
ABSTRACT
PDPK1 (3-phosphoinositide dependent protein kinase 1) is a phosphorylation-regulated kinase that plays a central role in activating multiple signaling pathways and cellular processes. Here, this study shows that PDPK1 turns on macroautophagy/autophagy as a SUMOylation-regulated kinase. In vivo data demonstrate that the SUMO modification of PDPK1 is a physiological feature in the brain and that it can be induced by viral infections. The SUMOylated PDPK1 regulates its own phosphorylation and subsequent activation of the AKT1 (AKT serine/threonine kinase 1)-MTOR (mechanistic target of rapamycin kinase) pathway. However, SUMOylation of PDPK1 is inhibited by binding to PIK3C3 (phosphatidylinositol 3-kinase catalytic subunit type 3). The nonSUMOylated PDPK1 then tethers LC3 to the endoplasmic reticulum to initiate autophagy, and it acts as a key component in forming the autophagic vacuole. Collectively, this study reveals the intricate molecular regulation of PDPK1 by post-translational modification in controlling autophagosome biogenesis, and it highlights the role of PDPK1 as a sensor of cellular stress and regulator of autophagosome biogenesis.
Abbreviations: AKT1: AKT serine/threonine kinase 1; ATG14: autophagy related 14; Co-IP: co-immunoprecipitation; ER: endoplasmic reticulum; hpi: hours post-infection; mAb: monoclonal antibody; MAP1LC3/LC3: microtubule associated protein 1 light chain 3; MOI: multiplicity of infection; MTOR: mechanistic target of rapamycin kinase; pAb: polyclonal antibody; PDPK1: 3-phosphoinositide dependent protein kinase 1; PI3K: phosphoinositide 3-kinase; PIK3C3: phosphatidylinositol 3-kinase catalytic, subunit type 3; RPS6KB1: ribosomal protein S6 kinase B1; SGK: serum/glucocorticoid regulated kinase; SQSTM1: sequestosome 1; SUMO: small ubiquitin like modifier; UBE2I/UBC9: ubiquitin conjugating enzyme E2 I; UVRAG: UV radiation resistance associated
KEYWORDS:
Introduction
Conjugation of the small ubiquitin-related modifier (SUMO) to substrates requires the sequential action of an E1 activating enzyme, an E2 conjugating enzyme, and an E3 protein ligase. SUMO1 (small ubiquitin like modifier 1) was reported to participate in connecting the RANGAP1 (Ran GTPase activating protein 1) to the RANBP2 (RAN binding protein 2) to promote the translocation of RANGAP1 from the cytosol to the nuclear pore [Citation1]. SUMO1 was also shown to accelerate the accumulation of autophagic vacuoles by promoting amyloid-beta production [Citation2]. SUMO modification (SUMOylation) was also shown to abolish the interaction between UBE2K (ubiquitin conjugating enzyme E2 K) and the ubiquitin E1 enzyme [Citation3]. Moreover, SUMOylation not only stimulates phosphorylation of the microtubule-associated protein [Citation4], but it also negatively regulates the tyrosine phosphorylation of PIK3R2 (phosphoinositide-3-kinase regulatory subunit 2) [Citation5]. Correspondingly, several deSUMOylation enzymes have been identified to remove SUMO from substrates, including Ubl-specific proteases and SUMO-specific peptidases (SENPs) [Citation6]. All of this shows that post-translational modification through SUMOylation is involved in various cellular processes, such as nuclear-cytosolic transport, transcriptional regulation, protein stability, stress response, and cell-cycle progression [Citation7].
Autophagy is a highly conserved process that involves the sequestration and transport of damaged organelles, misfolded and toxic proteins, and microorganisms to lysosomes for degradation [Citation8–10]. Autophagy process includes autophagosome formation and maturation. Phosphatidylinositol 3-phosphate (PtdIns3P), a key membrane marker for autophagosome formation, is produced when PIK3C3 (phosphatidylinositol 3-kinase catalytic subunit type 3) phosphorylates the 3-position of phosphatidylinositol [Citation11]. PtdIns3P then recruits autophagy-specific PtdIns3P effectors through its FYVE-binding domain and WD repeat-containing domain, and these effectors induce the nucleation of the phagophore [Citation12]. Following nucleation, autophagy-related proteins, such as WIPI2 (WD repeat domain, phosphoinositide interacting 2) and the ATG12 (autophagy related 12)–ATG5 (autophagy related 5) complex, are recruited to the membrane to promote the conversion of LC3-I to LC3-II [Citation13]. The formation of LC3-II enables the protein to associate with the phagophore. Assembly of LC3-II, together with its associated membrane, as well as several autophagy-related proteins, enables the growth and expansion of the autophagic membrane. However, how SUMOylation is involved in autophagosome formation remains poorly understood.
The PDPK1 exerts its kinase role in multiple signaling pathways by phosphorylating several other kinases, including RPS6KB/p70S6 kinase and RPS6KA/p90S6 kinase [Citation14–17], PRKC (protein kinase C) isoforms [Citation18–21], and the SGK1 (serum/glucocorticoid regulated kinase 1) [Citation22,Citation23]. phosphatidylinositol-3,4,5-triphosphate (PtdIns[3,4,5]P3) formation mediated by class I phosphoinositide 3-kinase (PI3K) recruits PDPK1 and AKT1 to the cell membrane [Citation24], and then PDPK1 phosphorylates AKT1 and activates the MTOR pathway. Phosphorylation of PDPK1 at Ser241 plays an important role in regulating the activity and function of the protein. Phosphorylation of Ser241 is thought to be constitutive and to be independent of growth factor stimulation. However, the mechanism regulating phosphorylation of PDPK1 at Ser241 remains unknown.
In this work, we showed that PDPK1 is SUMOylated. SUMOylation of PDPK1 promotes its phosphorylation and subsequent activation of the AKT1-MTOR pathway. However, the SUMOylation of PDPK1 is inhibited by PIK3C3 binding to SUMOylated/phosphorylated PDPK1. NonSUMOylated PDPK1 (PDPK1 without SUMO modification) induces autophagosome biogenesis by promoting LC3 lipidation. Thus, these intricate SUMOylation/nonSUMOylation modifications regulate the differential function of PDPK1 by binding to differently modified proteins.
Results
PDPK1 is SUMOylated under physiological conditions and during virus infection
To study the post-translational modification of PDPK1, we firstly measured PDPK1 from mouse tissues and different cells by western blotting. Two PDPK1 isoforms with molecular masses of approximately 210 kDa (termed PDPK1*S) and 70 kDa (PDPK1) were isolated with the polyclonal antibody (pAb) to PDPK1 in the brain of mice, but only the 70 kDa form was seen in the lung, kidney, and spleen (). Interestingly, the level of PDPK1*S band was only increased in the brain of mice treated with INS (insulin; an activator of PI3K), but not LY294002 (an inhibitor of PI3K) or rapamycin (an inhibitor of MTOR) (). Consistent with this, when different cell lines were treated with INS, LY294002, or rapamycin, the PDPK1*S bands could be enhanced in INS-treated cells ( and S1A). Moreover, the PDPK1*S band was also confirmed in wild type (WT) and PDPK1-silenced cells transfected with FLAG-tagged PDPK1 upon INS treatment, but not with LY294002 or rapamycin treatment ( and S1B), indicating that PDPK1 is post-translationally modified in vivo and in vitro. By contrast, rapamycin or starvation treatment is able to counteract the effect of INS on induction of modified PDPK1 (Figure S1C). To further test whether virus infection also induces the post-translational modification of PDPK1, we prepared RNA virus and DNA virus-infected cells. Notably, the post-translational modification of PDPK1 was also observed in both RNA virus and DNA virus-infected cells (). These results clearly demonstrate that PDPK1 has the same post-translational modification under physiological conditions and during viral infection.
Figure 1. SUMOylation of PDPK1. (A) PDPK1 with a molecular weight of 210 kDa in the brain of mice. Lysates from the spleen, lung, kidney and brain of BALB/c mice were performed by in vivo SUMOylation detection assay described in material and method section and WB assay with using indicated antibodies. (B) PDPK1*S levels in the brain of drug-treated mice. Mice were intraperitoneally injected with rapamycin (0.4 mg/kg), LY294002 (1.5 mg/kg), INS (1 IU/kg), or DMSO. Lysates from the spleen, lung, kidney and brain of BALB/c mice were performed by in vivo SUMOylation detection assay described in material and method section and WB assay with using indicated antibodies. (C and D) PDPK1*S levels in wild-type and PDPK1-transfected cells. HEK293T cells and FLAG-PDPK1 transfected HEK293T cells were separately treated with rapamycin (R, 5 μM), LY294002 (L, 10 μg/ml), INS (I, 100 nM), or DMSO (D) for 24 h followed by in vivo SUMOylation detection assay described in material and method section and WB assay with using indicated antibodies (# indicates nonspecific bands). (E) The 210-kDa form of PDPK1 was formed upon RNA or DNA virus infections. RNA virus infections included IBDV infection (MOI = 10) in DF-1 cells for 7 h, influenza virus H7N9 (MOI = 10) and PR8 (MOI = 8) infections in A549 cells for 9 h, and VSV infection (MOI = 10) in HEK293T cells for 9 h. DNA virus infections contained PCV2 (MOI = 10) and PRV (MOI = 1) infections in PK15 cells for 72 h. The resultant samples are followed an in vivo SUMOylation detection described in material and method section with using indicated antibodies in immunoblotting analysis (# indicates nonspecific bands). (F) HEK293T cells co-transfected with FLAG-PDPK1 and PCI-NEO-UBE2I and empty vector HA, HA-SUMO1, HA-SUMO2, or HA-SUMO3 for 24 h is followed an in vivo SUMOylation detection by using indicated antibodies in immunoblotting analysis. (G) SUMOylation of PDPK1 is detected with indicated antibodies in immunoblotting analysis, after HEK293T cells were co-transfected with FLAG-PDPK1 and ShCon. or ShUBE2I for 20 h and then treated with INS for 24 h. (H) Colocalization between PDPK1 and SUMO1 in A549 cell lines. Scale bar: 10 μm. (I) HEK293T cells were co-transfected with PCI-NEO-UBE2I, HA-SUMO1 and empty vector FLAG, FLAG-SENP1, FLAG-SENP3, or MYC-PDPK1 for 24 h, and in vivo SUMOylation (described in material and method section) of PDPK1 is detected with using indicated antibodies in immunoblotting assay. (J) Map of SUMOylation sites and SUMO interaction sites in alignment of PDPK1 protein sequence from different species. (K) HEK293T cells were co-transfected with PCI-NEO-UBE2I and FLAG-PDPK1, FLAG-PDPK1CD, FLAG-PDPK1ID, FLAG-PDPK1SD, MYC-SUMO1, or empty vector MYC for 48 h. Co-IP assays were performed by using indicated antibodies. (L) HEK293T cells were co-transfected with PCI-NEO-UBE2I, MYC-SUMO1 and FLAG-PDPK1 or FLAG-PDPK1CD, FLAG-PDPK1ID, empty vector FLAG for 48 h, respectively. Co-IP assays with indicated antibodies. (M) HIS-PDPK1 and HIS-PIK3C3 were expressed in BL21 and purified. In vitro SUMOylation assay was then performed. Immunoblotting analysis of PDPK1 by using indicated antibodies. (N) SUMOylation analysis of PDPK1 by nickel beads affinity-isolation assay under denaturing condition. *, p < 0.05; **, p < 0.01; ***, p < 0.001
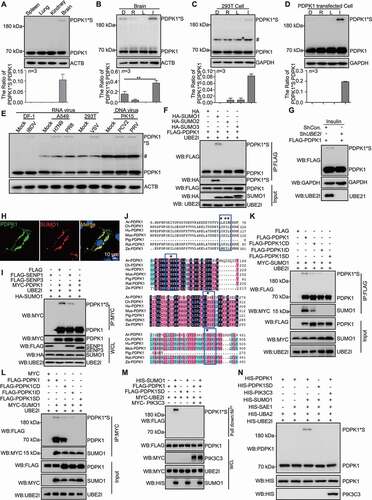
To determine whether the post-translational modification is SUMOylation of PDPK1, cells were separately co-transfected with PDPK1 and UBE2I/UBC9 (ubiquitin conjugating enzyme E2 I), SUMO1, or SUMO2, or SUMO3 and analyzed by immunoprecipitation (IP) assay. SUMO1-conjugated PDPK1, but not SUMO2 or SUMO3, was detected in the co-transfected cells using an anti-FLAG mAb (). Consistently, PDPK1*S was not detected in UBE2I-silenced cells () and PDPK1 colocalized with SUMO1 in transfected cells (), revealing that PDPK1 was SUMOylated with SUMO1. Notably, a significant decrease in PDPK1*S was seen in cells overexpressing the deSUMOylases SENP1 and SENP3 (). These results verify that the PDPK1*S was a SUMO1-modified PDPK1.
To further identify the SUMO modification and interaction sites, we predicted the SUMO conjugated or interacted consensus motif of PDPK1 using online SUMOylation prediction tools (http://sumosp.biocuckoo.org/) and identified the predicted sites of SUMO modification and interaction by biochemical and mass spectrometry (MS) analyzes. Bioinformatics analysis showed that lysine (K)207, K296 and K495 might be SUMOylation sites of PDPK1 and the motif 110IKIL114E might be the SUMO-interaction domain of PDPK1 ( and S1D). To indeed validate these SUMOylation sites of PDPK1, we performed the mass spectrometry (MS) identification of SUMO modification sites. Mass spectrometry analysis displayed that the residues K207, K296 and K495 of PDPK1 were identified as SUMOylation sites (Figure S1E), showing that PDPK1 is a protein with SUMOylation modification. We subsequently generated various substitution mutants of PDPK1, including PDPK1 with K296R and K495 R (PDPK1K296,495R); PDPK1 with K207R and K495R (PDPK1K207,495R); PDPK1 with K207R and K296R (PDPK1K207,296R); PDPK1 with I110R, I112R, and L113R (PDPK1ID); PDPK1 with I110R, I112R, L113R, K296R and K495R (IDK296,495R); PDPK1with I110R, I112R, L113R, K207R and K495R (IDK207,495R); PDPK1 with I110R, I112R, L113R, K207R and K296R (IDK207,296R); PDPK1 with K207, K296R, and K495R (PDPK1CD); and PDPK1 with I110R, I112R, L113R, K207R, K296R, and K495R (PDPK1SD). An IP assay with FLAG-PDPK1 showed that all PDPK1 mutants could interact with SUMO1 except for PDPK1ID and PDPK1SD (), and only PDPK1SD lost the ability to produce SUMOylated PDPK1 completely ( and S1F). Moreover, in vitro SUMO modification assay simultaneously revealed that PDPK1 but not PDPK1SD was able to be SUMOylated (), suggesting that the residues I110, I112, L113 and K207, K296, K495 are critical for the SUMOylation of PDPK1. In order to test whether the conjugation is covalent linkage, we performed nickel-bead affinity isolation of HIS-SUMO1 under denaturing condition. The data showed that the 210-kD band could be detected using anti-FLAG antibody from extracts of 293 T cells co-expressing of HIS-SUMO1 and FLAG-PDPK1, rather than FLAG-PDPK1SD (), suggesting that SUMO1 was conjugated to PDPK1 in a covalent linkage manner.
To determine whether substitution of these residues to arginine (R) disrupts the structure of PDPK1, the structures of WT and mutant PDPK1 were remodeled and compared by utilizing bioinformatics tool Hhpred [Citation25]. As presented in Figure S2A, the structures of both the best model of WT and mutant PDPK1 showed good convergence, indicating that the mutations do not alter the overall structure of PDPK1. Subsequently, we also analyzed the relative stability of these structural models by calculating the energy score of top 10 models with the Rosetta program. Data shown in Figure S2B revealed that though the energy score of WT PDPK1 was more negative than that of the mutant, the mutant protein is still able to fold into a stable three-dimensional structure similar to that of WT protein, implying that the three-dimensional structure of mutant PDK1 is stable. In addition, in order to test the stability of PDPK1 experimentally, we firstly purified HIS-PDPK1 and HIS-PDPK1SD expressed in E. coli (Figure S2C) and applied them to HiPrep 16/60 Sephacryl S-300 HR for gel filtration by using ÄKTA™ start protein purification system. As shown in Figure S2D, WT PDPK1 and PDPK1SD appeared at the same peak, suggesting that the two proteins had very similar behaviors. We then tested thermal stability of WT PDPK1 and PDPK1SD by using Protein Thermal Shift™ Dye Kit. The results showed that both melting temperature values of WT PDPK1 and PDPK1SD was at about 45°C, suggesting that the thermal stability of the mutant is similar to that of WT PDPK1 (Figure S2E). The above data proved that the structure of PDPK1SD was stable.
Collectively, our data clearly demonstrate that PDPK1 is able to be SUMOylated under physiological conditions and in response to virus infection.
The SUMO modification of PDPK1 is critical for activating PDPK1-AKT1-MTOR pathway
The phosphorylation of PDPK1 is critical for the activation of its substrates, such as AKT1 and RPS6KB1 [Citation14,Citation24]; thus, we tested whether SUMOylation of PDPK1 affects phosphorylation of itself or its downstream substrates. First, we investigated if SUMOylation regulates phosphorylation of PDPK1. PDPK1 knockdown (KD) cells were transfected with vectors including FLAG-PDPK1, FLAG-PDPK1CD, FLAG-PDPK1ID, and FLAG-PDPK1SD. Immunoblotting analysis against the Ser241-phosphorylated PDPK1 showed that the phosphorylated PDPK1 was not detectable in PDPK1-silenced cells transfected with PDPK1SD, PDPK1ID or PDPK1CD (), respectively, and an IP assay showed that less phosphorylated PDPK1 was detected in cells expressing PDPK1SD and PDPK1ID when compared to cells expressing PDPK1 or PDPK1CD (Figure S3A). The fusion of SUMO to the substrates has been widely used to mimic the activity of SUMO conjugation [Citation26–29]. Therefore, we constructed vectors expressing fusion protein PDPK1-SUMO1 (FLAG-P-S1) or PDPK1SD-SUMO1 (FLAG-PSD-S1) to further validate that SUMOylation promotes PDPK1 phosphorylation. As shown in , we observed that SUMOylation-deficient PDPK1 fused with SUMO1 rescued the function on phosphorylating the PDPK1 Ser241 site, confirming that SUMOylation is required for the Ser241 phosphorylation of PDPK1. Next, we explored whether SUMOylated PDPK1 phosphorylates nonSUMOylated PDPK1. PDPK1-KD 293 T cells were co-transfected with FLAG-P-S1 and MYC-PDPK1 or MYC-PDPK1SD. Anti-MYC IP showed that MYC-PDPK1, but not MYC-PDPK1SD, was recognized by anti-phospho-PDPK1 antibody (Figure S3B), suggesting that only SUMOylated PDPK1 is phosphorylated.
Figure 2. SUMOylation of PDPK1 is critical for activating the PDPK1-AKT1-MTOR pathway. (A) Phosphorylation analysis of RPS6KB1, AKT1, MTOR and PDPK1 in PDPK1 KD cells. PDPK1 KD HEK293T cells were separately transfected with vectors expressing FLAG-PDPK1, FLAG-PDPK1CD, FLAG-PDPK1ID, or FLAG-PDPK1SD for 48 h. Cell lysates were immunoblotted using indicating antibodies. (B) Vectors expressing FLAG-PDPK1, FLAG-PDPK1-SUMO1 (FLAG-P-S1), and FLAG-PDPK1SD-SUMO1 (FLAG-PSD-S1) were separately transfected into PDPK1 knocked down cells. The resultant cells were then lysed and subjected to immunoblotting analyses by using indicated antibodies. (C) PDPK1 KD HEK293T cells were transiently transfected with FLAG, FLAG-PDPK1, FLAG-PDPK1S241A, FLAG-PDPK1S241D, FLAG-PDPK1SD, FLAG-PDPK1SD and S241A, or FLAG-PDPK1SD and S241D for 48 h. Phosphorylation of MTOR, AKT1, and ULK1 is detected using indicated antibodies. *, p < 0.05; **, p < 0.01; ***, p < 0.001
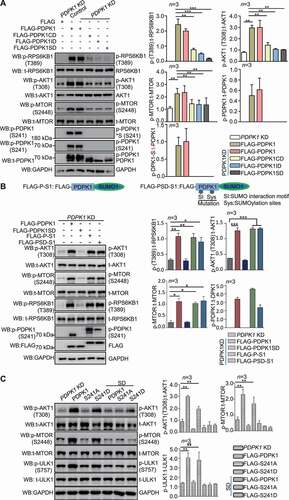
Because SUMOylation affected on phosphorylation of PDPK1, next, we test whether SUMOylation regulates PDPK1’s function on phosphorylating its substrates. SUMOylation site K495 localized at PH domain of PDPK1. This domain is shown to have little or moderate effect on lipid biding [Citation30], and is critical for phosphorylation of AKT1-MTOR [Citation31]. Therefore, we tested whether K495R mutation is sufficient to affect phosphorylation of AKT1 or MTOR. As shown in Figure S3C, K495R mutation had no effect on activation of AKT1-MTOR pathway. Subsequently, we detected the effect of SUMOylation-deficient PDPK1 on phosphorylation of AKT1-MTOR, as well as RPS6KB1. In PDPK1 KD cells expressing SUMOylation-deficient PDPK1, we found the phosphorylation level of AKT1, MTOR, and RPS6KB1 decreased in comparison with PDPK1 KD cells expressing WT PDPK1 (). Consistent with this, immunoblotting analysis showed that phosphorylated AKT1, MTOR, and ULK1 (unc-51 like autophagy activating kinase 1) levels increased significantly in cells transfected with WT and phosphomimetic PDPK1 compared to cells transfected with the nonSUMOylated mutants (). Moreover, as shown in , SUMOylation-deficient PDPK1 fused with SUMO1 rescued the function on phosphorylating AKT1-MTOR and RPS6KB1. These data suggest that SUMOylated/phosphorylated PDPK1 indeed facilitates the activity of AKT1, MTOR, and RPS6KB1.
To understand how AKT1, MTOR and RPS6KB1 are phosphorylated, we examined the PDPK1*S interaction with AKT1 and RPS6KB1. Indeed, in SUMO1-transfected cells, the PDPK1*S interacted with AKT1 or RPS6KB1 (Figure S3D and S3E). Consistently, the affinity of the nonSUMOylated PDPK1 mutant to AKT1 and RPS6KB1 decreased (Figure S3F and S3G), and INS treatment exhibited the strengthened interaction between PDPK1, AKT1 and RPS6KB1 (Figure S3H and S3I), suggesting that SUMOylation of PDPK1 promoted its interaction with AKT1, RPS6KB1. All these results all demonstrate that both SUMOylation and phosphorylation of PDPK1 are essential for activation of the AKT1-MTOR pathway.
The SUMO modification of PDPK1 is critical for regulating autophagy flux
Since SUMOylation of PDPK1 is critical for regulating MTOR signal pathway, which negatively regulates autophagy, we further investigated whether PDPK1 SUMOylation affects autophagy. Interestingly, immunoblotting analysis showed that SQSTM1 increased greatly in PDPK1 KD cells expressing WT and phosphomimetic PDPK1, but decreased significantly when expressing the nonSUMOylated mutants (), suggesting that SUMOylation of PDPK1 is necessary for inhibition of SQSTM1 degradation. However, surprisingly, expression of WT and phosphomimetic PDPK1 enhances lipidation of LC3 slightly, while expression of three SUMOylation-deficient PDPK1 mutants promotes it significantly and reduced the level of SQSTM1. In order to determine that the increase of LC3-II level resulted from a decrease of autophagy flux or an increase of newly formed lipidated LC3, cells transfected with various constructs were treated with CQ, which inhibits the autophagic degradation in lysosome. As shown in , expression of WT and phosphomimetic PDPK1, and three SUMOylation-deficient PDPK1 indeed increase the level of LC3-II, strongly suggesting that SUMOylation-deficient PDPK1 promoted the newly formation of LC3-II. Collectively, the above results suggested that SUMOylation of PDPK1 inhibits autophagy flux, while SUMOylation-deficient PDPK1 promotes the lipidation of LC3 and autophagy flux.
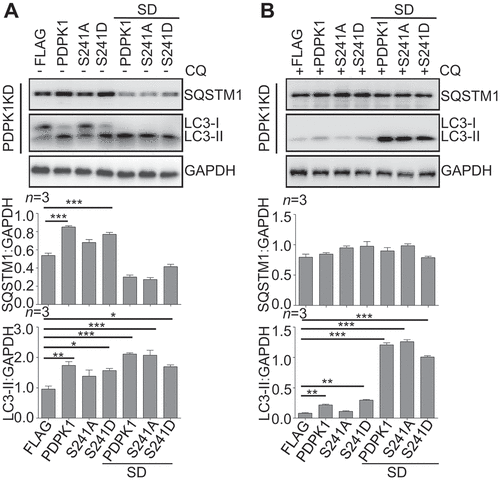
Figure 3. SUMOylation of PDPK1 is critical for regulating autophagy flux. Expression of PDPK1, PDPK1S241A, PDPK1S241D, PDPK1SD, PDPK1SD and S241A and PDPK1SD and S241D in PDPK1 knockdown cells treated with (A) or without (B) of CQ (50 μM) for 4 h. Cell lysis samples were subjected to immunoblotting analysis by using indicated antibodies. *, p < 0.05; **, p < 0.01; ***, p < 0.001
SUMOylation of PDPK1 promotes its interaction with PIK3C3
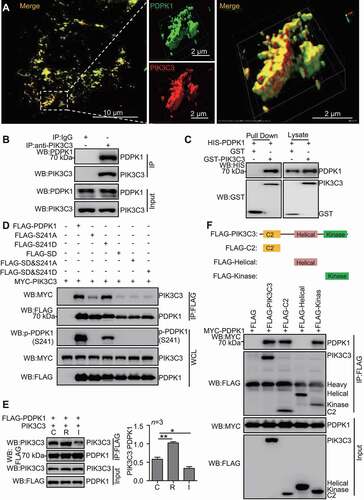
PIK3C3 is essential for autophagy [Citation32]. We then test whether PDPK1 interacts with PIK3C3. Indeed, as shown in , PDPK1 colocalized with PIK3C3 in A549 cells. Consistently, anti-PIK3C3 IP showed that endogenous PIK3C3 interacted with endogenous PDPK1 (). Affinity-isolation assay confirmed that purified PDPK1 binds to PIK3C3 directly (). Furthermore, in comparison to WT PDPK1 and PDPK1SD, PDPK1-SUMO1 interaction to PIK3C3 strengthened significantly (Figure S4A), suggesting that SUMOylation promotes interaction between PDPK1 and PIK3C3. Interestingly, binding of the PDPK1S241A to PIK3C3 exhibited marked reduction in comparison with that of WT PDPK1 and PDPK1S241D mutation (), indicating that phosphorylation of PDPK1 (S241) enhances interaction between PDPK1 and PIK3C3. Notably, rapamycin treatment enhanced obviously the interaction of PIK3C3 with PDPK1, while INS treatment diminished PDPK1 binding to PIK3C3 (). To further identify the PIK3C3 binding domain for PDPK1, we constructed a set of FLAG-tagged PIK3C3 mutants. As shown in , the C2 and kinase domains of PIK3C3 were necessary for interaction of PIK3C3 with PDPK1. PIK3R4 is critical for lipid kinase activity of PIK3C3 [Citation33]. Next, we explored whether lipid kinase activity of PIK3C3 affects PIK3C3 interaction with PDPK1 in the absence of PIK3R4. As shown in Figure S4B, PIK3R4 knockdown had no effect on the interaction between PIK3C3 and PDPK1, suggesting that binding of PIK3C3 to PDPK1 was not dependent on lipid kinase activity. Moreover, we also determine whether PDPK1 interacts with PIK3C3 complex. Anti-FLAG (FLAG-PDPK1) IP was performed and immunoblotting analysis by using anti-ATG14, -PIK3R4, -BECN1 and -UVRAG antibodies, as well as FLAG-tag antibody. As shown in Figure S4C, endogenous ATG14, PIK3R4, BECN1 and UVRAG could interact with PDPK1, suggesting that PDPK1 interacts with PIK3C3 complexes. Collectively, these results confirmed that PIK3C3 preferentially binds to SUMOylated and phosphorylated PDPK1.
Figure 4. SUMOylation of PDPK1 is required for its interaction with PIK3C3. (A) The colocalization analysis between PDPK1 and PIK3C3. A549 cells were transiently transfected with FLAG-PDPK1 and MYC-PIK3C3 for 48 h, the cells were fixed and immunostained with anti-FLAG and anti-MYC antibodies and then observed under super-resolution microscope (SIM). Scale Bar: 2 μm. (B) Endogenous PDPK1 interaction with PIK3C3. 293 T cells were lysed and then performed anti-PIK3C3 IP analysis of endogenous PDPK1 and PIK3C3 by using indicated antibodies. (C) Verification of direct interaction between PIK3C3 and PDPK1 in GST-affinity-isolation assay by using recombinant HIS-PDPK1 and GST-PIK3C3 or GST proteins purified from E.coli BL21. (D) SUMOylated and phosphorylated PDPK1 interacted strongly with PIK3C3. HEK293T cells were transiently transfected with FLAG, FLAG-PDPK1, FLAG-PDPK1S241A, FLAG-PDPK1S241D, FLAG-PDPK1SD, FLAG-PDPK1SD and S241A, or FLAG-PDPK1SD and S241D together with MYC-PIK3C3 for 48 h. Anti-FLAG IP analysis was conducted to detect MYC-PIK3C3, FLAG-PDPK1, and phosphorylated PDPK1. (E) Binding of PDPK1 to PIK3C3 was enhanced in rapamycin- but not in INS-treated cells. HEK293T cells were transiently co-transfected with FLAG-PDPK1 and PIK3C3 for 24 h. The resultant cells were treated with rapamycin (R, 5 μM), INS (I, 100 nM), or DMSO (D) for another 24 h, respectively. Anti-FLAG IP of FLAG-PDPK1 and PIK3C3 is performed by using indicated antibodies. (F) Mapping interaction domain of PIK3C3 with PDPK1. Co-IP and immunoblotting assays were performed with anti-MYC and anti-FLAG antibodies in extracts of HEK293T cells transfected with MYC-PDPK1 together with empty vector FLAG, FLAG-PIK3C3, FLAG-SH2, FLAG-Helical, or FLAG-Kinase for 48 h
PIK3C3 negatively regulated PDPK1-AKT1-MTOR pathway by inhibiting the SUMOylation of PDPK1
Since PIK3C3 interacted with PDPK1 (), we next evaluated the role of PIK3C3 on the post-translational modification of PDPK1. When PIK3C3 was overexpressed, SUMOylation of PDPK1 presented a marked decrease (). Conversely, PIK3C3 KO cells exhibited the increase of SUMOylation of PDPK1 (), demonstrating that the expression of PIK3C3 inhibits the SUMOylation of PDPK1. Consistently, in vitro SUMO modification assay and nickel-bead affinity-isolation assay under denaturing condition also revealed that PIK3C3 impaired SUMO modification of PDPK1 (). Moreover, PIK3R4 knockdown had no effect on PIK3C3-mediated deSUMOylation of PDPK1, suggesting the role of PIK3C3 on inhibiting PIK3C3 was not dependent on lipid kinase (Figure S5).
Figure 5. PIK3C3 inhibits SUMOylation of PDPK1. (A) PIK3C3 overexpression downregulated SUMOylated PDPK1. HEK293T cells were transiently co-transfected with MYC-PIK3C3 together with FLAG-PDPK1, PCI-UBE2I, and HA-SUMO1 for 48 h. FLAG precipitation and immunoblot analyses of FLAG-PDPK1 were performed by using indicated antibodies. (B) PIK3C3 deletion promoted SUMOylation of PDPK1. PIK3C3 KO or WT HEK293T cells were co-transfected with HA-SUMO1 and PCI-UBE2I together with FLAG-PDPK1 for 48 h. Anti-FLAG IP and immunoblot analyses were performed by using indicated antibodies. (C) PDPK1 SUMOylation is inhibited by PIK3C3. HEK293T cells were transiently co-transfected with FLAG-PDPK1 and PCI-UBE2I together with increasing amounts of MYC-PIK3C3 for 48 h. Anti-IgG or -FLAG IP and immunoblot analyses were performed by using indicated antibodies. (D) HEK293T cells were transiently co-transfected with FLAG-PDPK1 and MYC-SENP3 together with or without HA-PIK3C3 for 48 h. Anti-IgG or -FLAG IP and immunoblotting analyses were conducted by using indicated antibodies. (E) The expression of PIK3C3 inhibited phosphorylation of PDPK1, AKT1 and MTOR. HEK293T cells were transiently transfected with empty vector or FLAG-PIK3C3 for 48 h. Cell lysates were subjected to western blotting with indicated antibodies. (F) Knockout of PIK3C3 increased the phosphorylation of PDPK1, AKT1 and MTOR. Lysates of WT and PIK3C3-null HEK293T cells was subjected to immunoblot analysis by using indicated antibodies
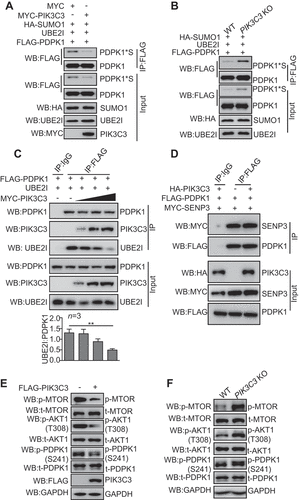
Given that UBE2I is the only SUMO E2 enzyme required for the transfer of SUMO molecules to the target substrates, we tested whether the decrease of SUMOylated PDPK1 resulted from reduced affinity to UBE2I. As expected, we found that the affinity of UBE2I to PDPK1 was impaired in a dose-dependent manner when PIK3C3 was overexpressed (), further proving that PIK3C3 inhibited the SUMOylation of PDPK1 by competing with UBE2I for binding to PDPK1. Meanwhile, we observed that PIK3C3 had no effect on interaction between SENP3 and PDPK1 (). Consistently, the phosphorylated PDPK1, AKT1 and MTOR displayed a visual downregulation in PIK3C3-overexpressed cells, and a detectable upregulation in PIK3C3 knockout cells (), showing that PIK3C3 negatively regulated the phosphorylation of PDPK1, AKT1 and MTOR.
Taken together, our data clearly demonstrate that PIK3C3 plays a core role in negatively regulating PDPK1 SUMOylation and activity of PDPK1-AKT1-MTOR pathway.
NonSUMOylated PDPK1 is critical for the formation of autophagosomes
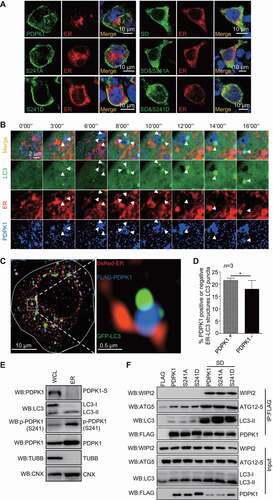
Overlapping analysis showed that nonSUMOylated PDPK1 translocated to ER when compared to SUMOylated PDPK1 (). Considering ER subdomains provide sites for autophagosome initiation and PDPK1 interaction with PIK3C3, we investigated the involvement of PDPK1 in autophagosome formation. Live-cell time-lapse images showed that initially, PDPK1 accumulated close to the ER subdomain, and then the aggregated masses of GFP-LC3 molecules gradually moved toward the PDPK1 ( and Movie 1). SIM colocalization analysis further showed that PDPK1-ER colocalized with a subset of LC3 puncta, and PDPK1 acted like a bridge that links the ER subdomain with LC3 (). Quantitative analysis showed that PDPK1-positive ER-LC3 structures were more than PDPK1-negative ER-LC3 structures (). Consistent with this, LC3-II and phosphorylated PDPK1 without SUMOylation were the main modified isoforms detected in the ER fraction in an immunoblotting assay (). Moreover, IP and affinity-isolation assays also showed a strong and direct interaction between nonSUMOylated PDPK1 and LC3-II in comparison with WT PDPK1 and S241A or S241D mutant PDPK1 ( and S6A), indicating that nonSUMOylated PDPK1 was the main isoform that binds to LC3-II. Additionally, in comparison to WT PDPK1, nonSUMOylated PDPK1 also recruited more WIPI2 and ATG12–ATG5 complex (). Our data clearly demonstrated that the nonSUMOylated PDPK1 tethers LC3 to the ER by binding to LC3-II and forming a PDPK1-LC3-II complex with WIPI2 and ATG12–ATG5 (PDPK1-LC3-II complex).
Figure 6. NonSUMOylated PDPK1 located to the ER facilitates autophagosome biogenesis. (A) The ER co-localization with nonSUMOylated PDPK1. 293 T cells were transiently co-transfected with DsRed-ER and FLAG-PDPK1, FLAG-PDPK1S241D, FLAG-PDPK1S241A, FLAG-PDPK1SD, FLAG-PDPK1SD and S241A, or FLAG-PDPK1SD and S241D for 48 h. Cells were immunostained with anti-FLAG antibodies and observed under a confocal microscope. Scale bar: 10 μm. (B) Live-cell time-lapse images of Vero cells expressing PDPK1 (blue), GFP-LC3 (green), and DsRed-ER (red). Cells were observed under a Nikon A1R/A1 laser-scanning confocal microscope. White arrowheads indicate LC3 co-localization with PDPK1. Scale bar: 2 μm. (C) Three-dimensional image of PDPK1 co-localization with LC3 and the ER. HEK293T cells were transiently co-transfected with DsRed2-ER, GFP-LC3, or FLAG-PDPK1 for 24 h, and twenty-one cells immunostained with anti-FLAG antibody were observed by SIM from each treatment. Scale Bar: 0.5 μm. (D) Quantitative analysis of ER-PDPK1-LC3 structures or ER-LC3 structures relative to LC3 puncta in (C). Results is shown as mean ± SD. (E) LC3-II and phosphorylated PDPK1 were main modified isoforms in the subcellular ER fraction. ER was obtained from HEK293T cells by using the ER0100 ER isolation kit and then subjected to immunoblot analysis by using indicated antibodies. (F) NonSUMOylated PDPK1 interacted with endogenous LC3-II, WIPI2, as well as ATG12-ATG5. HEK293T cells were transiently transfected with FLAG, FLAG-PDPK1, FLAG-PDPK1S241D, FLAG-PDPK1S241A, FLAG-PDPK1SD, FLAG-PDPK1SD and S241A, or FLAG-PDPK1SD and S241D for 48 h. Anti-FLAG IP and immunoblotting analyses were performed by using indicated antibodies
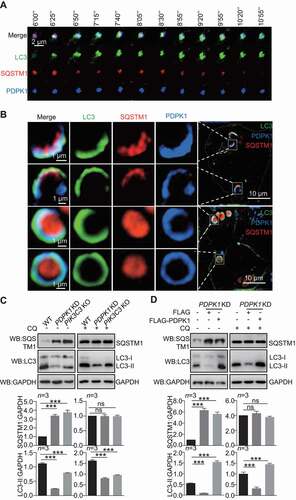
In order to evaluate whether autophagosome decorated with PDPK1 is an activated form, we tested if the PDPK1-LC3-II complex could capture the autophagic substrate SQSTM1. Live-cell time-lapse images showed that SQSTM1 adhered to the PDPK1-LC3-II complex and then disappeared gradually ( and Movie 2). Moreover, SIM analysis () showed that SQSTM1 localized at the inner side of cup-shaped structures decorated with PDPK1-LC3 complex. Subsequently, PDPK1-LC3-II complex structures expanded finally formed a closed ring-like autophagic vacuole that packaged SQSTM1 masses. To further determine which forms of PDPK1 participated in forming autophagic vacuoles, we performed SIM analysis to observe the relationship between PDPK1 mutants, SQSTM1, and LC3 (Figure S6B). Wild-type, dephosphorylated, and phosphomimetic PDPK1 were mainly distributed in the plasma membrane and partially colocalized with GFP-LC3 or SQSTM1. In contrast, nonSUMOylated PDPK1, nonSUMOylated/dephosphorylated PDPK1 and nonSUMOylated/phosphomimetic PDPK1, were mainly expressed in the cytoplasm and overlapped with LC3 to package SQSTM1. Collectively, these data demonstrate that the PDPK1-LC3-II complex packaged with SQSTM1 is critical for the morphogenesis of autophagosomes.
Figure 7. NonSUMOylation of PDPK1 is critical for lipidation of LC3. (A) Live-cell time-lapse images of Vero cells expressing PDPK1 (blue), GFP-LC3 (green), and DsRed-SQSTM1 (red) for 24 h. Cells were observed under a Nikon A1R/A1 laser-scanning confocal microscope. (B) Three-dimensional dynamic image of autophagosome formation. HEK293T cells were transiently transfected with FLAG-PDPK1, GFP-LC3, and DsRed-SQSTM1 for 24 h and then observed by SIM. Fifty cells in each treatment were recorded. (C) Immunoblot analysis of lysates from WT-PDPK1, PDPK1 KD, or PIK3C3 KO HEK293T cell lines in the presence or absence of CQ by using indicated antibodies. (D) Immunoblot analysis of lysates from WT-PDPK1 or PDPK1 KD HEK293T cell lines overexpressing FLAG or FLAG-PDPK1 in presence or absence of CQ by using indicated antibodies
To verify that PDPK1 regulates the formation of autophagic vacuoles, we analyzed the conversion of LC3-I to LC3-II when the expression of PDPK1 or PIK3C3 was inhibited. As shown in , the conversion of LC3-I to LC3-II was inhibited in PDPK1-silenced and PIK3C3 knockout cells and recovered significantly when expression of PDPK1 in PDPK1-silenced cells (P < 0.001), in presence or absence of CQ. As expected, number of GFP-LC3 puncta decreased greatly in PDPK1-silenced cells (Figure S6C).
Collectively, our results clearly indicated that PDPK1 is critical for the biogenesis of autophagosomes.
Discussion
Phosphorylation of PDPK1 controls multiple signaling pathways by phosphorylating the downstream protein kinases, such as AKT1 and RPS6KB1 [Citation14,Citation24]. However, whether other post-translational modifications regulate the kinase function of PDPK1 is unknown. In this study, we suggested a novel mechanism for the regulation of autophagosome biogenesis by nonSUMOylation of PDPK1. To our knowledge, this is the first report that the SUMO modification of PDPK1 regulates the autophagic signaling pathway.
SUMOylation of PDPK1 links PIK3C3 to the AKT1-MTOR pathway
SUMO-interacting motif (SIM) is reported to promote the SUMO modification of substrates (i.e., Srs2 SUMOylation) through binding to SUMO-charged UBE2I [Citation34]. In our study, either the mutation of SUMOylation sites at K207, K296, and K495 of PDPK1 (PDPK1CD) or of SIM at I110, I112, and L113 of PDPK1 (PDPK1ID) did not completely eliminate the SUMOylation. Only the mutation of both SIM and SUMOylation sites of PDPK1 (PDPK1[SD]) abolished completely SUMOylation of PDPK1; meanwhile, both SUMO1 and UBE2I was critical for the SUMOylation of PDPK1 (). Therefore, a reasonable hypothesis is that PDPK1 SUMOylation could be due to the combined effect of SUMOylation site and SIM.
The Ser241 phosphorylation of PDPK1 is necessary for phosphorylating AKT1 and activating the MTOR pathway, which negatively regulates autophagosome formation by phosphorylating ULK1 [Citation35,Citation36]. In our study, we showed that SUMOylation of PDPK1 is critical for generating Ser241 phosphorylation of PDPK1 (). SUMOylated/phosphorylated PDPK1 is required for phosphorylating AKT1, MTOR, and ULK1, and it inhibits the conversion of LC3-I to LC3-II, indicating that SUMOylation of PDPK1 inhibits autophagy by activating the AKT1-MTOR-ULK1 pathway.
PIK3C3, a class III PI3K, is a positive regulator of autophagy [Citation32,Citation37]. Conversely, the class I PI3K-PDPK1-AKT1-MTOR pathway negatively regulates autophagy [Citation38]. However, the relationship between PIK3C3 and the class I PI3K-PDPK1-AKT1-MTOR pathway is unknown. A previous study suggested that specific amino acids mediate the MTOR pathway through activation of PIK3C3 [Citation39], but the molecular mechanism involved in PIK3C3 signaling in the MTOR pathway is not clear. In our study, we found that PDPK1 binds to AKT1 and RPS6KB1 as well as to PIK3C3 ( and S2). SUMOylated/phosphorylated PDPK1 activates AKT1 and RPS6KB1 by phosphorylation, resulting in inhibition of autophagy flux (). However, binding of PIK3C3 to SUMOylated/phosphorylated PDPK1 induces the nonSUMOylation of PDPK1 (). NonSUMOylation of PDPK1 leads to its translocation from the cell membrane and inhibition of AKT1 activity, which results in inactivation of the AKT1-MTOR pathway. Thus, our experimental evidence suggests the role of negative feedback of PIK3C3 in inactivating the PDPK1-AKT1-MTOR pathway by interacting with SUMOylated/phosphorylated PDPK1 to inhibit the SUMOylation and phosphorylation of PDPK1, indicating a direct connection between PIK3C3 and the PDPK1-AKT1-MTOR pathway.
PIK3C3 induces autophagy through nonSUMOylation of PDPK1
Several studies have shown how PIK3C3 functions in inducing autophagy in response to autophagy stimulation [Citation32,Citation40,Citation41]. Our data show that SUMOylation of PDPK1 is inhibited by competitively binding of PIK3C3 to PDPK1 with SUMO1 in a dose-dependent manner (), and nonSUMOylated PDPK1 forms a complex by interacting with LC3, WIPI2 and the ATG12–ATG5 complex for further lipidation of LC3 in the ER (). Moreover, the formation of LC3-II increased significantly in PDPK1-silenced cells with expression of nonSUMOylated PDPK1, indicating that LC3-II is newly formed, and this process is induced by nonSUMOylated PDPK1 (.) Therefore, we conclude that autophagy is induced by PIK3C3- mediated nonSUMOylation of PDPK1.
NonSUMOylated PDPK1 plays a role in delivering lipids to expanding phagophores
ER-membrane structure interaction sites, such as ER-mitochondrial contact sites [Citation42], ER-Golgi intermediate compartments [Citation43], and ER-plasma membrane contact sites [Citation44], play critical roles in autophagosome formation. ER subdomains provide sites for autophagosome initiation [Citation45] and lipids for autophagosome membrane expansion [Citation46,Citation47]. Interestingly, we observed that PDPK1 with different modifications shuttled between the plasma membrane, ER membrane, and autophagosome membrane. SUMOylated PDPK1 was localized to the cell membrane (); however, nonSUMOylated PDPK1 acted as a bridge between the ER and LC3 () and at the expanding autophagosome membrane (). Together with verification that nonSUMOylated PDPK1 interacts with LC3-II, the hypothesis is that nonSUMOylated PDPK1 might assist in transferring lipids to the phagophore to support the expansion of the phagophore.
PIK3C3 inhibits SUMOylation of PDPK1
The small ubiquitin-related protein family plays vital roles in post-translational modification and regulates diverse cellular processes such as transcription [Citation48], replication [Citation49], chromosome segregation [Citation50], and DNA repair [Citation51]. The E1 activating enzyme, E2 conjugating enzyme, and E3 protein ligase function sequentially in the conjugation of SUMO to proteins [Citation7], and several other enzymes, including Ubl-specific proteases and sentrin-specific proteases, play roles in the reversibility of SUMO conjugation [Citation7]. Besides, SUMOylation could be inhibited by disruption of interaction between substrates and UBE2I. For example, Zhang et al. suggested that MEL-18 inhibits the SUMOylation of HSF2 by interacting with HSF2 and UBE2I [Citation52]. PIK3C3 is capable of SUMOylation [Citation53]. In our study, PIK3C3 specifically inhibited SUMOylation of PDPK1 through direct physical interaction ().
The possible role of PDPK1 SUMOylation in brain
PI3K-PDPK1-AKT1-MTOR signaling cascades play a central role in normal brain development and contribute to mediating various cellular processes [Citation54]. Dysregulation of this signaling pathway has been linked to several neurodevelopmental diseases. Especially, PDPK1 is critical for AKT1-dependent or -independent mediation of neuronal responses and brain development [Citation55]. Conditional deletion of Pdpk1 in the forebrain results in neuron loss during cortical development [Citation56]. AKT1 activation regulated by PDPK1 also plays a protective role in melatonin’s neuroprotective after focal cerebral ischemia in mice [Citation57]. Interestingly, UBE2I expression increases SUMO1 conjugation mostly, SUMO2 and SUMO3 to a lesser extent, in mice brain and allows robust resistance to brain ischemia compared to WT mice [Citation58]. In addition, SUMOylation is well known to regulate brain development, neuronal morphology and spinogenesis [Citation59,Citation60]. In our study, PDPK1 in the brain, but not in other tissues, was SUMOylated at a high level, raising the possibility that SUMOylation regulated brain development or neuron protection through PDPK1 modification. Therefore, we should pay more attention to the role of SUMOyltion on neuron protection via PDPK1, as well as its downstream pathway in future work.
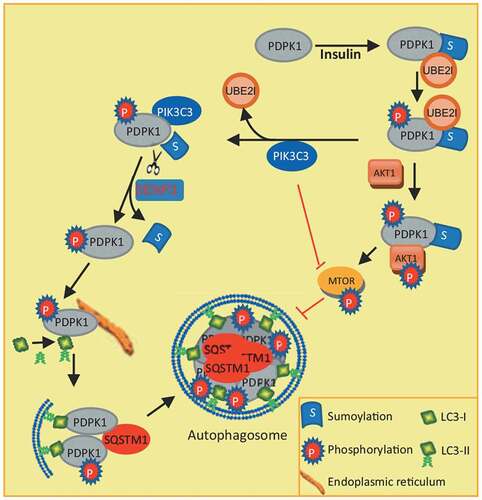
In conclusion, our study reveals a novel mechanism of autophagy induction by binding of PIK3C3 to SUMOylated PDPK1 and provides a connection between PIK3C3 (class III PI3K) and the class I PI3K-AKT1-MTOR pathway (). Inhibition of PDPK1 SUMOylation by PIK3C3 interaction negatively regulates PDPK1-AKT1-MTOR pathway and further facilitates autophagosome biogenesis by promoting LC3 lipidation.
Figure 8. Model for how PDPK1 regulates autophagosome formation. SUMOylation of PDPK1 induces its phosphorylation. Subsequently, SUMOylated and phosphorylated PDPK1 activates the AKT1-MTOR pathway and negatively regulates autophagy. In turn, the PDPK1-AKT1-MTOR pathway is inactivated by PIK3C3 binding to SUMOylated and phosphorylated PDPK1. PIK3C3 results in deSUMOylation of PDPK1 by disrupting PDPK1 interaction with UBE2I. NonSUMOylated PDPK1 links LC3 to the ER subdomain and promotes the lipidation of LC3. Subsequently, nonSUMOylated PDPK1 facilitates the expansion of the phagophore to form the enclosed autophagic vacuole
Materials and methods
Cells, antibodies, reagents, and viruses
HEK293T (ATCC, ACS-4500) and A549 (ATCC, CCL-185) cells were cultured in Dulbecco’s modified Eagle medium (DMEM; Life Technologies, 12100–038) supplemented with 10% fetal bovine serum (Sigma Life Technologies, F2442). Anti-phospho-AKT1 (Ser473; 4060), anti-phospho-AKT1 (Thr308; 4056), anti-phospho-RPS6KB1/p70 S6 kinase (Thr389; 9205), anti-phospho-ULK1 (Ser757; 14202), and anti-ULK1 (8054) rabbit monoclonal antibodies (mAb; 9234), anti-phospho-PDPK1 (Ser241; 3061), anti-PIK3C3 (3811), anti-LC3B (2775), anti-ATG14 (5504), and anti-SUMO1 (4930) rabbit polyclonal antibodies (pAb) were purchased from Cell Signaling Technology. Anti-PDPK1 (A1665) was purchased from ABclonal, anti-AKT1 (ab32505), anti-phospho-MTOR (S2448; ab109268), anti-SQSTM1 (ab109012), anti-WIPI2 (ab131271), and anti-ATG5 (ab108327) rabbit mAb and anti-MTOR rabbit pAb (ab83495) were purchased from Abcam. Anti-UBE2I/UBC9 rabbit pAb (51018-2-AP) was purchased from Proteintech. Anti-FLAG (F1804), anti-HA (H9658), and anti-MYC (M5546) mouse mAb for IP were purchased from Sigma-Aldrich. Anti-MYC (R1208-1) and anti-ACTB/β-actin (R1207-1) rabbit pAb were purchased from Hangzhou HuaAn Biotechnology. Anti-GAPDH (glyceraldehyde-3-phosphate dehydrogenase) rabbit pAb (AB-P-R001) was purchased from GoodHere Technology. DAPI (10236276001) was purchased from Roche. Anti-HsPIK3C3 pAb (Z-R015) for IP was purchased from Echelon Biosciences Inc. Dylight 405-conjugated goat anti-mouse secondary antibody (A23110) was obtained from Abbkine. Rapamycin (R8781), dimethyl sulfoxide (DMSO; 472301), and chloroquine phosphate (1118000) were purchased from Sigma-Aldrich. LY294002 (S1737) and NP-40 lysis buffer (P0013 F) were purchased from Beyotime. INS (insulin; 099–06473) was purchased from Wako. Protein A/G plus agarose (sc-2003) was purchased from Santa Cruz Biotechnology. ExFectTM transfection reagent (517051) was purchased from Vazyme Biotech Co. PMSF (P8340) was purchased from Solarbio. The endoplasmic reticulum isolation kit (ER0100) was purchased from Sigma-Aldrich.
Infectious bursal disease virus (IBDV) was propagated in the DF-1 cell line (ATCC, CRL-12203). Influenza A viruses (A/Hangzhou/1/2013 [H7N9]) and (A/PR8/34[H1N1]) were grown in 10-d-old embryonated chicken eggs. Vesicular stomatitis (VSV) was generated in HEK293T cells (ATCC, ACS-4500). Porcine circovirus 2 and pseudorabies virus were generated in PK15 cells (ATCC, CCL-33).
In vivo detection of SUMOylated PDPK1
Female BALB/c mice aged 6–8 weeks were purchased from the Model Animal Research Center of Nanjing University (Nanjing, China) and were housed under specific pathogen-free conditions for one week. Tissues, including the spleen, lung, kidney, and brain, were collected from BALB/c mice. Stimulation and inhibition of autophagy followed by the previously reported method [Citation61], the mice were separately treated with rapamycin (0.4 mg/kg), LY294002 (0.4 mg/kg), or INS (insulin; 1 U/kg) for 14 h by intraperitoneal injection before sampling, and then treated once more for 2 h. A similar volume of DMSO vehicle was injected as a negative control. The tissues were ground using liquid nitrogen and then homogenized using PMSF buffer (100 ml, including 5 ml 1 M Tris, pH 7.4, 1 ml Triton X-100 [Sigma-Aldrich, T9284], 1 g deoxycholate [Merck, D6750], 3 ml 5 M NaCl, 200 μl 0.5 M EDTA, pH 8.0, 20 mM N-ethylmaleimide [NEM; Sigma-Aldrich, E3876], 1 ml 10% sodium dodecyl sulfate [SDS; Merk, L3771]). The solutions were then centrifuged at 1,000 × g for 10 min. The protein concentration in the supernatant was measured using the bicinchoninic acid assay (Thermo Fisher Scientific/Pierce, 23225).
Cells were lysed using NP-40 buffer with complete protease inhibitors and 20 mM NEM. The lysates were centrifuged at 13,523 × g for 10 min at 4°C. The supernatants from tissues or cells were incubated with anti-FLAG M2 Magnetic Beads (Sigma-Aldrich, M8823) at 4°C overnight. The beads were washed using cold phosphate-buffered saline (PBS; 137 mM NaCl [Sinopharm Chemical Reagent Co.,Ltd, 10019318], 2.7 mM KCl [Sinopharm Chemical Reagent Co.,Ltd, 10016318], 10 mM Na2HPO4 [Sinopharm Chemical Reagent Co.,Ltd, 10020318], 1.8 mM KH2PO4 [Sinopharm Chemical Reagent Co.,Ltd, 10017618], pH 7.4) for five times followed by an additional washing with PBS twice. The immunoprecipitates were resolved in lysis buffer containing 2% SDS and denatured by heating for 10 min. The supernatants were diluted with NP-40 buffer until the concentration of SDS was decreased to 0.1%, followed by re-immunoprecipitation with the FLAG M2 Magnetic Beads. The immunoprecipitates were analyzed by immunoblotting using the PDPK1 antibody.
Homology-based structural models analysis
The sequences of PDPK1 (both WT and mutant) were uploaded to HHpred [Citation25] tool of MPI Bioinformatics Toolkit (https://toolkit.tuebingen.mpg.de) to detect the homologs of known structures. The results from HHpred were subsequently submitted to MODELER [Citation62] module of HHpred to generate homology-based structural models. To measure the stability of the models, we performed relax application [Citation63] in Rosetta suite [Citation64] version 2016.20 to generate the simplified protein structures. One hundred models were generated for each protein, and the top ten models with the lowest total energy scores were selected as candidates for subsequent analysis. The best models with the lowest energy scores of WT and mutant PDPK1 were rendered and structurally aligned in the UCSF Chimera software [Citation65] version 1.13.
Virus infection
DF-1 cells were infected with avibinavirus (multiplicity of infection [MOI] = 10) for 7 h. A549 cells were infected with H7N9 (MOI = 10) or PR8 (MOI = 8) for 9 h. HEK293T cells were infected with VSV (MOI = 10) for 6 h. PK15 cells were infected with porcine circovirus 2 (PCV2, MOI = 10) for 72 h or pseudorabies virus (PRV, MOI = 1) for 72 h. The lysates from these cells were then analyzed by western blotting.
Generation of PIK3C3 knockout cells
The PIK3C3 gene target sequence, 5ʹ-TAACTTACCATAGACATCTG-3ʹ, was inserted into the guide RNA expression plasmid PX459 (Addgene, 62988, depositing lab: Feng Zhang), a vector also expressing Cas9 enzyme. The construct was transfected into HEK293T cells for 48 h and then selected under puromycin (4 μg/ml) (InvivoGen, ant-pr) for another 72 h. The cells were then diluted to 50 cells/100 ml and inoculated into 96-well plates for colony formation. Each colony was separately transferred into 24-well plates. Knockout of PIK3C3 was confirmed by western blot.
Generation of PDPK1 knockdown cells
According to the PDPK1 gene target sequence, 5ʹ-ACGCCTAACAGGACGTATTAT-3ʹ, vectors expressing shPDPK1 were generated by using plasmid pGreenPuro™ shRNA (Biovector Science Lab, Inc., SI505A-1). The construct was transfected into HEK293T cells for 48 h and then selected under puromycin (4 μg/ml) for another 72 h. The cells were then diluted to 50 cells/100 ml and inoculated into 96-well plates for colony formation. Each colony was separately transferred into 24-well plates. The knockdown of PDPK1 was confirmed by western blot.
Immunoblotting analysis
Equal amounts of total protein from different samples were separated by SDS-PAGE, and the protein bands were transferred onto nitrocellulose blotting membrane (GE Healthcare Life Science, 10600001). After blocking with 5% nonfat dry milk containing 0.1% Tween 20 (Sigma-Aldrich, P1379-500 ML) for 30 min at 37°C, the membranes were incubated with primary antibody for 2 h at 37°C, followed by horseradish peroxidase-conjugated anti-mouse/rabbit IgG (Kirkegaard & Perry Laboratories, Inc., 074–1506) and visualized using a SuperSignal West Femto Substrate Trial Kit (Thermo Fisher Scientific/Pierce, 34095).
Immunofluorescence staining
Cells were plated on 35-mm glass-bottom cell culture dishes at a density of 6 × 104 cells/ml. Within 24 h, the cells were transfected with the indicated plasmids and treated with the indicated drugs, or cultured in starvation medium. After 24 h or 48 h, the cells were fixed with 4% paraformaldehyde in PBS for 10 min at room temperature and then blocked in 5% nonfat milk for 1 h. After washing three times with PBST (137 mM NaCl [Sinopharm Chemical Reagent Co.,Ltd, 10019318], 2.7 mM KCl [Sinopharm Chemical Reagent Co.,Ltd, 10016318], 10 mM Na2HPO4 [Sinopharm Chemical Reagent Co.,Ltd, 10020318], 1.8 mM KH2PO4 [Sinopharm Chemical Reagent Co.,Ltd, 10017618], 0.1% Tween 20 [Sigma-Aldrich, P1379-500 ML], pH 7.4), the cells were incubated with the indicated antibodies. The cells were washed with PBS and then incubated with Dylight 405 (Invitrogen, A31553), Alexa Fluor 488 dye (Invitrogen, A32723 and A32731), Alexa Fluor 546 dye (Invitrogen, A10040), or Alexa Fluor 647 dye (Invitrogen, A32733) for 1 h. Finally, the cells were observed under a Nikon A1R/A1 laser-scanning confocal microscope (Nikon) or under a SIM super-resolution microscope (Nikon). Only cells with at least five GFP dots or ring-like structures were scored as positive cells.
In vitro sumoylation assay and mass spectrometry analysis
SAE1, UBA2/SAE2, UBE2I/UBC9, PDPK1, PIK3C3 genes were amplified by PCR from HEK 293 T cells cDNA using the following primer pairs: SAE1-S:GGGAATTCATGGTGGAGAAGGAGGAGGCTG, SAE1-R:GGGTCGACTCACTTGGGGCCAAGGCACT;UBA2-S:GGGAATTCATGGCACTGTCGCGGGGGCTG,UBA2-R:GGGTCGACTCAATCTAATGCTATGACATC;UBE2I-S:GGGAATTCATGTCGGGGATCGCCCTCAGCAG,UBE2I-R:GGGTCGACTTATGAGGGCGCAAACTTCTT;PDPK1-S:GGGAATTCATGGCCAGGACCACCAGCCAGC,PDPK1-R:TCACTGCACAGCGGCGTCCGGG;PIK3C3-S:GGGAATTCATGGGGGAAGCAGAGAAGTT,PIK3C3-R:GGGTCGACTCATTTTCTCCAGTACTGGGC, and then separately cloned into pET-28A (Novagen, 69864). Especially, pET-28A-SUMO1T95R-expressing mutated SUMO1 with substitution of T95R on the C terminus was constructed. These enzymes or substrate proteins were expressed in E. coli and purified using nickel beads (Bio-Rad Laboratories, 7800801). PDPK1/PDPK1SD (5 μM), 100 nM SAE1, 100 nM UBE2, 1 μM UBE2I, 10 μM SUMO1, with or without 5 μM PIK3C3 were add to reaction buffer (1 mM ATP [Merck, A2383-1 G], 20 U/ml creatine phosphokinase [Merck, C7886-3.5KU], 5 mM phosphocreatine [Merck, P7936-1 G], 0.6 mg/ml inorganic pyrophosphatase [Merck, 10108987001], 20 mM HEPES-KOH, pH 7.3, 110 mM potassium acetate, 2 mM magnesium acetate, 1 mM dithiothreitol) for the final 20 μL reaction system. Reaction were conducted at 37°C for 1 h and were then subject to immunoblotting analysis.
The samples were also resolved on 10% SDS-PAGE gels, which were silver-stained. The band that SUMOylated PDPK1 accumulated was excised and subjected to Gel-LS-MS/MS sequencing and data analysis. LC-MS/MS analysis was performed at Shanghai Applied Protein Technology and AIMS Scientific Corporation (Shang Hai, China). The SUMOylation sites were identified using MASCOT engine (Matrix Science, London, UK; version 2.2) embedded into Proteome Discoverer 1.4 (Thermo Electron, San Jose, CA.).
Nickel-bead affinity-isolation assays
To detect conjugation of PDPK1 with HIS-SUMO1, vectors expressing FLAG-PDPK1 or HIS-SUMO1 were separately transfected into HEK293T cells. Purification and affinity-isolation assay was performed as described [Citation66]. Briefly, cells were lysed in a denaturing buffer (10 mM Tris-HCl, pH 8.0, 6 M guanidine hydrochloride (Thermo Fisher Scientific, G3272), 100 mM Na2HPO4-NaH2PO4, 5 mM imidazole (Thermo Fisher Scientific, 15513), 10 mM beta-mercaptoethanol (Thermo Fisher Scientific, M6250), and incubated with Ni2+-NTA beads (Bio-Rad Laboratories, 7800801) for 4 h at room temperature. The beads were washed with a washing buffer 1 (10 mM Tris-HCl, pH 8.0, 8 M urea [Thermo Fisher Scientific, U5378], 100 mM Na2PO4-NaH2PO4, 10 mM beta-mercaptoethanol), and washing buffer 2 (10 mM Tris–HCl, pH 6.3, 8 M urea, 100 mM Na2PO4-NaH2PO4 and 10 mM beta-mercaptoethanol). HIS-SUMO1 conjugated PDPK1 was eluted from the beads with elution buffer (150 mM Tris–HCl, pH 6.7, 200 mM imidazole, 30% glycerol [Thermo Fisher Scientific, G5516], 0.72 M beta-mercaptoethanol, 5% SDS). The eluted proteins were subjected to immunoblotting analysis using anti-FLAG or PDPK1 antibodies.
Co-IP assay
Cell lines were separately co-transfected with the indicated recombinant vectors and then lysed using NP-40 lysis buffer. Cellular lysates were incubated with anti-FLAG antibody and Protein A/G beads for 4 h at 4°C. After centrifugation, the supernatant was removed, and the pellets were resuspended in washing buffer. Centrifugation and resuspension of the pellets in fresh washing buffer were performed five times. Finally, the pellets were lysed in lysis buffer for western blot analysis.
Affinity-isolation assay
For the GST affinity isolation, PDPK1 was cloned into the pET-28A plasmid. PIK3C3, LC3 and SQSTM1 were separately cloned into the pGEX4T-1 plasmid (Merck, GE28-9545-49). These constructs were respectively transformed into E. coli BL21 cells for expression. HIS-PDPK1 was purified using Ni-NTA (QIAGEN, 30230). GST-PIK3C3, GST-LC3 and GST-SQSTM1 were purified using PierceTM glutathione agarose (Thermo Fisher Scientific, 16100) and washed by adding 1 ml cold 1× PBS per 10 mg of reduced glutathione. To prepare the bait proteins, purified GST and GST fusion proteins were immobilized on glutathione agarose beads, while HIS-PDPK1 was used as the prey protein. A total of 0.5 mg of bait proteins was added to the corresponding prey proteins and incubated for 4 h at 4°C and then washed with NP-40 lysis buffer and boiled in sample buffer. Finally, the samples were subjected to 12% SDS-PAGE and immunoblotted with mouse anti-HIS mAb and mouse anti-GST mAb (Huaan Biological Technology, M0807-1).
Live-cell imaging
The indicated Vero cells were plated on 35 mm glass-bottom cell culture dishes at a density of 6 × 104 cells/ml. Within 24 h, the cells were transfected with the indicated plasmids and treated with the indicated drugs. After 4 h or 48 h, the glass bottom dish was mounted onto the microscope stage, which was equipped with a Live Cell Station that maintained the dish at 37°C with 5% CO2. Images were acquired using a Nikon A1R/A1 laser-scanning confocal microscope (Nikon).
Endoplasmic reticulum isolation
ER was isolated using Endoplasmic Reticulum Isolation Kit (Sigma-Aldrich, ER0100). Briefly, the supernatant from centrifugation of cells at 600 × g for 5 min was removed by aspiration. The post-mitochondrial fraction was obtained following the instructions of the Endoplasmic Reticulum Isolation kit and was ultracentrifuged for 1 h at 100,000 × g at 4°C. The ER was obtained by density gradient centrifugation prior to western blotting.
Melting temperature experiments
PDPK1 (WT PDPK1) and PDPK1SD (mutant) were cloned into the PET-28A plasmid. These constructs were respectively transformed into Escherichia coli BL21 cells (Beyotime, D0339) for expression. HIS-PDPK1 and HIS-PDPK1SD were purified using Ni-NTA Agarose (QIAGEN, 30230). Briefly, the Ni-NTA Agarose conjugated with HIS-PDPK1 and HIS-PDPK1SD were washed using Tris-buffer (60 mM Tris-base, 0.3 M NaCl, pH 8.0) with 20 mM or 40 mM imidazole separately. Subsequently, HIS-PDPK1 and HIS-PDPK1SD were eluted using Tris buffer with 150 mM imidazole. Finally, the imidazole in buffer with purified proteins was removed by ultrafiltration using Tris buffer. The purified proteins HIS-PDPK1 and HIS-PDPK1SD were both diluted to 1 mg/ml. The Protein Thermal ShiftTM Dye Kit (Thermo Fisher Scientific, 4461146) was used to detect the stability of HIS-PDPK1 and HIS-PDPK1SD. In order to carry out the melting temperature experiment, the reactions were performed by using ABI 7500 fast real-time PCR system, and the melt curves and melting temperature values of the proteins were analyzed with Protein Thermal ShiftTM software (Thermo Fisher Scientific, 4466037).
Gel filtration
The purified HIS-PDPK1 and HIS-PDPK1 SD were concentrated with ultrafiltration tube to reduce volume to 1 ml. Then the proteins were separately applied to the HiPrep 16/60 Sephacryl S-300 HR (GE Healthcare, 17–1167-01) and were eluted with elution buffer (60 mM Tris, pH 8.0, 0.3 M NaCl). At the same time, the peak of proteins was separately detected and analyzed by using ÄKTA™ start protein purification system (GE Healthcare Life Sciences, 29022094-ECOMINSSW).
Statistical analysis
The protein bands were relatively quantified from western blot analysis. Briefly, mean gray value of protein bands within the linear range and background was measured by using ImageJ software, and the quantification will reflect the relative amounts as a ratio of each net band value relative to the net loading control. Data statistical significance was calculated by one-way ANOVA analysis. All data from three independent biological experiments are presented as mean ± SEM (*, p < 0.05; **, p < 0.01; ***, p < 0.001).
Supplemental Material
Download Zip (4.3 MB)Acknowledgments
We are grateful to Dr. Xufei Feng from College of Veterinary Medicine at Yangzhou University for editing movies.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
Related Research Data
References
- Sakin V, Richter SM, Hsiao HH, et al. Sumoylation of the GTPase ran by the RanBP2 SUMO E3 ligase complex. J Biol Chem. 2015;290(39):23589–23602.
- Cho MH, Cho K, Kang HJ, et al. Autophagy in microglia degrades extracellular beta-amyloid fibrils and regulates the NLRP3 inflammasome. Autophagy. 2014;10(10):1761–1775.
- Pichler A, Knipscheer P, Oberhofer E, et al. SUMO modification of the ubiquitin-conjugating enzyme E2-25K. Nat Struct Mol Biol. 2005;12(3):264–269.
- Luo HB, Xia YY, Shu XJ, et al. SUMOylation at K340 inhibits tau degradation through deregulating its phosphorylation and ubiquitination. Proc Natl Acad Sci U S A. 2014;111(46):16586–16591.
- De La Cruz-Herrera CF, Baz-Martinez M, Lang V, et al. Conjugation of SUMO to p85 leads to a novel mechanism of PI3K regulation. Oncogene. 2016;35(22):2873–2880.
- Schuldt A. Post-translational modification: a SUMO protease for stress protection. Nat Rev Mol Cell Biol. 2013;14(5):263.
- Gareau JR, Lima CD. The SUMO pathway: emerging mechanisms that shape specificity, conjugation and recognition. Nat Rev Mol Cell Biol. 2010;11(12):861–871.
- Yu L, Chen Y, Tooze SA. Autophagy pathway: cellular and molecular mechanisms. Autophagy. 2018;14(2):207–215.
- Yang Z, Klionsky DJ. Mammalian autophagy: core molecular machinery and signaling regulation. Curr Opin Cell Biol. 2010;22(2):124–131.
- Mizushima N. Autophagy: process and function. Genes Dev. 2007;21(22):2861–2873.
- Dall’armi C, Devereaux KA, Di Paolo G. The role of lipids in the control of autophagy. Curr Biol. 2013;23(1):R33–45.
- Lamb CA, Yoshimori T, Tooze SA. The autophagosome: origins unknown, biogenesis complex. Nat Rev Mol Cell Biol. 2013;14(12):759–774.
- Carlsson SR, Simonsen A. Membrane dynamics in autophagosome biogenesis. J Cell Sci. 2015;128(2):193–205.
- Alessi DR, Kozlowski MT, Weng QP, et al. 3-Phosphoinositide-dependent protein kinase 1 (PDK1) phosphorylates and activates the p70 S6 kinase in vivo and in vitro. Curr Biol. 1998;8(2):69–81.
- Frodin M, Jensen CJ, Merienne K, et al. A phosphoserine-regulated docking site in the protein kinase RSK2 that recruits and activates PDK1. Embo J. 2000;19(12):2924–2934.
- Jensen CJ, Buch MB, Krag TO, et al. 90-kDa ribosomal S6 kinase is phosphorylated and activated by 3-phosphoinositide-dependent protein kinase-1. J Biol Chem. 1999;274(38):27168–27176.
- Pullen N, Dennis PB, Andjelkovic M, et al. Phosphorylation and activation of p70s6k by PDK1. Science. 1998;279(5351):707–710.
- Balendran A, Hare GR, Kieloch A, et al. Further evidence that 3-phosphoinositide-dependent protein kinase-1 (PDK1) is required for the stability and phosphorylation of protein kinase C (PKC) isoforms. FEBS Lett. 2000;484(3):217–223.
- Chou MM, Hou W, Johnson J, et al. Regulation of protein kinase C zeta by PI 3-kinase and PDK-1. Curr Biol. 1998;8(19):1069–1077.
- Dutil EM, Toker A, Newton AC. Regulation of conventional protein kinase C isozymes by phosphoinositide-dependent kinase 1 (PDK-1). Curr Biol. 1998;8(25):1366–1375.
- Le Good JA, Ziegler WH, Parekh DB, et al. Protein kinase C isotypes controlled by phosphoinositide 3-kinase through the protein kinase PDK1. Science. 1998;281(5385):2042–2045.
- Biondi RM, Kieloch A, Currie RA, et al. The PIF-binding pocket in PDK1 is essential for activation of S6K and SGK, but not AKT. Embo J. 2001;20(16):4380–4390.
- Park J, Leong ML, Buse P, et al. Serum and glucocorticoid-inducible kinase (SGK) is a target of the PI 3-kinase-stimulated signaling pathway. Embo J. 1999;18(11):3024–3033.
- Wang X, Hills LB, Huang YH. Lipid and protein co-regulation of PI3K effectors AKT and itk in lymphocytes. Front Immunol. 2015;6:117.
- Soding J, Biegert A, Lupas AN. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 2005;33(WebServer issue):W244–8.
- Holmstrom S, Van Antwerp ME, Iniguez-Lluhi JA. Direct and distinguishable inhibitory roles for SUMO isoforms in the control of transcriptional synergy. Proc Natl Acad Sci U S A. 2003;100(26):15758–15763.
- Yurchenko V, Xue Z, Sadofsky MJ. SUMO modification of human XRCC4 regulates its localization and function in DNA double-strand break repair. Mol Cell Biol. 2006;26(5):1786–1794.
- Ross S, Best JL, Zon LI, et al. SUMO-1 modification represses Sp3 transcriptional activation and modulates its subnuclear localization. Mol Cell. 2002;10(4):831–842.
- Wen D, Wu J, Wang L, et al. SUMOylation promotes nuclear import and stabilization of polo-like kinase 1 to support its mitotic function. Cell Rep. 2017;21(8):2147–2159.
- Komander D, Fairservice A, Deak M, et al. Structural insights into the regulation of PDK1 by phosphoinositides and inositol phosphates. Embo J. 2004;23(20):3918–3928.
- Gao X, Lowry PR, Zhou X, et al. PI3K/AKT signaling requires spatial compartmentalization in plasma membrane microdomains. Proc Natl Acad Sci U S A. 2011;108(35):14509–14514.
- Jaber N, Dou Z, Chen JS, et al. Class III PI3K Vps34 plays an essential role in autophagy and in heart and liver function. Proc Natl Acad Sci U S A. 2012;109(6):2003–2008.
- Panaretou C, Domin J, Cockcroft S, et al. Characterization of p150, an adaptor protein for the human phosphatidylinositol (PtdIns) 3-kinase. Substrate presentation by phosphatidylinositol transfer protein to the p150.Ptdins 3-kinase complex. J Biol Chem. 1997;272(4):2477–2485.
- Kolesar P, Sarangi P, Altmannova V, et al. Dual roles of the SUMO-interacting motif in the regulation of Srs2 sumoylation. Nucleic Acids Res. 2012;40(16):7831–7843.
- Casamayor A, Morrice NA, Alessi DR. Phosphorylation of Ser-241 is essential for the activity of 3-phosphoinositide-dependent protein kinase-1: identification of five sites of phosphorylation in vivo. Biochem J. 1999;342(Pt 2):287–292.
- Russell RC, Tian Y, Yuan H, et al. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat Cell Biol. 2013;15(7):741–750.
- Jaber N, Zong WX. Class III PI3K Vps34: essential roles in autophagy, endocytosis, and heart and liver function. Ann N Y Acad Sci. 2013;1280:48–51.
- Yu X, Long YC, Shen HM. Differential regulatory functions of three classes of phosphatidylinositol and phosphoinositide 3-kinases in autophagy. Autophagy. 2015;11(10):1711–1728.
- Nobukuni T, Joaquin M, Roccio M, et al. Amino acids mediate mTOR/raptor signaling through activation of class 3 phosphatidylinositol 3OH-kinase. Proc Natl Acad Sci U S A. 2005;102(40):14238–14243.
- Kim J, Kim YC, Fang C, et al. Differential regulation of distinct Vps34 complexes by AMPK in nutrient stress and autophagy. Cell. 2013;152(1–2):290–303.
- Stjepanovic G, Baskaran S, Lin MG, et al. Vps34 kinase domain dynamics regulate the autoPhagic PI 3-kinase complex. Mol Cell. 2017;67(3):528–534 e3.
- Hamasaki M, Furuta N, Matsuda A, et al. Autophagosomes form at ER-mitochondria contact sites. Nature. 2013;495(7441):389–393.
- Ge L, Melville D, Zhang M, et al. The ER-Golgi intermediate compartment is a key membrane source for the LC3 lipidation step of autophagosome biogenesis. Elife. 2013;2:e00947.
- Nascimbeni AC, Giordano F, Dupont N, et al. ER-plasma membrane contact sites contribute to autophagosome biogenesis by regulation of local PI3P synthesis. Embo J. 2017;36(14):2018–2033.
- Nishimura T, Tamura N, Kono N, et al. Autophagosome formation is initiated at phosphatidylinositol synthase-enriched ER subdomains. Embo J. 2017;36(12):1719–1735.
- Tooze SA. Current views on the source of the autophagosome membrane. Essays Biochem. 2013;55:29–38.
- Turco E, Martens S. Insights into autophagosome biogenesis from in vitro reconstitutions. J Struct Biol. 2016;196(1):29–36.
- Rosonina E, Akhter A, Dou Y, et al. Regulation of transcription factors by sumoylation. Transcription. 2017;8(4):220–231.
- Dou H, Huang C, Singh M, et al. Regulation of DNA repair through deSUMOylation and SUMOylation of replication protein A complex. Mol Cell. 2010;39(3):333–345.
- Wan J, Subramonian D, Zhang XD. SUMOylation in control of accurate chromosome segregation during mitosis. Curr Protein Pept Sci. 2012;13(5):467–481.
- Cremona CA, Sarangi P, Zhao X. Sumoylation and the DNA damage response. Biomolecules. 2012;2(3):376–388.
- Zhang J, Goodson ML, Hong Y, et al. MEL-18 interacts with HSF2 and the SUMO E2 UBC9 to inhibit HSF2 sumoylation. J Biol Chem. 2008;283(12):7464–7469.
- Yang Y, Fiskus W, Yong B, et al. Acetylated hsp70 and KAP1-mediated Vps34 SUMOylation is required for autophagosome creation in autophagy. Proc Natl Acad Sci U S A. 2013;110(17):6841–6846.
- Wang L, Zhou K, Fu Z, et al. Brain development and AKT signaling: the crossroads of signaling pathway and neurodevelopmental diseases. J Mol Neurosci. 2017;61(3):379–384.
- Cordon-Barris L, Pascual-Guiral S, Yang S, et al. Mutation of the 3-phosphoinositide-dependent protein kinase 1 (PDK1) substrate-docking site in the developing brain causes microcephaly with abnormal brain morphogenesis independently of AKT, leading to impaired cognition and disruptive behaviors. Mol Cell Biol. 2016;36(23):2967–2982.
- Xu C, Yu L, Hou J, et al. Conditional deletion of PDK1 in the forebrain causes neuron loss and increased apoptosis during cortical development. Front Cell Neurosci. 2017;11:330.
- Kilic U, Caglayan AB, Beker MC, et al. Particular phosphorylation of PI3K/AKT on Thr308 via PDK-1 and PTEN mediates melatonin’s neuroprotective activity after focal cerebral ischemia in mice. Redox Biol. 2017;12:657–665.
- Lee YJ, Mou Y, Maric D, et al. Elevated global SUMOylation in Ubc9 transgenic mice protects their brains against focal cerebral ischemic damage. PLoS One. 2011;6(10):e25852.
- Gwizdek C, Casse F, Martin S. Protein sumoylation in brain development, neuronal morphology and spinogenesis. Neuromolecular Med. 2013;15(4):677–691.
- Martin S, Wilkinson KA, Nishimune A, et al. Emerging extranuclear roles of protein SUMOylation in neuronal function and dysfunction. Nat Rev Neurosci. 2007;8(12):948–959.
- Naito T, Kuma A, Mizushima N. Differential contribution of insulin and amino acids to the mTORC1-autophagy pathway in the liver and muscle. J Biol Chem. 2013;288(29):21074–21081.
- Eswar N, Webb B, Marti-Renom MA, et al. Comparative protein structure modeling using modeller. Curr Protoc Bioinf. 2006;Chapter 5 Unit–5 6.
- Conway P, Tyka MD, Dimaio F, et al. Relaxation of backbone bond geometry improves protein energy landscape modeling. Protein Sci. 2014;23(1):47–55.
- Huang PS, Ban YE, Richter F, et al. RosettaRemodel: a generalized framework for flexible backbone protein design. PLoS One. 2011;6(8):e24109.
- Pettersen EF, Goddard TD, Huang CC, et al. UCSF Chimera–a visualization system for exploratory research and analysis. J Comput Chem. 2004;25(13):1605–1612.
- Yang Y, Fu W, Chen J, et al. SIRT1 sumoylation regulates its deacetylase activity and cellular response to genotoxic stress. Nat Cell Biol. 2007;9(11):1253–1262.