
ABSTRACT
Neutrophils infected with Mycobacterium tuberculosis (Mtb) predominate in tuberculosis patients’ lungs. Neutrophils phagocytose the pathogen, but the mechanism of pathogen elimination is controversial. Macroautophagy/autophagy, a crucial mechanism for several neutrophil functions, can be modulated by immunological mediators. The costimulatory molecule SLAMF1 can act as a microbial sensor in macrophages being also able to interact with autophagy-related proteins. Here, we demonstrate for the first time that human neutrophils express SLAMF1 upon Mtb-stimulation. Furthermore, SLAMF1 was found colocalizing with LC3B+ vesicles, and activation of SLAMF1 increased neutrophil autophagy induced by Mtb. Finally, tuberculosis patients’ neutrophils displayed reduced levels of SLAMF1 and lower levels of autophagy against Mtb as compared to healthy controls. Altogether, these results indicate that SLAMF1 participates in neutrophil autophagy during active tuberculosis.
Abbreviations: AFB: acid-fast bacilli; BafA1: bafilomycin A1; CLL: chronic lymphocytic leukemia; DPI: diphenyleneiodonium; EVs: extracellular vesicles; FBS: fetal bovine serum; HD: healthy donors; HR: high responder (tuberculosis patient); IFNG: interferon gamma; IL1B: interleukin 1 beta; IL17A: interleukin 17A; IL8: interleukin 8; LR: low responder (tuberculosis patient); mAb: monoclonal antibody; MAP1LC3/LC3: microtubule associated protein 1 light chain 3; MAPK: mitogen-activated protein kinase; MAPK1/ERK2: mitogen-activated protein kinase 1; MAPK14/p38: mitogen-activated protein kinase 14; Mtb: Mycobacterium tuberculosis; Mtb-Ag: Mycobacterium tuberculosis, Strain H37Rv, whole cell lysate; NETs: neutrophils extracellular traps; PPD: purified protein derivative; ROS: reactive oxygen species; PIK3C3/VPS34: phosphatidylinositol 3-kinase catalytic subunit type 3; SLAMF1: signaling lymphocytic activation molecule family member 1; TB: tuberculosis; TLR: toll like receptor
Introduction
Tuberculosis (TB) is the leading cause of death from an infectious microorganism. Actually, Mycobacterium tuberculosis (Mtb) produces nearly 10 million new cases and 1.5 million deaths per year [Citation1]. The successful establishment of Mtb infection depends on its primary interaction with host macrophages, dendritic cells, neutrophils and NK cells [Citation2]. Although great progresses have been made in the characterization of the acquired cellular responses in TB patients, it remains to be elucidated what exactly constitutes a protective response [Citation3]. Furthermore, how Mtb is able to evade host immune surveillance and persist, particularly inside phagocytes, remains to be completely understood.
Neutrophils arrive first at the site of infection and are the cells predominantly infected with Mtb in patients’ lungs [Citation4]. Neutrophils’ ability to phagocytose Mtb has been demonstrated but its capacity to eliminate the bacteria remains controversial. These cells can sequester Mtb in neutrophils extracellular traps (NETs) [Citation5]; cooperate with other cells and release extracellular vesicles (EVs) upon activation. Particularly, EVs charged with Mtb activate macrophages, promoting autophagy induction and Mtb clearance [Citation6]. However, many studies agree about the fact that neutrophils trigger a hyper-inflammatory response that leads to tissue destruction and mediates damage and lung disease during active TB. In fact, human blood transcriptional profiling performed in TB patients indicated that the TB signature was dominated by a neutrophil-driven interferon (IFN)-inducible gene profile, reflecting the central participation of these cells in active disease [Citation7]. Therefore, a deeper investigation into the biology of neutrophils during Mtb infection is crucial for the identification of specific targets to be used in host-directed therapies.
Macroautophagy/autophagy is a central homeostatic mechanism that plays a role in innate and adaptive immunity against intracellular pathogens, including Mtb [Citation8]. Furthermore, in macrophages, enhanced autophagy mediates elimination of intracellular Mtb through lytic and antimicrobial properties unique to autolysosomes [Citation9]. Additionally, it has been demonstrated that host autophagy coordinates successful antimicrobial responses to mycobacteria during chemotherapy [Citation10]. Moreover, autophagy is essential for major neutrophil functions, including degranulation, reactive oxygen species (ROS) production, and release of NETs [Citation11].
It has also been reported that autophagy can be modulated by different immunological mediators [Citation12]. Accordingly, we have demonstrated that IFNG and IL17A regulate autophagy in Mtb-infected monocytes from TB patients in correlation with disease severity [Citation13,Citation14]. SLAMF1 (signaling lymphocytic activation molecule family member 1), a type I glycoprotein from the SLAM subfamily of the CD2-like family of proteins, is a costimulatory molecule involved in host immunity regulation of innate and adaptive responses. We have demonstrated that SLAMF1 contributes to Th1 cytokine responses in human TB [Citation15]. However, SLAMF1 is not only a costimulatory molecule but also a bacterial sensor that helps to remove Gram-negative bacteria by macrophages [Citation16]. Actually, after entering the phagosome by interaction with bacterial outer proteins, SLAMF1 recruits several molecules to the phagosome, including the autophagic protein BECN1/Beclin-1, thus making the connection to the cellular machinery that controls bacterial killing [Citation16,Citation17]. Interestingly, it has been recently shown that SLAMF1 regulates autophagy in B cells from chronic lymphocytic leukemia (CLL) patients, affecting CLL cells responses to autophagy-activating therapeutic agents [Citation18]. However, the expression of SLAMF1 in human neutrophils and its function during autophagy in these cells have not been previously demonstrated. Therefore, to gain insight into the mechanisms that operate during immune responses to Mtb, here we studied the participation of SLAMF1 as a regulator of the autophagy process in neutrophils from TB patients.
Results
Initially, we analyzed the frequency of neutrophils in peripheral blood from patients with active TB. Our results indicated that the number of neutrophils was significantly higher in patients with severe radiological lesions (bilateral and massive affectation with multiple cavities) as compared to patients with moderate lesions ()). Previously we demonstrated that TB patients could be classified based on in vitro lymphocyte responses to Mtb antigens. Briefly, high responder (HR) patients are individuals displaying significant proliferative responses, IFNG production and an increased SLAMF1 expression on T lymphocytes upon Mtb whole cell lysate (Mtb-Ag) stimulation, whereas low responder (LR) patients exhibit low proliferative responses, IFNG production and SLAMF1 expression. Furthermore, LR patients have a more severe pulmonary disease, lower leukocyte counts and a more prolonged illness, compared to HR individuals [Citation15]. Nevertheless, when we examined the number of neutrophils in high and low responder TB patients, a similar number of these cells were detected ()). Additionally, we observed a marked increase in neutrophil counts in patients with acid-fast bacilli (AFB)-positive smears ()) as well as a positive correlation between the number of neutrophils and the time of disease evolution ()). Consistent with previous reports [Citation19–21], our findings suggest that increasing numbers of neutrophils might contribute to TB severity. In fact, it was reported that TB cavities contain more neutrophils and fewer lymphocytes compared to nondestructive pulmonary infiltrates and radiologically unaffected lobes of the lungs [Citation19]. Moreover, we observed that patients undergoing advanced stages of anti-TB treatment displayed lower numbers of neutrophils than those undertaking the first week of chemotherapy (6714 ± 468 neutrophils vs. 5657 ± 710 neutrophils, respectively).
Figure 1. Neutrophil counts according to clinical and immunological parameters of tuberculosis patients. Neutrophil numbers per mm3 in blood from TB patients recruited at Hospital Muñiz, Buenos Aires. Each dot represents the number of neutrophils (per mm3) for each individual, analyzed according to: (A) the severity of the TB disease as determined by the radiological lesions (Severe, n = 88; Moderate, n = 48; Mild, n = 14); (B) an immunological classification based on in vitro lymphocyte responses to Mtb-Ag [Citation15] (HR, High responder, n = 65; and LR Low responder TB patients, n = 97); (C) Acid-fast bacilli (AFB) in sputum smear (Ziehl-Neelsen staining; BAAR-, n = 13; BAAR+, n = 88; BAAR++, n = 24; BAAR+++, n = 37); (D) Time of disease evolution (months with clinical symptoms previous to hospital admission of the individual; one month or less, n = 58; more than one month, n = 104). Statistical differences were calculated using the nonparametric Mann-Whitney test for unpaired samples. n.s., not significant differences. * p < 0.05; ** p < 0.01; One-way ANOVA with post hoc Dunnett’s multiple comparisons test; Correlation factor (R) and pwere calculated using the nonparametric Spearman correlation test
![Figure 1. Neutrophil counts according to clinical and immunological parameters of tuberculosis patients. Neutrophil numbers per mm3 in blood from TB patients recruited at Hospital Muñiz, Buenos Aires. Each dot represents the number of neutrophils (per mm3) for each individual, analyzed according to: (A) the severity of the TB disease as determined by the radiological lesions (Severe, n = 88; Moderate, n = 48; Mild, n = 14); (B) an immunological classification based on in vitro lymphocyte responses to Mtb-Ag [Citation15] (HR, High responder, n = 65; and LR Low responder TB patients, n = 97); (C) Acid-fast bacilli (AFB) in sputum smear (Ziehl-Neelsen staining; BAAR-, n = 13; BAAR+, n = 88; BAAR++, n = 24; BAAR+++, n = 37); (D) Time of disease evolution (months with clinical symptoms previous to hospital admission of the individual; one month or less, n = 58; more than one month, n = 104). Statistical differences were calculated using the nonparametric Mann-Whitney test for unpaired samples. n.s., not significant differences. * p < 0.05; ** p < 0.01; One-way ANOVA with post hoc Dunnett’s multiple comparisons test; Correlation factor (R) and pwere calculated using the nonparametric Spearman correlation test](/cms/asset/df2bcfe2-246b-45bb-9bba-de5cb9e3d633/kaup_a_1825273_f0001_oc.jpg)
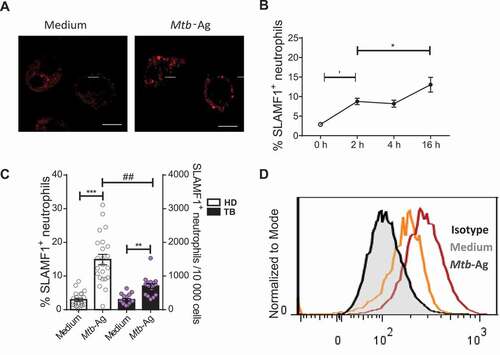
SLAMF1 expression has been demonstrated not only in lymphocytes but in myeloid cells (monocytes and macrophages) [Citation22] as well. However, the expression of SLAMF1 in human neutrophils has not been evaluated so far. Thus, we initially performed bioinformatic analysis finding evidence of SLAMF1 RNA expression in unstimulated mature neutrophils from healthy donors (HD) and TB patients (Fig. S1A, B). In line with those initial analyses, in this work, we observed the presence of SLAMF1 in neutrophils from HD and TB patients both by confocal microscopy ()) and flow cytometry ()). Furthermore, after 2 h of Mtb-Ag stimulation, we detected an increase in SLAMF1 levels on neutrophils’ membrane ()) and an accumulation of localized signal at the intracellular level ()). In fact, Mtb-Ag challenge significantly elevated the percentage of SLAMF1+ neutrophils as detected by flow cytometry ()). Interestingly, upon Mtb-Ag stimulation, HD displayed greater percentages of SLAMF1+ neutrophils as compared to TB patients () and Fig. S2A). Thus, these results show for the first time to our knowledge that SLAMF1 is expressed in human neutrophils and that it increases upon Mtb-Ag stimulation. Notably, comparable elevated surface levels of SLAMF1 were detected in both subjects’ populations after a robust neutrophil activation with phorbol myristate acetate/PMA (Fig. S2B).
Figure 2. Expression of SLAMF1 in human neutrophils. Neutrophils from HD and TB patients were stimulated with or without Mtb-Ag (10 µg/ml) during different times, and SLAMF1 expression was determined by (A) confocal microscopy and (B, C, D) flow cytometry. (A) Neutrophils from TB patients were stimulated ± Mtb-Ag during 2 h. Representative images from ten independent experiments are shown. Scale bars: 5 μm. (B) Kinetics of SLAMF1 surface expression as measured by flow cytometry. Each point represents the mean values of the percentage of SLAMF1+ neutrophils ± SEM at 0, 2, 4 or 16 h after Mtb-Ag stimulation (n = 5). (C) SLAMF1 surface expression in neutrophils from HD (n = 23) and TB patients (n = 18) was determined by flow cytometry. Bars represent mean values of the percentage of SLAMF1+ neutrophils ± SEM (left axis) and dots represent the number of SLAMF1 neutrophils/10,000 cells (right axis). (D) A representative histogram of flow cytometry is shown. Statistical differences were calculated using the Wilcoxon signed-rank test for paired samples. * p < 0.05, ** p < 0.01, *** p < 0.001, ## p < 0.01; Mann-Whitney nonparametric test for unpaired samples
In order to determine the nature of the mycobacterial components involved in the induction of SLAMF1 surface expression on neutrophils, we compared SLAMF1 levels after stimulation with various stimuli. Interestingly, the highest levels of SLAMF1 were induced by compounds that included several proteins, such as purified protein derivative (PPD, )) or culture filtrate proteins (data not shown). Single proteins such as ESAT-6 or CFP-10 did not induce significant amounts of SLAMF1. Moreover, total Mtb lipids induced a moderate proportion of SLAMF1+ neutrophils ()). Furthermore, additional antigens with different chemical compositions, such as the lipoglycan ManLam or the 19-kDa Mtb lipoglycoprotein (also known as LpqH), both significantly increased the percentage of SLAMF1+ neutrophils as well, in comparison with non-stimulated cells ()). Altogether, our results indicate that diverse bacterial compounds might be recognized by human neutrophils during Mtb-Ag stimulation and induce SLAMF1 surface expression. In particular, compounds including several proteins or lipo/glycans acted as the predominant stimulators of SLAMF1 expression on human neutrophils.
Figure 3. Mechanisms of induction of SLAMF1 expression in humans. (A) Human purified neutrophils from HD (n = 12) were stimulated either with Mtb-Ag (10 µg/ml), total Mtb lipids, purified protein derivative (PPD, 10 µg/ml), ESAT-6 (10 µg/ml), CFP-10 (10 µg/ml), ManLam (10 µg/ml) or 19kDa lipoprotein (10 µg/ml) during 2 h. Finally, SLAMF1 surface expression was evaluated by flow cytometry as previously described. (B) Neutrophils from HD (n = 7) were pre-incubated with PD98059 (50 µM), a MEK1/ERK MAPK inhibitor, or with SB202190 (10 µM), a MAPK14/p38 inhibitor, for 30 min. Cells were then stimulated with or without Mtb-Ag (10 µg/ml) during 2 h. Afterward SLAMF1 surface expression was determined by flow cytometry. (C) Neutrophils from HD (n = 4) were stimulated with or without Mtb-Ag (10 µg/ml) in the presence or absence of diphenyleneiodonium (DPI, 10 µM) during 2 h. Afterward, SLAMF1 surface expression was determined by flow cytometry. (A, B, C) Bars represent the mean values of the percentage of SLAMF1+ neutrophils ± SEM. Statistical differences were calculated using one-way ANOVA and post hoc Dunnett multiple comparison test. * p < 0.05, **p < 0.01, ***p < 0.001
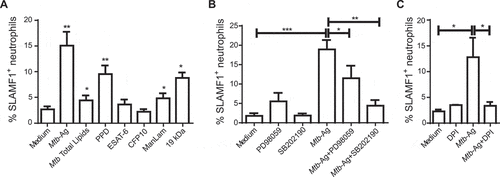
MAPKs (mitogen-activated protein kinases) have been described to be involved in SLAMF1 expression in lipopolysaccharide (LPS)-stimulated monocytes [Citation23]. Thus, to get insight into the molecular mechanisms that lead to SLAMF1 surface expression on human neutrophils, we next investigated the potential role of the MAPK14/p38 and MAPK1/ERK2-MAPK3/ERK1 pathways. Purified cells were pre-incubated with PD98059 or SB202190 inhibitors for 30 min, and then those neutrophils were stimulated with Mtb-Ag for 2 additional hours. Afterward, SLAMF1 expression was evaluated by flow cytometry. As shown in ), the analysis clearly revealed that inhibition of both MAPK/ERK and MAPK14/p38 kinases significantly diminished the percentage of SLAMF1+ neutrophils upon antigen stimulation. Therefore, these findings suggest that these MAPKs participate in the signaling pathway that triggers SLAMF1 surface expression on Mtb-Ag-stimulated neutrophils.
Oxidants produced upon NOX (NADPH oxidase) activation play a central role in diverse neutrophil functions [Citation24,Citation25]. Thus, we then analyzed their possible participation in SLAMF1 surface expression. Purified neutrophils were stimulated with or without Mtb-Ag in the presence or absence of the NOX inhibitor diphenyleneiodonium (DPI) and, after 2 h, SLAMF1 expression was evaluated using flow cytometry. As shown in ), inhibition of reactive oxygen species (ROS) generation significantly diminished the percentage of SLAMF1+ neutrophils. Therefore, our results suggest that expression of SLAMF1 in Mtb-Ag-stimulated neutrophils is dependent on ROS production.
Recently, Bologna et al. reported that reduced SLAMF1 expression in B lymphocytes from CLL patients regulated autophagy affecting drug responses [Citation18]. Additionally, the interaction of SLAMF1 with PIK3C3/VPS34 and BECN1, two proteins involved in the initiation of the autophagy process, had previously been described [Citation16]. In the present work, we observed that Mtb-Ag stimulation significantly augmented the percentage of MAP1LC3A/B-II/LC3A/B-II+ neutrophils from TB patients and HD ()), denoting the presence of more autophagosomes by the recognition of mycobacterial antigens. Given that autophagy is a dynamic process and a proportion of autophagosome-anchored-LC3-II is degraded when it reaches the lysosome, the autophagy flux in the presence or absence of bafilomycin A1 (Baf A1) was analyzed. It is important to notice that the observed increment corresponds to an elevated autophagic flux (Fig. S3A), as showed by a significant increase in the percentage of Mtb-Ag-stimulated LC3A/B-II+ neutrophils treated with Baf A1 compared with cells stimulated with Mtb-Ag alone. Interestingly, we detected significantly greater autophagy levels in neutrophils from HD as compared to neutrophils from TB patients ()). Moreover, we observed that HR TB patients’ neutrophils displayed elevated autophagy levels in comparison to LR TB patients’ cells (Fig. S3B).
Figure 4. Role of SLAMF1 in neutrophil autophagy. (A) Neutrophils from HD (n = 12) and TB patients (n = 13) were stimulated with or without Mtb-Ag (10 µg/ml) during 2 h, and intracellular saponin-resistant LC3A/B-II determination was performed by flow cytometry. (B, C) In separate experiments, neutrophils from HD and TB patients were stimulated with or without Mtb-Ag (10 µg/ml) and treated with a SLAMF1 agonist antibody (anti-SLAMF1, 10 µg/ml). Then, autophagy levels in neutrophils were evaluated by (B) immunofluorescence against LC3B in neutrophils (n = 4) and (C) flow cytometry against intracellular saponin-resistant LC3A/B-II (n = 16). (D) Before autophagy determination, bafilomycin A1 (Baf A1) (100 nM) was added for the last 60 min of culture. Then, autophagy levels in neutrophils were evaluated by immunofluorescence against LC3B. Bars represent the mean values of LC3 puncta per cell ± SEM. (E) Fluorescence distribution of SLAMF1 and LC3B in Mtb-Ag-stimulated neutrophils. Neutrophils were fixed after 1 h of Ag stimulation. Then, cells were permeabilized and stained using specific antibodies anti-SLAMF1 (red) and anti-LC3B (green). Images were acquired with a confocal microscope. Representative images of one out of five TB patients are shown. (A, C) Bars represent the mean values of the percentage of LC3A/B-II+ neutrophils ± SEM. (B, D) Representative images of one experiment are shown. Scale bars: 5 μm. Statistical differences were calculated using one-way ANOVA and post hoc Dunnett multiple comparison test. * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001, # p < 0.05; Mann-Whitney nonparametric test for unpaired samples
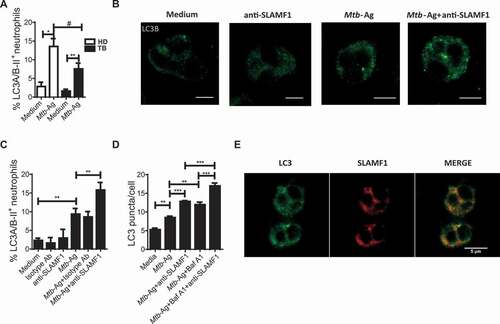
Considering our data showing expression of SLAMF1 on Mtb-stimulated neutrophils, we next evaluated the potential role of this molecule during the autophagy process in neutrophils from TB patients. Isolated neutrophils were stimulated with Mtb-Ag and, at the same time, cultured in the presence or absence of an agonistic anti-SLAMF1 antibody. As shown in ), we were able to confirm by confocal microscopy an accumulation of localized LC3B foci in Mtb-Ag-treated neutrophils. Moreover, activation of SLAMF1 in Ag-stimulated neutrophils significantly increased endogenous LC3B aggregation ()). Furthermore, stimulation with anti-SLAMF1 significantly augmented the percentage of LC3A/B-II+ neutrophils as measured by flow cytometry ()). Importantly, treatment of neutrophils from TB patients with anti-SLAMF1 in the absence of Mtb-Ag had no effect on autophagy levels.
We next investigated the autophagy flux in the presence or absence of Baf A1. Anti-SLAMF1 significantly increased LC3B puncta/cell levels in neutrophils from TB patients stimulated with Mtb-Ag when Baf A1 was present in the culture ()), suggesting that SLAMF1 activation induces autophagosome formation and a functional autophagy pathway. Furthermore, to support that SLAMF1 regulates autophagy in neutrophils, and because neutrophils are terminally differentiated cells that cannot be transfected, we silenced SLAMF1 by using PLB985 cells, a human myeloid cell line that can be differentiated to a neutrophilic profile. Notably, SLAMF1 silencing markedly reduced the percentage of LC3A/B-II+ cells as compared to PLB985 cells transfected with non-targeting siRNA when stimulated with Mtb-Ag (data not shown).
In addition, a positive correlation between the percentage of SLAMF1+ neutrophils and LC3A/B-II+ cells was found (Fig. S4A), further reinforcing the potential role of SLAMF1 during neutrophil autophagy in TB patients. Moreover, we performed a flow cytometry analysis in Mtb-Ag-stimulated neutrophils by gating on SLAMF1+ and SLAMF1− cells. This analysis revealed that autophagy levels detected after Mtb-Ag stimulation were mainly observed in SLAMF1+ cells (Fig. S4B). Besides, we observed colocalization of LC3B and SLAMF1 in Mtb-Ag-stimulated neutrophils ()). These findings suggest that upon neutrophil activation by Mtb-Ag, the triggering of autophagy flux would lead SLAMF1 to LC3+ vesicles.
Considering that ROS are critical intracellular signal transducers sustaining autophagy [Citation26] and our results described above, we next investigated the intracellular levels of ROS upon SLAMF1 activation. As shown in ), Mtb-Ag triggered ROS production. Moreover, signaling through SLAMF1 in Mtb-stimulated neutrophils significantly increased intracellular levels of ROS ()). These results, together with those of ), suggest that ROS are required for the increase in SLAMF1 membrane expression, and, in turn, SLAMF1 activation promotes ROS production induced by Mtb-Ag stimulation.
Figure 5. SLAMF1 ligation increases ROS levels in human neutrophils. (A) Neutrophils from HD (n = 8) were incubated with 2ʹ,7ʹ-dichlorofluorescein diacetate (DCFDA, 50 µM) during 15 min and then stimulated with or without Mtb-Ag (10 µg/ml) ± a SLAMF1 agonist antibody (anti-SLAMF1, 10 µg/ml) during 60 min. Finally, DCFDA fluorescence was evaluated to monitor ROS production by flow cytometry. Left panel: Bars represent the mean values of the mean fluorescence intensity (MFI) of neutrophils of 8 HD ± SEM. Right panel: a representative histogram is shown (Control: non-stained cells). (B) Neutrophils from HD (n = 5) were stimulated or not with Mtb-Ag (10 µg/ml) ± a SLAMF1 agonist antibody (anti-SLAMF1, 10 µg/ml) and treated or not with diphenyleneiodonium (DPI, 10 µM), a NOX inhibitor, for 2 h. Then, neutrophil autophagy levels were evaluated by intracellular saponin-resistant LC3A/B-II immunostaining and analyzed by flow cytometry. Bars represent the mean values of the percentage of LC3A/B-II+ neutrophils ± SEM. No statistical differences were found between Mtb-Ag-stimulated neutrophils treated with DPI compared to Mtb-Ag-stimulated cells treated with both anti-SLAMF1 and DPI. Statistical differences were calculated using one-way ANOVA and post hoc Dunnett multiple comparison test. ** p < 0.01
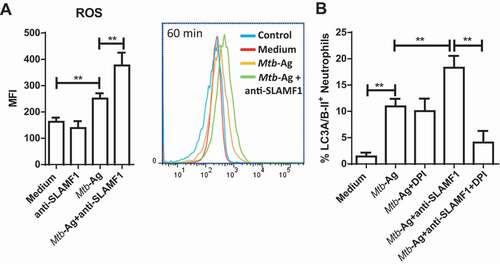
Because the membrane expression of SLAMF1 induced by Mtb-Ag was dependent on ROS production ()) and taking into account that SLAMF1 activation enhanced neutrophil autophagy induced by Mtb-Ag (), we then reasoned that DPI should inhibit the increase in autophagy levels upon SLAMF1 stimulation. To test this possibility, HD’s neutrophils were treated or not with DPI and then these cells were stimulated with Mtb-Ag ± anti-SLAMF1. Finally, autophagy levels were determined. ) shows that inhibition of ROS generation did not modulate autophagy levels in Mtb-Ag-stimulated cells but abrogated the increase in the percentage of LC3A/B-II+ cells observed when neutrophils were stimulated simultaneously with Mtb-Ag and anti-SLAMF1. These results, together with those shown in and Fig. S4A (showing the correlation between SLAMF1 levels and the percentage of LC3A/B-II+ neutrophils), further support the idea that SLAMF1 regulates neutrophil autophagy triggered by Mtb.
Discussion
The outcome of Mtb infection depends on the capacity of the host to develop an effective immunity together with its ability to balance the inflammatory responses. Neutrophils participate in granuloma formation but they also mediate tissue destruction and disease severity [Citation27]. These cells appear as multi-functional leukocytes with variable roles in host defense. In fact, the existence of a duality between neutrophils’ ability to clear Mtb infection and the contribution of increasing numbers of these cells to TB severity and mortality has been documented [Citation20].
SLAMF1 is a self-ligand costimulator that activates lymphocytes. We previously demonstrated that SLAMF1 expression on TB patients’ T cells correlates with responsiveness to Mtb-Ag [Citation15]. Most members of the SLAM subfamily are expressed on myeloid cells such as macrophages and monocytes [Citation17], but the expression of SLAMF1 in neutrophils has not been reported. In this work we demonstrated that Mtb-stimulation significantly induced SLAMF1 expression on human neutrophils. Moreover, we observed that diverse mycobacterial components might be recognized by human neutrophils during stimulation with Mtb-Ag and induce SLAMF1 surface expression. In particular, multiple-protein or lipo/glycans compounds were found as the predominant stimulators of SLAMF1 expression ()). These findings are in line with the reported increased SLAMF1 expression on monocytes and macrophages by various toll-like receptor (TLR) ligands such as Pam3Cys (TLR1-TLR2), synthetic FSL-1 (TLR2-TLR6), LTA and PGN from Staphylococcus aureus (TLR2), flagellin (TLR5) and R848 (TLR7 and TLR8) [Citation23,Citation28]. Indeed, for example, both TLR1-TLR2 and TLR2-TLR6 bind to ManLam [Citation29,Citation30] lipoglycan a major well-characterized Mtb virulence factor that regulates the intracellular trafficking network and the immune response of the infected host cell. Nevertheless, recognition by other types of receptors cannot be ruled out.
Additionally, by using MAPK14/p38 and MAP2K/MEK-MAPK/ERK inhibitors we demonstrated that these MAPKs participate in the signaling pathway induced by Mtb-Ag that leads to SLAMF1 surface expression on neutrophils ()). Previously, Farina et al. have described the participation of MAPK14/p38 signaling during transient SLAMF1 expression on LPS-stimulated monocytes [Citation23]. Besides, Boggaram et al. reported that ESAT-6, a protein secreted by Mtb, induces the expression of IL8 in lung epithelial cells by activating MAPK/ERK and MAPK14/p38 and a rapid induction of ROS production [Citation31]. Moreover, we found that NOX inhibition significantly diminished the percentage of SLAMF1+ neutrophils ()), suggesting that expression of SLAMF1 in response to Mtb is dependent on ROS production.
Of further interest was the fact that Mtb-Ag-stimulation induced higher numbers of SLAMF1+ neutrophils in HD than in TB patients ()). We have previously reported that Mtb-Ag-stimulation induced significantly lower levels of SLAMF1 in T lymphocytes from TB patients as compared to HD [Citation15]. In fact, we observed the highest levels of SLAMF1 in T cells from HD and the lowest levels of the protein in TB patients with the poorest immune response to Mtb-Ag (LR TB patients) [Citation15]. Besides, as mentioned previously, Bologna et al. have reported altered expression of SLAMF1 levels in cells from patients with CLL [Citation18]. In particular, they found that SLAMF1 is lost in CLL cells of patients characterized by a shorter overall survival. Therefore, considering all these previous studies, the reduced levels of SLAMF1 found in neutrophils from TB patients as compared to HD, might be related to imbalances of costimulatory molecules during certain human pathologies [Citation15,Citation18,Citation32–34]. Furthermore, it was suggested that TB disease dramatically alters neutrophils, leading to the accumulation of heterogeneous subpopulations of immature and activated dysfunctional cells [Citation27,Citation35,Citation36], although their occurrence during human TB and the role of SLAMF1 remains to be investigated. Besides, the development of TB is multifactorial, and genetic causes might influence the pathology and the immune responses against Mtb [Citation37–42]. Indeed, single nucleotide polymorphisms may influence the differential SLAMF1 expression on neutrophils from TB patients and HD.
Previous studies showed that SLAMF receptors react with self-proteins, measles virus proteins and bacterial proteins. Additionally, it was shown that SLAMF1 also interacts with the PIK3C3/VPS34-BECN1-UVRAG complex involved in autophagosome maturation [Citation17], and supports phagosome maturation by using the ubiquitous autophagy machinery [Citation16]. Moreover, SLAMF1 was reported to regulate an intracellular autophagic pathway. Actually, SLAMF1 ligation with a monoclonal antibody increases ROS accumulation and induces MAPK14/p38, MAPK8/JNK1-MAPK9/JNK2 and BCL2 phosphorylation, promoting autophagic flux in B cells [Citation18]. However, the role of SLAMF1 as a regulator of autophagy in the context of immunity against Mtb has not been described. Here we demonstrated that Mtb-Ag induced autophagy in human neutrophils from patients undergoing active TB; and SLAMF1 ligation further increased autophagy in these cells. In fact, a positive correlation between the percentage of surface SLAMF1+ neutrophils and LC3A/B-II+ cells was found, further reinforcing the potential role of SLAMF1 during neutrophil autophagy in TB patients.
Our findings indicated that the increased SLAMF1 expression induced by Mtb involves the MAPKs MAPK14/p38 and MAPK/ERK and NADPH-dependent ROS production. Accordingly, the inhibition of NADPH, by impairing SLAMF1 upregulation, abrogated the increase in autophagy observed upon SLAMF1 ligation in Mtb-stimulated cells. These findings, together with the fact that only SLAMF1-expressing neutrophils from TB patients underwent detectable autophagy levels, and the positive correlation found between neutrophil SLAMF1 expression and autophagy levels, support the role of SLAMF1 in regulating neutrophil autophagy upon Mtb stimulation. Interestingly, as reported by Bologna et al. [Citation18] in CLL cells, our findings also showed that SLAMF1 ligation increased ROS production. If these ROS further modulate SLAMF1 expression at later time points remains to be determined.
Most of the studies on mycobacterium-triggered autophagy were performed in mouse model cell lines or HD primary culture cells, but very limited information exists about autophagy in TB patients. Actually, we showed that Mtb-induced IFNG regulates autophagy in TB patients’ cells [Citation13]. Consequently, an important aspect of our study is the investigation of autophagy in neutrophils from patients with active TB.
Interestingly, we observed a significant decrease of autophagy levels in neutrophils from TB patients as compared to HD. Because TB patient’s displayed lower levels of SLAMF1 as compared to HD, our findings are in agreement with those previously reported in CLL patients, which showed that CLL SLAMF1 – cells exhibit an impairment in autophagy activation [Citation18]. Moreover, we further detected a direct correlation between neutrophil numbers and the severity of the disease, as previously demonstrated by other authors [Citation19–21]. In fact, it was reported that TB cavities contain more neutrophils and less lymphocytes compared to nondestructive pulmonary infiltrates and radiological unaffected lobes of the lungs [Citation19]. Hence, considering that autophagy protects against excessive inflammation during Mtb infection [Citation43], the reduced autophagy detected in patients’ neutrophils might be associated with the common detrimental inflammatory responses occurring during active disease.
Thus, published results [Citation16,Citation18] together with our present findings suggest that SLAMF1 constitutes a human regulator of the autophagy process. Then, if autophagy prevents over-exuberant inflammation, it could be a target for clinical purposes [Citation43]. Therefore, both neutrophil autophagy and SLAMF1 emerge as attractive targets for host-directed therapies either directly increasing the ability of the host to eliminate mycobacteria or limiting collateral tissue damage associated with infection. Because autophagy can suppress the activation of several inflammasomes by multiple mechanisms but we demonstrated that it also mediates neutrophil IL1B unconventional-secretion [Citation44], further investigations are underway to elucidate the functional consequences of this event.
In conclusion, we identified SLAMF1 as an innate receptor in human neutrophils that can regulate cellular responses against Mtb. Therefore, either inducing autophagy in myeloid cells or increasing Th1 responses, SLAMF1 would be a key receptor in human immunity against Mtb. Whether SLAMF1 might participate in clinical outcome affecting drug responses of TB patients remains to be elucidated. Altogether, our findings suggest that stimulation of SLAMF1 promotes neutrophil autophagy induced by Mtb, suggesting that it might participate in the anti-mycobacterial human response during active TB.
Material and methods
Subjects
HIV-uninfected patients with TB were diagnosed at the Servicio de Tisioneumonología Hospital F.J. Muñiz, Buenos Aires, Argentina, based on clinical and radiological data, together with the identification of acid-fast bacilli in sputum. All participating patients had received less than 1 week of anti-TB regular therapy. Table 1 summarizes the demographic and clinical characteristics of enrolled TB patients. Bacillus Calmette-Guerin (BCG)-vaccinated healthy control individuals (HD) from the community participated in this study. Peripheral blood was collected in heparinized tubes from each participant after obtaining informed consent. The local Ethics Committee approved all studies.
Exclusion criteria and patient classification
All subjects were 18–60 years old and had no history of illnesses that affect the immune system, such as HIV infection, a recent diagnosis of cancer, treatment with immunosuppressive drugs, hepatic or renal disease, pregnancy, or positive serology for other viral (e.g., hepatitis A, B or C), or bacterial (e.g., leprosy, syphilis) infections. Individuals with bleeding disorders or under anticoagulant medication that might be at an increased risk of bleeding during the procedure of obtaining the sample were excluded from the study. Individuals with latent infection were excluded from the present study by using the QuantiFERON-TB® GOLD PLUS kit (Qiagen, 622,120 and 622,526). TB patients were classified as high-responder (HR) or low responder patients (LR) as previously reported [Citation15], based on in vitro lymphocyte responses to Mtb-Ag. Briefly, HR patients are individuals displaying significant proliferative responses, IFNG production and an increased SLAMF1 expression against the antigen; whereas LR patients exhibit low proliferative responses, IFNG production and SLAMF1 expression. LR patients had more severe pulmonary disease, lower leukocyte counts, and a more prolonged illness, compared to HR individuals [Citation15].
Mtb whole cell lysate
In vitro stimulation of cells was performed with a cell lysate from the virulent Mycobacterium tuberculosis strain H37Rv, prepared by probe sonication (Mtb-Ag) (BEI Resources, NIAID, NIH: Mycobacterium tuberculosis, Strain H37Rv, whole cell lysate, NR-14,822).
Cell preparations and culture conditions
Neutrophils were isolated from heparinized blood by centrifugation on Ficoll-Paque (GE Healthcare, 17–1440-03), dextran sedimentation, and hypotonic lysis [Citation45]. Cells were suspended at 2 × 106 /mL in RPMI 1640 (Invitrogen, 22,400,071) supplemented with L-glutamine (2 mM; Sigma Aldrich, G6392), penicillin (100 U/mL; Gibco, 15,140,122), streptomycin (100 µg/mL; Gibco, 15,140,122), and 10% fetal bovine serum (FBS; Gibco, 10,437,028) and used immediately after isolation. Neutrophils were stimulated with Mtb-Ag (BEI Resources, NIH, 10 μg/ml) ± anti-SLAMF1 mAb (10 µg/ml, A12; Biolegend, 306,310). In some experiments, bafilomycin A1 (100 nM; Fermentek, 88,899–55-2), diphenyleneiodonium chloride (DPI, 100 nM; Sigma Aldrich, 4673–26-1), PD98059 (50 µM; Invivogen, tlrl-pd98) or SB202190 (10 µM; Sigma Aldrich, S7067) were added in different time points of culture.
Purity and viability of neutrophils
After isolation, neutrophil preparations were stained with an anti-CD14-PE (Biolegend, 325,608) antibody and analyzed with a FACS Aria II cytometer (Beckton Dickinson, San Jose, CA, USA) to guarantee that monocyte contamination was <0.5% (Fig. S5). Viability was corroborated by staining with propidium iodide (PI; BD, 556,547) and flow cytometry analysis.
Flow cytometry
To determine the level of SLAMF1 membrane expression on neutrophils, cells stimulated with Mtb-Ag (10 μg/ml) were blocked in PBS (137 mM NaCl, 2.7 mM KCl, 8 mM Na2HPO4, 2 mM KH2PO4, pH 7.4)-FBS 5% for 15 min and then stained for surface expression with fluorophore-marked antibodies against SLAMF1 (BD, 559,572) before and after culture.
Intracellular staining of endogenous saponin-resistant LC3 was performed as described [Citation46]. Briefly, neutrophils were washed with PBS and then permeabilized with PBS containing 0.05% saponin (Sigma-Aldrich, 47,036). In this protocol, the cells are not fixed, therefore LC3-I is washed out of the cell because, unlike LC3-II, it is not anchored to the autophagosome [Citation46]. Cells were then incubated with mouse anti-human LC3A/B antibody (MBL International, M152-3) for 20 min, rinsed with PBS, incubated with anti-mouse secondary antibody conjugated to fluorescein isothiocyanate (eBioscience, 62–6511) for 20 min and rinsed twice with PBS. Negative control samples were incubated with an irrelevant isotype-matched monoclonal antibody (Biolegend, 400,140). Samples were analyzed on a FACSAria II flow cytometer (BD Biosciences).
Confocal microscopy
Treated neutrophils (seeded on poly-L-lysine coated coverslips) were fixed and permeabilized (cold methanol, 20 min) and stained with anti-LC3B (Cell Signaling Technology, 2775) and/or anti-SLAMF1 primary antibodies and then with the corresponding secondary antibodies (Alexa Fluor® 488 Goat Anti-Rabbit IgG, Abcam, 150,077; Alexa Fluor® 555 Donkey Anti-Mouse IgG, Invitrogen, A31570). Nuclei were counterstained with DAPI. The coverslips were mounted with PBS-mowiol (Sigma-Aldrich, 81,381) and imaged using an Olympus FV100 confocal microscope (objective 60/NA1.42, Olympus, Tokyo, Japan).
Image processing
All the images were processed using ImageJ software (Wayne Rasband, National Institutes of Health). After the image binarization using a defined threshold, the number of LC3 puncta was quantified using the Particle Analyzer plugin. Brightness and contrast were adjusted in all images belonging to the same individual, when needed.
ROS measurement
Neutrophils from HD were incubated with 2ʹ,7ʹ-dichlorofluorescein diacetate (DCFDA, 50 µM; Invitrogen, D399) during 15 min at 37°C. Then, cells were washed and stimulated with or without Mtb-Ag (10 µg/ml) ± a SLAMF1 agonist antibody (anti-SLAMF1, 10 µg/ml) for 60 min. Finally, DCFDA fluorescence was evaluated to monitor ROS production by flow cytometry.
Statistical analysis
Analysis of variance and post hoc Tukey’s multiple comparisons test were used as indicated in the figure legend. Mann-Whitney U test and Wilcoxon rank sum test were used for the analysis of unpaired and paired samples respectively. Correlations were calculated using the nonparametric Spearman correlation test. P values of < 0.05 were considered statistically significant.
Supplemental Material
Download MS Word (719.5 KB)Acknowledgments
We are thankful to Dr. Nicolás Anselmino, Licenciada Julieta Alcain, Licenciado Martín García Solá, Licenciada Candela Martín, Licenciado Guillermo Piazza and Dr. Maria Cecilia de Rossi for their constant support and technical assistance.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
References
- Harding E. WHO global progress report on tuberculosis elimination. Lancet Respir Med. 2020 Jan;8(1):19.
- Sia JK, Georgieva M, Rengarajan J. Innate immune defenses in human tuberculosis: an overview of the interactions between Mycobacterium tuberculosis and innate immune cells [Review]. J Immunol Res. 2015;2015:747543.
- Cooper AM. Editorial: be careful what you ask for: is the presence of IL-17 indicative of immunity? [Comment Editorial Research Support, N.I.H., Extramural]. J Leukoc Biol. 2010 Aug;88(2):221–223.
- Eum SY, Kong JH, Hong MS, et al. Neutrophils are the predominant infected phagocytic cells in the airways of patients with active pulmonary TB. Chest. 2010 Jan;137(1):122–128.
- Ramos-Kichik V, Mondragon-Flores R, Mondragon-Castelan M, et al. Neutrophil extracellular traps are induced by Mycobacterium tuberculosis. Tuberculosis. 2009 Jan;89(1):29–37.
- Alvarez-Jimenez VD, Leyva-Paredes K, Garcia-Martinez M, et al. Extracellular vesicles released from mycobacterium tuberculosis-infected neutrophils promote macrophage autophagy and decrease intracellular mycobacterial survival. Front Immunol. 2018;9:272.
- Berry MP, Graham CM, McNab FW, et al. An interferon-inducible neutrophil-driven blood transcriptional signature in human tuberculosis. Nature. 2010 Aug 19;466(7309):973–977.
- Gutierrez MG, Master SS, Singh SB, et al. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell. 2004 Dec 17;119(6):753–766.
- Ponpuak M, Davis AS, Roberts EA, et al. Delivery of cytosolic components by autophagic adaptor protein p62 endows autophagosomes with unique antimicrobial properties [Research Support, N.I.H., Extramural]. Immunity. 2010 Mar 26;32(3):329–341.
- Kim -J-J, Lee H-M, Shin D-M, et al. Host cell autophagy activated by antibiotics is required for their effective antimycobacterial drug action. Cell Host Microbe. 2012 May 17;11(5):457–468.
- Skendros P, Mitroulis I, Ritis K. Autophagy in neutrophils: from granulopoiesis to neutrophil extracellular traps. Front Cell Dev Biol. 2018;6:109.
- Djavaheri-Mergny M, Amelotti M, Mathieu J, et al. NF-kappaB activation represses tumor necrosis factor-alpha-induced autophagy [Research Support, Non-U.S. Gov’t]. J Biol Chem. 2006 Oct 13;281(41):30373–30382.
- Rovetta AI, Pena D, Hernandez Del Pino RE, et al. IFNG-mediated immune responses enhance autophagy against Mycobacterium tuberculosis antigens in patients with active tuberculosis. Autophagy. 2014;10(12):2109–2121.
- Tateosian NL, Pellegrini JM, Amiano NO, et al. IL17A augments autophagy in Mycobacterium tuberculosis-infected monocytes from patients with active tuberculosis in association with the severity of the disease. Autophagy. 2017 Jul 3;13(7):1191–1204.
- Pasquinelli V, Quiroga MF, Martinez GJ, et al. Expression of signaling lymphocytic activation molecule-associated protein interrupts IFN-gamma production in human tuberculosis. J Immunol. 2004 Jan 15;172(2):1177–1185.
- Berger SB, Romero X, Ma C, et al. SLAM is a microbial sensor that regulates bacterial phagosome functions in macrophages [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t]. Nat Immunol. 2010 Oct;11(10):920–927.
- Ma C, Wang N, Detre C, et al. Receptor signaling lymphocyte-activation molecule family 1 (Slamf1) regulates membrane fusion and NADPH oxidase 2 (NOX2) activity by recruiting a Beclin-1/Vps34/ultraviolet radiation resistance-associated gene (UVRAG) complex [Research Support, N.I.H., Extramural]. J Biol Chem. 2012 May 25;287(22):18359–18365.
- Bologna C, Buonincontri R, Serra S, et al. SLAMF1 regulation of chemotaxis and autophagy determines CLL patient response [Research Support, Non-U.S. Gov’t]. J Clin Invest. 2016 Jan;126(1):181–194.
- Barry S, Breen R, Lipman M, et al. Impaired antigen-specific CD4(+) T lymphocyte responses in cavitary tuberculosis. Tuberculosis. 2009 Jan;89(1):48–53.
- Kroon EE, Coussens AK, Kinnear C, et al. Neutrophils: innate effectors of TB resistance? [Review Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t]. Front Immunol. 2018;9:2637.
- Choi H, Chon HR, Kim K, et al. Clinical and laboratory differences between lymphocyte- and neutrophil-predominant pleural tuberculosis. PloS One. 2016;11(10):e0165428.
- Theil D, Farina C, Meinl E. Differential expression of CD150 (SLAM) on monocytes and macrophages in chronic inflammatory contexts: abundant in Crohn’s disease, but not in multiple sclerosis. J Clin Pathol. 2005 Jan;58(1):110–111.
- Farina C, Theil D, Semlinger B, et al. Distinct responses of monocytes to Toll-like receptor ligands and inflammatory cytokines [Research Support, Non-U.S. Gov’t]. Inter immunol. 2004 Jun; 16(6):799–809.
- Winterbourn CC, Kettle AJ, Hampton MB. Reactive oxygen species and neutrophil function [Review]. Annu Rev Biochem. 2016 Jun 2;85:765–792.
- Aratani Y. Myeloperoxidase: its role for host defense, inflammation, and neutrophil function [Research Support, Non-U.S. Gov’t Review]. Arch Biochem Biophys. 2018 Feb 15;640:47–52.
- Filomeni G, De Zio D, Cecconi F. Oxidative stress and autophagy: the clash between damage and metabolic needs [Research Support, Non-U.S. Gov’t Review]. Cell Death Differ. 2015 Mar;22(3):377–388.
- Lyadova IV. Neutrophils in tuberculosis: heterogeneity shapes the way? [Review]. Mediators Inflamm. 2017;2017:8619307.
- Yurchenko M, Skjesol A, Ryan L, et al. SLAMF1 is required for TLR4-mediated TRAM-TRIF-dependent signaling in human macrophages [Research Support, Non-U.S. Gov’t]. J Cell Biol. 2018 Apr 2;217(4):1411–1429.
- Yuan C, Qu ZL, Tang XL, et al. Mycobacterium tuberculosis mannose-capped lipoarabinomannan induces IL-10-producing B cells and hinders CD4(+)Th1 immunity. iScience. 2019 Jan 25;11:13–30.
- Mazurek J, Ignatowicz L, Kallenius G, et al. Divergent effects of mycobacterial cell wall glycolipids on maturation and function of human monocyte-derived dendritic cells [Research Support, Non-U.S. Gov’t]. PLoS One. 2012;7(8):e42515.
- Boggaram V, Gottipati KR, Wang X, et al. Early secreted antigenic target of 6 kDa (ESAT-6) protein of Mycobacterium tuberculosis induces interleukin-8 (IL-8) expression in lung epithelial cells via protein kinase signaling and reactive oxygen species [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t]. J Biol Chem. 2013 Aug 30;288(35):25500–25511.
- Jurado JO, Alvarez IB, Pasquinelli V, et al. Programmed death (PD)-1: PD-ligand1/PD-ligand 2 pathway inhibits T cell effector functions during human tuberculosis [Research Support, Non-U.S. Gov’t]. J Iimmunol. 2008 Jul 1;181(1):116–125.
- Quiroga MF, Pasquinelli V, Martinez GJ, et al. Inducible costimulator: a modulator of IFN-gamma production in human tuberculosis [Research Support, Non-U.S. Gov’t]. J Immunol. 2006 May 15;176(10):5965–5974.
- Yan X, Gu Y, Wang C, et al. Unbalanced expression of membrane-bound and soluble inducible costimulator and programmed cell death 1 in patients with myasthenia gravis [Research Support, Non-U.S. Gov’t]. Clin Immunol. 2019;207:68–78.
- Nancy Hilda J, Neutrophil DS. CD64, TLR2 and TLR4 expression increases but phagocytic potential decreases during tuberculosis [Research Support, Non-U.S. Gov’t]. Tuberculosis. 2018 Jul;111:135–142.
- Correa RDS, Rodrigues LS, Pereira LHL, et al. Neutrophil CD64 expression levels in IGRA-positive individuals distinguish latent tuberculosis from active disease. Memorias Do Instituto Oswaldo Cruz. 2019;114:e180579.
- Vannberg FO, Chapman SJ, Khor CC, et al. CD209 genetic polymorphism and tuberculosis disease [Research Support, Non-U.S. Gov’t]. PloS One. 2008 Jan 2;3(1):e1388.
- Wang FM, Zhang X, Lan L, et al. [Association of PD-1, TIM-3 and TREM-1 single nucleotide polymorphisms with pulmonary tuberculosis susceptibility]. Zhonghua Yi Xue Za Zhi. 2017 Nov 14;97(42):3301–3305.
- Rolandelli A, Hernandez Del Pino RE, Pellegrini JM, et al. The IL-17A rs2275913 single nucleotide polymorphism is associated with protection to tuberculosis but related to higher disease severity in Argentina [Research Support, Non-U.S. Gov’t]. Sci Rep. 2017 Jan 18;7:40666.
- Rolandelli A, Pellegrini JM, Hernandez Del Pino RE, et al. The non-synonymous rs763780 single-nucleotide polymorphism in IL17F gene is associated with susceptibility to tuberculosis and advanced disease severity in argentina [Research Support, Non-U.S. Gov’t]. Front Immunol. 2019;10:2248.
- Zhang X, Ji JM, Li Q, et al. [Association of ICOS and CD28 single nucleotide polymorphisms with pulmonary tuberculosis susceptibility]. Zhonghua Yi Xue Za Zhi. 2019 Nov 26;99(44):3466–3470.
- Ortega E, Hernandez-Bazan S, Sanchez-Hernandez B, et al. Single nucleotide polymorphisms in TLR4 affect susceptibility to tuberculosis in mexican population from the state of veracruz. J Immunol Res. 2020;2020:2965697.
- Deretic V, Levine B. Autophagy balances inflammation in innate immunity. Autophagy. 2018;14(2):243–251.
- Iula L, Keitelman IA, Sabbione F, et al. Autophagy Mediates Interleukin-1beta Secretion in Human Neutrophils. Front Immunol. 2018;9:269.
- Gabelloni ML, Sabbione F, Jancic C, et al. NADPH oxidase derived reactive oxygen species are involved in human neutrophil IL-1beta secretion but not in inflammasome activation. Eur J Immunol. 2013 Dec;43(12):3324–3335.
- Eng KE, Panas MD, Karlsson Hedestam GB, et al. A novel quantitative flow cytometry-based assay for autophagy [Research Support, Non-U.S. Gov’t]. Autophagy. 2010 Jul;6(5):634–641.