
ABSTRACT
TECPR2 (tectonin beta-propeller repeat containing 2) is a large, multi-domain protein comprised of an amino-terminal WD domain, a middle unstructured region and a carboxy-terminal TEPCR domain comprises of six TECPR repeats followed by a functional LIR motif. Human TECPR2 mutations are linked to spastic paraplegia type 49 (SPG49), a hereditary neurodegenerative disorder. Here we show that basal macroautophagic/autophagic flux is impaired in SPG49 patient fibroblasts in the form of accumulated autophagosomes. Ectopic expression of either full length TECPR2 or the TECPR domain rescued autophagy in patient fibroblasts in a LIR-dependent manner. Moreover, this domain is recruited to the cytosolic leaflet of autophagosomal and lysosomal membranes in a LIR- and VAMP8-dependent manner, respectively. These findings provide evidence for a new role of the TECPR domain in particular, and TECPR2 in general, in lysosomal targeting of autophagosomes via association with Atg8-family proteins on autophagosomes and VAMP8 on lysosomes.
Abbreviations: HOPS: homotypic fusion and vacuole protein sorting; LIR: LC3-interacting region; SPG49: spastic paraplegia type 49; STX17: syntaxin 17; TECPR2: tectonin beta-propeller repeat containing 2; VAMP8: vesicle associated membrane protein 8
Introduction
Autophagy is an evolutionarily conserved intracellular process for the delivery of proteins and organelles to lysosomal degradation thereby contributing to the maintenance of cell homeostasis [Citation1]. Dysregulation of this catabolic pathway has been implicated in numerous pathological conditions including neurodegenerative and metabolic diseases [Citation2–4]. Autophagy is initiated with the formation of the phagophore, a cup-shaped structure that elongates and enwraps parts of the cytoplasm including organelles, and seals itself to form a unique double-membrane vesicle termed autophagosome [Citation1]. This pathway is terminated by targeting and fusion of the autophagosome with the lysosomal membrane. The fusion machinery includes the membrane-bound lysosomal SNARE VAMP8 (vesicle associated membrane protein 8), which forms a trans-SNARE complex with the autophagosomal SNARE STX17 (syntaxin 17) and SNAP29 (synaptosome associated protein 29) [Citation5]. Tethering of the autophagosome to lysosome also requires the HOPS (homotypic fusion and vacuole protein sorting) complex that interacts with LC3 on the autophagosomal membrane [Citation6,Citation7]. The lysosome-associated multiprotein complex termed BLOC-1 related complex (BORC) was recently reported to regulate this process [Citation8]. Nevertheless, the regulation and targeting of autophagosome to lysosomal fusion remain largely obscure.
TECPR2 (tectonin beta-propeller repeat containing 2) is a large multi-domain protein, comprising 1411 amino acids in humans. It is predicted to contain an amino-terminal domain of multiple WD repeats (40 amino acids long, often terminating in a tryptophan-aspartic acid dipeptide), a carboxy-terminal domain of six TECPR repeats, and an LC3-interacting region (LIR) motif at the extreme carboxy terminus [Citation9]. Mutations in exon 8, 16 or both in the TECPR2 gene have been identified as the basis of a unique neuronal disorder, currently classified as SPG49 (spastic paraplegia 49; OMIM 615,031). SPG49 patients may present autosomal-recessive inheritance or compound heterozygotic inheritance with different mutation in each allele [Citation10]. TECPR2 was originally identified as an interactor of the ATG8 family proteins that play key roles in autophagy [Citation11]. While evidence for its involvement in autophagy came from a study in primary fibroblasts derived from SPG49 patients, suggesting that it may regulate targeting of autophagosomes to lysosomes [Citation9], its exact role in this process remains unclear. Recently, a more detailed interactome of TECPR2 was obtained, validating its interaction with Atg8-family proteins through its functional LIR motif as well as with two tethering proteins complexes that mediate autophagosomal-lysosomal membrane tethering, BLOC1 and HOPS [Citation12]. We therefore hypothesized that TECPR2 regulates autophagosome targeting and tethering to lysosome. In line with this hypothesis, TECPR1, the closest homologous protein of TECPR2 originally implicated in selective autophagy processes [Citation13,Citation14], was recently reported as a regulator of autophagosome-lysosome fusion [Citation15].
To better characterize the link between TECPR2 and autophagy we here characterize its role in primary fibroblasts derived from SPG49 patients or healthy individuals. Our findings clearly indicate that TECPR2 regulates targeting of autophagosomes to lysosomes, a process primarily mediated by its carboxy-terminal TECPR domain. We show that this domain associates with the lysosome through VAMP8, possibly through interaction with the HOPS complex, and with Atg8-family proteins on the autophagosomal membrane through the LIR motif at its extreme carboxy terminus.
Results
Inhibition of basal autophagy in primary fibroblasts derived from SPG49 patients
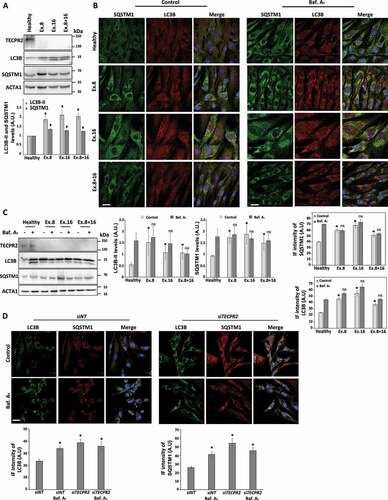
Autophagy is essential for post mitotic neurons functions, and its imbalance is associated with a variety of neurodegenerative diseases including ALS, FTD, PD, HD, BPAN, HSP, Vici syndrome and others [Citation16–18]. As TECPR2 has been previously linked to autophagy [Citation11,Citation12,Citation19], yet in a poorly understood manner, we characterized it in primary fibroblasts derived from SPG49 patients. First, we determined by western blot analysis the levels of autophagic marker proteins LC3B and SQSTM1/p62 (sequestosome 1) in primary fibroblasts from a healthy individual or carriers for different mutant TECPR2 alleles: Ex.8, Ex.16 and Ex.8 + 16. As depicted in , the basal levels of both proteins were significantly higher in SPG49-derived fibroblasts than for the healthy control, suggesting impaired autophagy. We next tested autophagic flux by treatment with the lysosomal inhibitor bafilomycin A1, followed by immunofluorescence analysis (). Whereas fibroblasts of the healthy individual accumulated LC3B- and SQSTM1-positive structures upon inhibition of lysosomal consumption, fibroblasts of SPG49 patients showed accumulation of both in untreated conditions which did not increase further upon treatment with bafilomycin A1, indicating a compromise in basal autophagic flux. Similar results were also observed by western blot analysis (). Fractionation of cell homogenates of healthy and Ex.8 fibroblasts revealed higher levels of LC3B and SQSTM1 in the pellet fractions of Ex.8 (Figure S1A). The contribution of TECPR2 to autophagic flux was also confirmed by a knockdown approach in a BJ (primary foreskin fibroblasts) cell line (). Unlike in our original study [Citation9], here we utilized a more efficient siRNA (see Materials and Methods) that led to a stronger and more prominent effect by efficient TECPR2 knockdown (Figure S1B). These results suggest a contribution of TECPR2 to autophagy under basal cellular maintenance conditions.
Figure 1. Basal accumulation of autophagosomes in SPG49 patient fibroblasts. (A) Total protein extracts from primary fibroblast cells derived from SPG49 patients (and healthy control) were analyzed by western blot for TECPR2, LC3B and SQSTM1 using corresponding antibodies. The accumulation of autophagosomes under basal conditions is indicated by higher ACTA1/actin-normalized levels of LC3B-II and SQSTM1, which were calculated and presented (lower panel) with the SEM of three independent experiments, *p < 0.05, determined by one-way ANOVA with post-hoc Dunnett’s Multiple Comparison Test. (B) Visual assessment of autophagy markers in primary fibroblasts cells derived from SPG49 patients (and healthy control) upon treatment with 0.1 µM bafilomycin A1 (Baf. A1 where indicated) for 4 h. Cells were fixed with methanol, immunostained for LC3B and SQSTM1 and analyzed by confocal microscopy. Scale bar: 20 μm. The accumulation of LC3B and SQSTM1 was calculated as relative intensity (lower panel) and presented with the SEM of three independent experiments, *p < 0.05, determined by one-way ANOVA with post-hoc Dunnett’s Multiple Comparison Test. (C) Primary fibroblast cells derived from SPG49 patients (and healthy control) were grown to confluency in complete medium and treated (where indicated) for the last 4 h with 0.1 µM bafilomycin A1. Total protein extracts were analyzed by western blotting for TECPR2, LC3B and SQSTM1. Levels of LC3B-II and SQSTM1 with the SEM of three independent experiments, *p < 0.05, determined by one-way ANOVA with post-hoc Dunnett’s Multiple Comparison Test. (D) Primary fibroblast BJ cells were transfected with nontargeting (control) siRNA (siNT) or TECPR2 siRNA using DharmaFECT1 transfection reagent for 72 h and treated (where indicated) for the last 4 h with 0.1 µM bafilomycin A1. Cells were fixed with methanol, immunostained for LC3B and SQSTM1 and analyzed by confocal microscopy. Scale bar: 20 μm. The accumulation of LC3B and SQSTM1 was calculated as relative intensity (lower panel) and presented with the SEM of three independent experiments, *p < 0.05, determined by one-way ANOVA (analysis of variance) with post-hoc Dunnett’s Multiple Comparison Test
Starvation rescues autophagic flux in fibroblasts of SPG49 patients
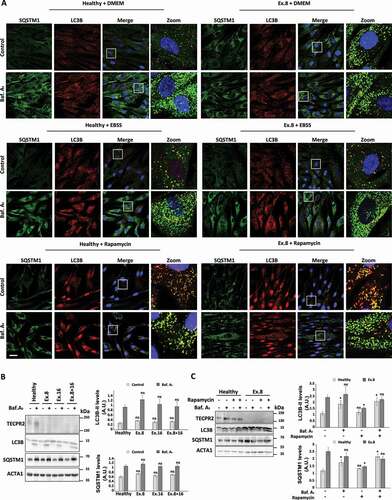
To test whether starvation-induced autophagy is also affected in SPG49-derived cells, we visualized the accumulation of LC3B and SQSTM1 by immunofluorescence in the healthy control and in Ex.8, Ex.16 and Ex.8 + 16 fibroblasts upon starvation (EBSS medium) or treatment with the MTOR inhibitor rapamycin, which induces starvation-like signaling. In contrast to significant accumulation of LC3B and SQSTM1 under basal conditions for TECPR2 mutants, accumulation upon starvation or treatment with rapamycin was only present upon further treatment with bafilomycin A1 for both healthy and SPG49-derived cells, indicating normal autophagic flux ( and S2A). Western blot analysis of the levels of LC3B and SQSTM1 further indicated recovery of flux upon starvation () or rapamycin ( and S2B) for the SPG49-derived cells. These findings suggest that TECPR2 is mainly important for autophagy under basal conditions but becomes largely dispensable for autophagy upon starvation.
Figure 2. Starvation rescues autophagosomes accumulation in fibroblasts of SPG49 patients. (A) Visual assessment of autophagy markers, LC3B and SQSTM1, in primary fibroblasts cells derived from SPG49 patients. Cells were maintained in complete medium (“DMEM”) under basal conditions or switched to starvation medium (“EBSS”), or treated with 0.1 µM rapamycin and in addition were treated (where indicate) with 0.1 µM bafilomycin A1 for 4 h. After 4 h, cells were fixed with methanol, immunostained for LC3B and SQSTM1 and analyzed by confocal microscopy. Scale bar: 20 μm. (B) Primary fibroblasts from SPG49 patients were switched to starvation medium (EBSS) for 4 h with 0.1 µM bafilomycin A1 (where indicated). Total protein extracts were analyzed by western blotting for TECPR2, LC3B and SQSTM1, and ACTA1/actin-normalized levels of LC3B-II and SQSTM1 were calculated and presented (right panel) with the SEM of three independent experiments, *p < 0.05, determined by one-way ANOVA with post-hoc Dunnett’s Multiple Comparison Test. (C) SPG49 Ex.8 mutant fibroblasts (and fibroblasts from healthy control) were grown to confluence and treated as indicated with 0.1 µM rapamycin and 0.1 µM bafilomycin A1 (where indicated) for 4 h. Total protein extracts were analyzed by western blotting for TECPR2, LC3B and SQSTM1. Levels of LC3B-II and SQSTM1 were calculated and presented (right panel) with SEM of three independent experiments, *p < 0.05, determined by one-way ANOVA with post-hoc Dunnett’s Multiple Comparison Test
Replenishment of TECPR2 alleviates basal accumulation of autophagosomes in SPG49 patient fibroblasts in a LIR-dependent manner
We next examined whether the exogenous expression of TECPR2 may reverse the accumulation of autophagic markers. The full length TECPR2 fused to a Flag tag or an empty Flag construct control were exogenously expressed in Ex.8, Ex.16 and Ex.8 + 16 SPG49 patient cells as well as in the healthy control cells, which were then analyzed by immunofluorescence for LC3B (to detect autophagic structures) and Flag (to identify transfected cells). Flag-positive TECPR2 mutant cells demonstrated LC3B levels similar to healthy control whereas empty Flag-positive or untransfected TECPR2 mutant cells, accumulated LC3B ( and S3).
Figure 3. Replenishment of TECPR2 alleviates basal accumulation of autophagosomes in SPG49 patient fibroblasts in a LIR-dependent manner. (A) Primary fibroblasts of SPG49 Ex.8 mutant patient (and healthy control) were transfected for expression of TECPR2-Flag, TECPR2[ΔLIR]-Flag or empty Flag construct control using JetPrime transfection reagent for 48 h. Cells were then fixed with methanol, immunostained for LC3B and Flag and analyzed by confocal microscopy. Scale bar: 20 μm. The level of LC3B was calculated as relative intensity and presented with the SEM of three independent experiments, *p < 0.05, determined by one-way ANOVA with post-hoc Dunnett’s Multiple Comparison Test. (B) Endogenous TECPR2 was immunoprecipitated from HeLa cells with anti-TECPR2 antibody by protein G beads and co-co-precipitation of LC3B was probed by anti-LC3B antibody. (C) Lysates derived from parental HeLa and LC3BendoHA cells were immunoprecipitated with anti-HA followed by immunoblot analysis with indicated antibodies
![Figure 3. Replenishment of TECPR2 alleviates basal accumulation of autophagosomes in SPG49 patient fibroblasts in a LIR-dependent manner. (A) Primary fibroblasts of SPG49 Ex.8 mutant patient (and healthy control) were transfected for expression of TECPR2-Flag, TECPR2[ΔLIR]-Flag or empty Flag construct control using JetPrime transfection reagent for 48 h. Cells were then fixed with methanol, immunostained for LC3B and Flag and analyzed by confocal microscopy. Scale bar: 20 μm. The level of LC3B was calculated as relative intensity and presented with the SEM of three independent experiments, *p < 0.05, determined by one-way ANOVA with post-hoc Dunnett’s Multiple Comparison Test. (B) Endogenous TECPR2 was immunoprecipitated from HeLa cells with anti-TECPR2 antibody by protein G beads and co-co-precipitation of LC3B was probed by anti-LC3B antibody. (C) Lysates derived from parental HeLa and LC3BendoHA cells were immunoprecipitated with anti-HA followed by immunoblot analysis with indicated antibodies](/cms/asset/1dfef7cd-d08c-4fef-a553-815d4295078b/kaup_a_1852727_f0003_oc.jpg)
A functional LIR motif at the extreme carboxy terminus of TECPR2 has been recently identified and characterized [Citation12]. To determine whether TECPR2 activity depends on this motif we expressed in cells as above a LIR-deficient full-length protein TECPR2[ΔLIR]. As depicted in and S3A, TECPR2[ΔLIR], unlike the wildtype TECPR2, failed to reverse the accumulation of LC3B in TECPR2 mutant cells. Consistent with previous reports on interactions of TECPR2 with proteins of the Atg8 family [Citation11,Citation12] endogenous TECPR2 co-immunoprecipitated LC3B (). The reciprocal immunoprecipitation of LC3BendoHA co-immunoprecipitated endogenous TECPR2 (). In addition, endogenous TECPR2 successfully co-immunoprecipitated with the HOPS complex proteins VPS41 and VPS16 (Figure S3B). The association of TECPR2 with the HOPS complex however, was independent of its interaction with the lysosomal SNARE protein, VAMP8 (see below) as knockdown of VAMP8 did not affect TECPR2 coimmunoprecipitation with VPS41 (Figures S3C and S3D). Taken altogether, these findings support a role for TECPR2 in basal autophagy through association with ATG8 proteins via its LIR motif.
Loss of TECPR2 compromises basal lysosomal consumption of autophagosomes
As TECPR2 was recently found to interact with protein complexes responsible for targeting autophagosomes to lysosomes [Citation12], we hypothesized that basal accumulation of autophagic markers upon loss of TECPR2 function in SPG49 patients may be attributed to compromised autophagosomal consumption due to dysfunctional lysosomal tethering. To examine this hypothesis, we colocalized autophagosomes and lysosomes by testing immunofluorescence for LC3B and LAMP1, respectively, (). LC3B and LAMP1 colocalized prominently, in healthy control cells, but to a lesser extent in the TECPR2 mutant cells – suggesting lysosomal mistargeting of autophagic structures. To further assay lysosomal consumption, we utilized the tandem fluorescent autophagy reporter mRFP-GFP-LC3B [Citation20]. As depicted in , most LC3B-positive structures in healthy control cells were red, indicating normal lysosomal consumption of autophagic structures. In contrast, these structures remained yellow in SPG49-derived cells, indicating compromised lysosomal consumption. We then employed the proteinase K protection assay to determine whether those mistargeted basal autophagic structures in TECPR2-deficient cells are sealed or open. In healthy control, the autophagic cargo SQSTM1 in the membrane fraction was protected in vesicles from proteinase K in untreated cells, and was further accumulated in these protected vesicles upon lysosomal inhibition by bafilomycin A1. In comparison, SQSTM1 in Ex. 8 patient cells already accumulated SQSTM1 in protected vesicles to similar extent in absence of bafilomycin A1, indicating accumulation of sealed autophagosomes in TECPR2-deficient cells. Unlike SQSTM1, TECPR2 remained sensitive to proteinase K (Figure S4A), indicating association with the cytosolic leaflet of the membrane. These results suggest that TECPR2 is not essential for formation and sealing of autophagosomes, but contributes to their downstream lysosomal targeting and consumption.
Figure 4. TECPR2 promotes basal lysosomal consumption of autophagosomes. (A) Primary fibroblasts from SPG49 patients (and healthy control) were treated (where indicated) with 0.1 µM bafilomycin A1 for 4 h. Cells were fixed with methanol, immunostained with LC3B and LAMP1 and analyzed by confocal microscopy. Scale bar: 20 μm and 2 μm for zoomed images. The colocalization events were calculated manually from at least three independent fields and presented with the SEM, ***p < 0.001, determined by one-way ANOVA with post-hoc Dunnett’s Multiple Comparison Test. (B) Primary fibroblasts from SPG49 Ex. 8 or Ex. 8 + 16 mutant patients (and healthy control) were transfected for mRFP-GFP-LC3B expression using JetPrime transfection reagent for 48 h. Cells were fixed with methanol and analyzed by confocal microscopy. Scale bar of original images: 20 μm and 2 μm for zoomed images. Large magnifications of stained cells are presented. The number of autophagosomes (yellow) and autolysosomes (red) was calculated and presented with the SEM, *p < 0.05, determined by one-way ANOVA with post-hoc Dunnett’s Multiple Comparison Test. (C) Homogenates of HeLa cells were floated over a sucrose gradient as described in Materials and Methods, proteins of collected fraction were immunoblotted for TECPR2, LC3B and VAMP8 and quantified by ImageJ software. (D) Endogenous VAMP8 from HeLa cells was immunoprecipitated with anti-VAMP8 antibody and co-precipitation of TECPR2 was probed by anti-TECPR2 antibody
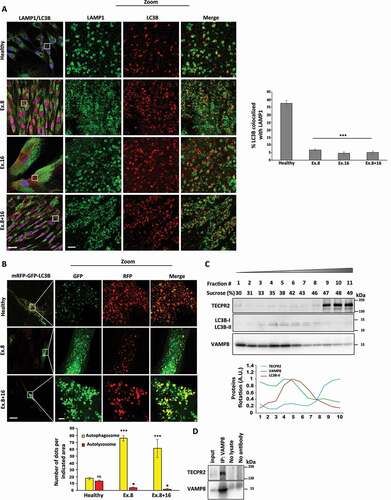
To determine the subcellular site of this contribution, HeLa cell homogenates were fractionated on a sucrose gradient (). While most TECPR2 remained soluble (fractions #9-11), indicating cytosolic distribution, a minor, albeit discernible, portion co-fractionated with LC3B and VAMP8, indicating autophagosomal and lysosomal association, respectively. To further establish a putative lysosomal association of TECPR2 we immunoprecipitated the endogenous VAMP8. Indeed, as shown in , TECPR2 co-precipitated with VAMP8. Overall, TECPR2 was found to associate with both autophagosomes and lysosomes and play a prominent role in autophagosomal targeting and consumption under basal conditions.
The TECPR domain of TECPR2 associates with autophagosomes and lysosomes
TECPR2 is predicted to consist of several domains [Citation9,Citation11,Citation12]. To identify the structural determinants for association of TECPR2 with subcellular membranes, amino-terminal mCherry fusions to different TECPR2 domains () were expressed in HeLa cells (Figure S5A). While the TECPR domain localized to puncta in a LIR-independent manner, the full-length TECPR2 and its other domains showed diffuse cytosolic distribution (Figure S5B). Immunofluorescence analysis upon either the presence or absence of bafilomycin A1 revealed a colocalization of mCherry-TECPR with LC3B and LAMP1 (), indicating the association of the TECPR domain with autophagic structures and lysosomes, respectively. To biochemically establish the association of the TECPR domain with membranes, homogenates from these cells were fractionated on sucrose gradients. In contrast to the endogenous TECPR2 that was mostly cytosolic (), most mCherry-TECPR proteins co-fractionated with autophagic membranes (LC3B) and lysosomes (VAMP8) ().
Figure 5. The TECPR domain of TECPR2 associates with autophagosomes and lysosomes. (A) Schematic presentation of TECPR2 domains fused to mCherry. (B) HeLa cells were transfected for mCherry-TECPR2 expression using JetPrime reagent for 24 h. Cells were fixed with methanol, immunostained for LC3B and LAMP1 and analyzed by confocal microscopy. Scale bar: 20 μm and 2 μm for zoomed images. (C) Proteins extracted from cells transfected as (B) were floated on a sucrose gradient as described in Materials and Methods, collected fraction proteins were immunoblotted for mCherry, LC3B and VAMP8. (D) HeLa cells were transfected with siRNA targeting VAMP8 and/or STX17, or a non-targeting (siNT) control, using DharmaFECT1 transfection reagent. After 24 h cells were transfected for mCherry-TECPR or mCherry-TECPR[ΔLIR] expression using JetPrime reagent for additional 24 h. Cells were fixed with methanol, immunostained for LC3B and LAMP1 and examined by confocal microscopy. Scale bar: 2 μm. Large magnifications of stained cells are presented. Colocalization of structures positive for mCherry-TECPR2 or mCherry-TECPR2[ΔLIR] with LC3B, LAMP1 or both was quantified manually
![Figure 5. The TECPR domain of TECPR2 associates with autophagosomes and lysosomes. (A) Schematic presentation of TECPR2 domains fused to mCherry. (B) HeLa cells were transfected for mCherry-TECPR2 expression using JetPrime reagent for 24 h. Cells were fixed with methanol, immunostained for LC3B and LAMP1 and analyzed by confocal microscopy. Scale bar: 20 μm and 2 μm for zoomed images. (C) Proteins extracted from cells transfected as (B) were floated on a sucrose gradient as described in Materials and Methods, collected fraction proteins were immunoblotted for mCherry, LC3B and VAMP8. (D) HeLa cells were transfected with siRNA targeting VAMP8 and/or STX17, or a non-targeting (siNT) control, using DharmaFECT1 transfection reagent. After 24 h cells were transfected for mCherry-TECPR or mCherry-TECPR[ΔLIR] expression using JetPrime reagent for additional 24 h. Cells were fixed with methanol, immunostained for LC3B and LAMP1 and examined by confocal microscopy. Scale bar: 2 μm. Large magnifications of stained cells are presented. Colocalization of structures positive for mCherry-TECPR2 or mCherry-TECPR2[ΔLIR] with LC3B, LAMP1 or both was quantified manually](/cms/asset/f21124f0-1678-4dd7-bc38-3db04af877c3/kaup_a_1852727_f0005_oc.jpg)
To challenge the autophagosomal and lysosomal association of the TECPR domain, we knocked down the autophagosomal SNARE STX17, which drives autophagosome-lysosome membrane fusion [Citation5], the lysosomal SNARE VAMP8, or both. The efficiency of knockdowns was determined by immunoblot of STX17 and VAMP8 (Figure S5C). The knockdown of STX17 led to the expected accumulation of LC3B-positive autophagosomes [Citation21], accompanied by a more pronounced autophagosomal colocalization of mCherry-TECPR ( left panel). On the other hand, the knockdown of VAMP8 abolished lysosomal colocalization of mCherry-TECPR. Importantly, the combined STX17 and VAMP8 knockdown maintained the colocalization of the TECPR domain with autophagosomes but not lysosomes (, and S5D (left panels)).
To determine whether subcellular distribution of the TECPR domain depends on its LIR motif, we applied the same analysis as above to the LIR-deficient mCherry-TECPR[ΔLIR]. This TECPR2 truncation lost its autophagosomal colocalization, while its lysosomal colocalization was maintained, but further lost upon the knockdown of VAMP8 in a STX17-independent manner ( and S5D (right panels)). These observations strongly suggest that the carboxy-terminal TECPR domain of TECPR2 tethers LIR-associated autophagosomes to lysosomal VAMP8.
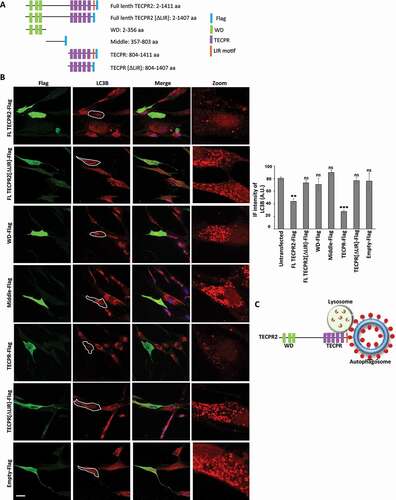
The TECPR domain of TECPR2 is sufficient for alleviating basal accumulation of autophagosomes in SPG49 patient fibroblasts
Based on the role we ascribed to the TECPR domain of TECPR2 in lysosomal consumption of autophagosomes under basal conditions, we further examined the possibility that this region alone is sufficient to reverse the accumulation of autophagic structures in the primary fibroblast cells derived from SPG49 patients. To this end carboxy-terminal Flag tag fusions to different TECPR2 domains were constructed () and successfully expressed in HeLa cells (Figure S6A). As for the mCherry fusions above, only the TECPR domain localized to puncta (in a LIR-independent manner), while the full-length TECPR2 and the other domains fused to Flag tag showed diffuse cytosolic distribution (Figure S6B). We then transfected Ex.8 mutant fibroblast cells with indicated constructs and immunostained for LC3B (to detect autophagic structures) and Flag (to identify transfected cells). Cells positive for expression of either full length TECPR2 or the TECPR domain Flag fusions demonstrated reduced levels of LC3B, compared to those expressing the WD, middle or LIR-deficient TECPR[ΔLIR] Flag fusions (). This indicates that the TECPR domain can substitute the role of the full-length TECPR2 in lysosomal consumption of autophagosomes in a LIR-dependent manner, and that introduction of this domain to cells of SPG49 patients may alleviate the impairment in autophagy due to mutations in TECPR2.
Figure 6. The TECPR domain of TECPR2 is sufficient for alleviating basal accumulation of autophagosomes in SPG49 patient fibroblasts. (A) Schematic presentation of TECPR2 variants fused to Flag tag. (B) Primary fibroblasts of SPG49 Ex.8 mutant were transfected for expression of Flag tag-fused TECPR2 variants (or empty Flag cassette control) using JetPrime transfection reagent for 48 h. Cells were fixed with methanol, immunostained for Flag and LC3B and analyzed by confocal microscopy. Scale bar: 20 μm. The accumulation of LC3B was calculated as relative intensity and presented with the SEM of three independent experiments, *p < 0.05, determined by one-way ANOVA with post-hoc Dunnett’s Multiple Comparison Test. (C) Model – the TECPR domain of TECPR2 mediates autophagosome-lysosome fusion
According to data gathered throughout this study, we propose a model in which under basal conditions the carboxy-terminal TECPR domain of TECPR2 interacts with autophagosomal ATG8 proteins via its LIR motif and also associates with the lysosomal SNARE protein VAMP8, thereby promoting efficient targeting of autophagosomes to the lysosome and consequent lysosomal consumption ().
Discussion
Autophagy plays a vital role in neuronal regulation, and its impairment has been shown to lead to various neurodegenerative pathologies such as Alzheimer, Parkinson and Huntington diseases, amyotrophic lateral sclerosis, Vici syndrome, HSP, and others [Citation4,Citation16–18]. TECPR2 has been previously linked to autophagy [Citation11,Citation12,Citation19], though its role in this process is still poorly understood.
Here we present data implicating TECPR2 in targeting of autophagosomes formed under basal conditions, to lysosome. We show that this activity is primarily mediated by its C-terminal six TECPR repeats, and a functional LIR located at the very end of the protein. We show that this domain is targeted to both lysosomal and autophagosomal membrane in VAMP8- and LIR-depended manner. It has been previously shown that TECPR2 interacts with the HOPS complex that directly interact with VAMP8 [Citation22]. Consistently, we demonstrated that endogenous TECPR2 co-immunoprecipitated with VPS41 and VPS16, members of the HOPS complex. Moreover, Stadel et al. originally identified a functional LIR at TECPR2 C terminus end that interact with several Atg8-family members [Citation12]. The data presented in the present study indicate that both TECPR2 interaction with autophagosome and its overall activity depends on this LIR, by interaction with LC3 or other Atg8 family members. The interaction with the lysosomal membrane depends on VAMP8 and does not require the LIR motif. Based on these findings a model for TECPR2 C-terminal domain in tethering autophagosome to lysosome is shown in the scheme presented in . Consistent with the notion that TECPR2 mediates targeting of autophagosome to lysosomes, our accompanied study of tecpr2−/− mouse, detected a neuroaxonal dystrophy accompanied by a large accumulation of autophagosomes, but not autolysosomes [Citation23].
Targeting and fusion between autophagosome and lysosome requires diverse targeting proteins including autophagosomal SNARE molecules STX17, SNAP29 and lysosomal SNARE VAMP8, as well as tethering proteins HOPS and BORC. The tethering machinery also incorporates adaptors such as EPG5 and TECPR1 [Citation15,Citation24]. TECPR1, closest homolog of TECPR2, was also localized to the lysosomal membranes and its depletion led to accumulation of LC3 and SQSTM1, indicating its role in autophagosomes targeting to the lysosomes [Citation15]. Moreover, the fact that TECPR2 is mostly distributed in the cytosol and only a small fraction is localized to the lysosomal and autophagosomal membrane in steady state, indicate a transient association with the membrane. Our finding that TECPR repeats region is mostly associated with the membranal fraction may indicate that the N terminal and the mid regions of the protein regulate this process. The exact function(s) of these regions await clarification by future studies.
TECPR2 being a large multidomain protein was recently reported to interact with multiple complexes most of which implicated in different intracellular trafficking processes [Citation12]. An interaction with SEC24D, a COPII vesicle component, was shown to affect secretion of collagen, thus indicating a possible role of TECPR2 in early secretion stages [Citation12]. Alternatively, this interaction may also indicate a role for TECPR2 in autophagosome formation. However, our findings showing the formation of fully sealed autophagosomes in SPG49-derived cells are consistent with the notion that TECPR2 plays important role in late stages of autophagosome targeting to lysosomes. Support to the notion that TECPR2 is needed late along the autophagy process comes from the fact that autophagosomes accumulate in a recently established TECPR knockout mouse [Citation23] as well as in post mortem analysis of brain sections obtained from Spanish Water dogs with mutation in TECPR2 [Citation25].
Our results indicate that TECPR2 is mainly needed for autophagosomal delivery under conditions that autophagy is not induced. Upon starvation or in the presence of rapamycin however, TECPR2 becomes dispensable. The exact reason for its dispensability remains unclear, however we raise two hypothesis that should be examined in future studies. The first implies that the targeting machinery of autophagosomes formed under basal conditions is entirely different from this formed upon stress. Accordingly, it is expected that under stress a particular tethering complex and/or its regulators will mediate targeting and docking of autophagosome to the lysosomal membrane. Alternatively, and not mutually exclusive, the other hypothesis predicts that while a particular set of tethering complex mediates targeting of selective autophagosome formed under basal conditions, upon stress multiple tethering factors are incorporated to the autophagosomal and lysosomal membranes, thus introducing some redundancy to assure an efficient targeting. Indeed, multiple factors including: HOPS, EPG5, ATG14, TECPR1, GORASP2/GRASP55, BIRC6/BRUCE and RUFY4 were recently implicated in this process [Citation22,Citation26]. The essentiality of each of these factors to different selective autophagy processes remained to be determined.
The fact that starvation or treating cells with rapamycin overcome the autophagy defects observed in fibroblasts derived from all three SPG49 patients, opens the possibility for new therapeutic strategies. Future studies on different model systems such as the newly characterized tecpr2−/- mouse [Citation23] may help determine the power of such treatments. Finally, our findings that ectopic expression of TECPR2 in general and particularly its six C terminal TECPR repeats prevents the autophagy inhibition shown in SPG49-derived cells and also raise possibilities of potential treatments of these patients by gene therapy.
Materials and Methods
Study approval
Primary fibroblasts cells were obtained from Sheba Medical Center in accordance with the Helsinki Declaration of 1975.
Cell cultures and treatments
HeLa cells (strain JW; obtained from the Weizmann Institute Cell-Line Core) were grown on alpha minimum essential media (aMEM; Biological Industries, 01–042-1A) supplemented with 10% fetal calf serum (FBS; Invitrogen, 10,270,106) at 37°C in 5% CO2. LC3BendoHA [Citation27] were grown on Dubeco’s modified Eagle’s medium (DMEM) plus GlutaMAX-I (Gibco, 10,566,016) supplemented with 10% fetal bovine serum (FBS) and 1 mM sodium pyruvate (Gibco, 11,360,070) and grown at 37°C and 5% CO2. Primary fibroblast cells were grown on Dubeco’s modified Eagle’s medium (DMEM; Gibco, 41,965–039) supplemented with 20% fetal calf serum (FCS) and 1% L-glutamine (Sigma, G5763) at 37°C in 5% CO2. For induction of autophagy cells were treated with 100 nM rapamycin (Sigma, R0395) or starved by washing twice with phosphate-buffered saline (PBS; Biological Industries, 02–023-1A) and incubated in Earle’s Balanced Salt Solution (EBSS; Biological Industries, 02–011-1A). Lysosomal degradation was inhibited by 100 nM bafilomycin A1 (LC Laboratories, B-1080) for 4 h. Plasmids were transfected using JetPrime transfection reagent (TAMAR Laboratories Supplies Ltd, 114–07). All cell lines were routinely inspected for mycoplasma contamination on a monthly basis.
Plasmids and siRNA
The cloning of full-length TECPR2 and its fragments into mCherry- or Flag-labeled plasmid was performed using the restriction free transfer PCR technique. The mCherry plasmid was a gift from Prof. Reuveny from the Weizmann Institute of Science. The origin of this plasmid is pEGFP, where EGFP was replaced by mCherry or by Flag tag. For TECPR2 knockdown, cells were transfected with siRNA oligo GUGCUGAGUUGGAAUGAAU using DharmaPHECT reagent (Dharmacon, T-2001-03) (this oligo was found to be more efficient than a smart pool that was used in our previous studies). The mRFP-GFP-LC3B plasmid was a generous gift from Yoshimori lab [Citation20].
Western blot analyses
Total cellular protein extracts were prepared in RIPA buffer (0.1 M NaCl [Bio-Lab Ltd, 21,955], 5 mM EDTA [J.T. Baker, 8993], 0.1 M sodium phosphate [Sigma, 342,483], pH 7.5, 1% Triton X-100 [Sigma, X100], 0.5% sodium deoxycholate [Sigma, D6750], 0.1% sodium dodecyl sulfate [Sigma, L4509]) with a protease inhibitor cocktail (PIC; Merck, 539,134). The extracts were centrifuged at 16,000 x g for 15 min at 4°C and protein concentrations were determined using Bio-Rad Protein Assay Dye Reagent Concentrate (Bio-Rad, 500–0006). Total proteins (30 µg) were separated by SDS−PAGE (12% polyacrylamide) and transferred to a polyvinylidene difluoride (PVDF) membrane (Bio-Rad, 1,704,157). The membrane was blocked in PBS with 5% skim milk for 1 h at room temperature, and then incubated with the appropriate primary antibody overnight at 4°C. It was then washed three times with PBS-TWEEN 20 (0.1%; Sigma, P1379) and incubated with the secondary antibody (goat anti-mouse or goat anti-rabbit) for 1 h at room temperature. Finally, the membrane was washed three times and specific proteins were visualized using the Enhanced ChemiLuminescence (ECL) detection system (Biological Industries, 20–500-120). The antibodies employed for this assay are described in .
Table 1. Antibodies used in this study
Membrane flotation assay
Cells were homogenized with a Balch homogenizer (HGM Precision Engineering, 218,024/212). Homogenates (2 mg protein) were adjusted to 2 M sucrose (J.T. Baker, 4072), placed at the bottom of rotor tubes, overlaid with 1.75, 1.5, 1.25, 1 and 0.75 M sucrose and centrifuged in the SW-28 rotor (Beckman, 28,000 rpm) at 102,000 x g rpm (slow acceleration and deceleration) overnight, 4°C. Fractions from the top of the gradient were collected and sucrose densities were estimated from their refractive indices. Proteins from each fraction (180 µl) were precipitated by 10% trichloroacetic acid (TCA; Merck, T6399), boiled in sample buffer and immunoblotted.
Immunoprecipitation
Cells were washed twice with cold PBS, collected and resuspended in NP40 buffer (25 mM Tris, pH 7.5 [Bio-Lab Ltd, 20,097,759], 50 mM KCl [Sigma, P9333] and 0.5% NP-40 [Sigma, I3021]). Lysates were centrifuged at 20,000 x g for 15 min at 4°C and the supernatant was incubated overnight at 4°C with the indicated antibodies which absorbed to protein G coated beads (Santa Cruz Biotechnology, sc-2002). Total cell lysate samples constituted 5% of the lysate. For immunoprecipitation of LC3BendoHA cell from 4x10-cm cell culture plates were harvested and lysed for 30 min in NP40 buffer (50 mM Tris, pH 7.4, 150 mM NaCl, 1 mM EDTA, 0.5% NP-40, 1x protease inhibitor, 1x phosphatase inhibitor [Merck, 539,134]) at 4°C. Lysates were cleared from cell debris by centrifugation (20,000 x g, 10 min) followed by adjustment of protein concentrations between the samples. Immunoprecipitation was performed for 1 h at room temperature with pre-equilibrated anti-HA-agarose (Sigma, A2095). Agarose beads were washed five times with NP40 buffer and boiled with 3x loading buffer (200 mM Tris-HCl, pH 6.8, 6% SDS [Sigma, L3771], 20% glycerol [Sigma, G5516], 0.1 g/ml DTT [Sigma, D0632], 0.1 mg Bromophenol Blue [Sigma, 0126]) at 95°C. Then samples were subjected to western blot analyses.
Proteinase K protection assay
Cells cultured and treated in 15-cm dishes were detached by trypsin, washed (PBS x 3), resuspended in 4 pellet volumes of homogenization buffer (10 mM Tris, pH 7.4, 0.25 M sucrose) supplemented by protease inhibitors and homogenized on ice with a Balch homogenizer. Unbroken cells and nuclei were removed (700 x g, 5 min, 4°C) and equal amounts of homogenate were centrifuged using a TLA 120.2 rotor 352,000 x g, 30 min, 4°C for cytosol and membrane fractions, and the pellets were resuspended in homogenization buffer. Each fraction was then divided into equal volumes for treatment with (30 min, 37°C) proteinase K (10 µg/ml, Merck, 1,245,680) or Triton X-100 (0.4% [v:v]) with proteinase K, as indicated. Treatments were terminated by addition of phenylmethylsulfonyl fluoride (PMSF; Sigma, 78,830; 200 mM, 10 min on ice), and proteins were precipitated by 10% TCA, boiled in sample buffer and immunoblotted.
Immunostaining
Cells cultured on sterile coverslips (13 mm) and treated as indicated were fixed and permeabilized by 100% methanol (Bio-Lab Ltd, 136,805) for 10 min at −20°C, washed three times with PBS, blocked (10% FCS in PBS, 30 min at room temperature), and incubated with primary antibody (1 h at room temperature), washed (as above) and incubated with secondary antibody (30 min at room temperature).
Confocal fluorescence microscopy
Immunostained cells were observed under a confocal laser scanning microscope (Zeiss) and images were analyzed by Zen software.
Statistical analysis
Where appropriate, statistical significance between data sets was analyzed by t-tests or Analysis of variance one way (Anova) using GraphPad Prism. N.S., non-significant; *p < 0.05, **p < 0.01, ***p < 0.001.
Supplemental Material
Download Zip (36 MB)Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
Related Research Data
References
- Weidberg H, Shvets E, Elazar Z. Biogenesis and cargo selectivity of autophagosomes. Annu Rev Biochem. 2011;80:125–156.
- Ravikumar B, Sarkar S, Davies JE, et al. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol Rev. 2010 Oct;90(4):1383–1435.
- Abada A, Elazar Z. Getting ready for building: signaling and autophagosome biogenesis. EMBO Rep. 2014 Aug;15(8):839–852.
- Fraiberg M, Elazar Z. Genetic defects of autophagy linked to disease. Prog Mol Biol Transl Sci. 2020;172:293–323.
- Itakura E, Kishi-Itakura C, Mizushima N. The hairpin-type tail-anchored SNARE syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell. 2012 Dec 7;151(6):1256–1269.
- Manil-Segalen M, Lefebvre C, Jenzer C, et al. The C. elegans LC3 acts downstream of GABARAP to degrade autophagosomes by interacting with the HOPS subunit VPS39. Dev Cell. 2014 Jan 13;28(1):43–55.
- McEwan DG, Popovic D, Gubas A, et al. PLEKHM1 regulates autophagosome-lysosome fusion through HOPS complex and LC3/GABARAP proteins. Mol Cell. 2015 Jan 8;57(1):39–54.
- Jia R, Guardia CM, Pu J, et al. BORC coordinates encounter and fusion of lysosomes with autophagosomes. Autophagy. 2017 Oct 3;13(10):1648–1663.
- Oz-Levi D, Ben-Zeev B, Ruzzo E, et al. Mutation in TECPR2 reveals a role for autophagy in hereditary spastic paraparesis. Am J Hum Genet. 2012 Dec 7;91(6):1065–1072.
- Heimer G, Oz-Levi D, Eyal E, et al. TECPR2 mutations cause a new subtype of familial dysautonomia like hereditary sensory autonomic neuropathy with intellectual disability. Eur J Paediatr Neurol. 2016 Jan;20(1):69–79.
- Behrends C, Sowa ME, Gygi SP, et al. Network organization of the human autophagy system. Nature. 2010 Jul 1;466(7302):68–76.
- Stadel D, Millarte V, Tillmann K, et al. TECPR2 cooperates with LC3C to regulate COPII-dependent ER export. Mol Cell. 2015 Oct 1;60(1):89–104.
- Ogawa M, Sasakawa C. The role of Tecpr1 in selective autophagy as a cargo receptor. Autophagy. 2011 Nov;7(11):1389–1391.
- Wetzel L, Blanchard S, Rama S, et al. TECPR1 promotes aggrephagy by direct recruitment of LC3C autophagosomes to lysosomes. Nat Commun. 2020 Jun 12;11(1):2993.
- Chen D, Fan W, Lu Y, et al. A mammalian autophagosome maturation mechanism mediated by TECPR1 and the Atg12-Atg5 conjugate. Mol Cell. 2012 Mar 9;45(5):629–641.
- Menzies FM, Fleming A, Caricasole A, et al. Autophagy and neurodegeneration: pathogenic mechanisms and therapeutic opportunities. Neuron. 2017 Mar 8;93(5):1015–1034.
- Cullup T, Kho AL, Dionisi-Vici C, et al. Recessive mutations in EPG5 cause Vici syndrome, a multisystem disorder with defective autophagy. Nat Genet. 2013 Jan;45(1):83–87.
- Vantaggiato C, Crimella C, Airoldi G, et al. Defective autophagy in spastizin mutated patients with hereditary spastic paraparesis type 15. Brain. 2013 Oct;136(Pt 10):3119–3139.
- Ebrahimi-Fakhari D, Saffari A, Wahlster L, et al. Congenital disorders of autophagy: an emerging novel class of inborn errors of neuro-metabolism. Brain. 2016 Feb;139(Pt 2):317–337.
- Kimura S, Noda T, Yoshimori T. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy. 2007 Sep-Oct;3(5):452–460.
- Uematsu M, Nishimura T, Sakamaki Y, et al. Accumulation of undegraded autophagosomes by expression of dominant-negative STX17 (syntaxin 17) mutants. Autophagy. 2017 Aug 3;13(8):1452–1464.
- Lorincz P, Juhasz G. Autophagosome-Lysosome Fusion. J Mol Biol. 2020 Apr 3;432(8):2462–2482.
- Tamim-Yecheskel BC, Fraiberg M, Kokabi K, et al. A TECPR2 knockout mouse exhibits age-dependent neuroaxonal dystrophy associated with autophagosome accumulation. Autophagy. 2020 Nov 20. doi:https://doi.org/10.1080/15548627.2020.1852724. Online ahead of print.
- Wang Z, Miao G, Xue X, et al. The Vici syndrome protein EPG5 is a rab7 effector that determines the fusion specificity of autophagosomes with late endosomes/lysosomes. Mol Cell. 2016 Sep 1;63(5):781–795.
- Hahn K, Rohdin C, Jagannathan V, et al. TECPR2 associated neuroaxonal dystrophy in Spanish water dogs. PLoS One. 2015;10(11):e0141824.
- Kriegenburg F, Ungermann C, Reggiori F. Coordination of autophagosome-lysosome fusion by Atg8 family members. Curr Biol. 2018 Apr 23;28(8):R512–R518.
- Eck F, Phuyal S, Smith MD, et al. ACSL3 is a novel GABARAPL2 interactor that links ufmylation and lipid droplet biogenesis. J Cell Sci. 2020 Sep 16;133:18.