, suggesting that peroxisomal β-oxidation is a major source of this metabolite in the liver. Mechanistically, we showed that this peroxisome-derived pool of acetyl-CoA is involved in acetylation of RPTOR, which is a component of the MTOR (mechanistic target of rapamycin kinase) complex 1 (MTORC1), a key growth and metabolism regulatory complex that inhibits autophagy. Consequently, lysosomal localization and activation of MTOR is impaired, resulting in decreased autophagy-inhibitory Ser757 phosphorylation of ULK1. Increasing acetyl-CoA levels using dichloroacetic acid restores RPTOR acetylation and MTOR activation, inhibits autophagy, and increases hepatic triglycerides in acox1-LKO mice [Citation1]. Together, these data suggest that ACOX1-mediated fatty acid oxidation in the liver maintains a basal level of MTORC1 activation and limits autophagy activation, even in the fasted state. This might explain why autophagy/lipophagy is normally not sufficient to prevent fatty liver.
Although ACOX1 is required for oxidation of VLCFA, the dramatic decrease in total acetyl-CoA in acox1-LKO mice suggests that ACOX1 might be able to catabolize a broader range of fatty acids. An interesting possibility is that fatty acids released by lipophagic hydrolysis of triglycerides are oxidized in peroxisomes by ACOX1 to generate acetyl-CoA, potentially reflecting a negative feedback mechanism to limit mobilization of intracellular lipid stores. Future research will be required to test this hypothesis. Additional work will also be required to determine how acetyl-CoA derived from peroxisomal β-oxidation is specifically channeled to lysosomes to mediate MTORC1 activation.
In summary, our results suggest that acetyl-CoA derived from peroxisomal fatty acid oxidation mediates RPTOR acetylation and MTOR activation, revealing a novel mechanism to restrict autophagic degradation of lipid droplets. Induction of lipophagy via inhibition of peroxisomal acetyl-CoA production might lead to a novel treatment option for NAFLD.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Reference
- He A, Chen X, Tan M, et al. Acetyl-CoA derived from hepatic peroxisomal β-oxidation inhibits autophagy and promotes steatosis via MTORC1 activation. Mol Cell. 2020 Jul 2;79(1):30–42.e4.