
ABSTRACT
Ion exchange between intracellular and extracellular spaces is the basic mechanism for controlling cell metabolism and signal transduction. This process is mediated by ion channels and transporters on the plasma membrane, or intracellular membranes that surround various organelles, in response to environmental stimuli. Macroautophagy (hereafter referred to as autophagy) is one of the lysosomal-dependent degradation pathways that maintains homeostasis through the degradation and recycling of cellular components (e.g., dysfunctional proteins and damaged organelles). Although autophagy-related (ATG) proteins play a central role in regulating the formation of autophagy-related member structures (e.g., phagophores, autophagosomes, and autolysosomes), the autophagic process also involves changes in expression and function of ion channels and transporters. Here we discuss current knowledge of the mechanisms that regulate autophagy in mammalian cells, with special attention to the ion channels and transporters. We also highlight prospects for the development of drugs targeting ion channels and transporters in autophagy.
Introduction
The term “autophagy” (Greek for “self-eating”), first used by Christian de Duve in 1963, describes an evolutionarily conserved cellular pathway that responds to environmental stress to remove and degrade cytoplasmic waste by lysosomes [Citation1,Citation2]. Degradation products can be recycled for macromolecular synthesis and energy production. Under different conditions, autophagy functions as a “double-edged sword” by promoting cell survival or mediating cell death. Accordingly, autophagy plays a dual role in many diseases [Citation3].
As an important part of homeostasis, the movement of ions across the plasma membrane (PM) and organelle membranes affects various cellular processes. This dynamic modulation is mediated by several types of transport proteins, especially ion channels or transporters, which broadly distribute in impermeable cellular membranes, including those of the mitochondria, the endoplasmic reticulum (ER), Golgi apparatus (GA), endosomes, and lysosomes. Ion channels and transporters allow the ions (e.g., calcium [Ca2+], potassium [K+], sodium [Na+], and chloride [Cl−]) to influx and efflux across lipid membrane barriers [Citation4]. Ion channels are integral pore-forming membrane proteins, which enable passive transport of ions through the membrane to reduce their electrochemical gradient [Citation5]. In contrast, ion transporters actively move ions through primary active transporters (also called pumps, which generate an electrochemical gradient in an ATP-dependent manner) or secondary active transporters (also known as co-transporters/anti-porters/exchangers, which obtain energy from the electrochemical gradient of ions [usually Na+]) [Citation6].
In recent years, we have begun to understand how ion channels and transporters play context-dependent roles in the induction and regulation of autophagy in different cell types [Citation7]. Moreover, impaired autophagy caused by dysfunctional ion channels and transporters is implicated in various diseases, such as cancers, diabetes, cardiovascular diseases, and neurological diseases. Therefore, the study of the regulation mechanisms of ion channels and transporters in autophagy may not only reveal unknown biological processes, but also guide the development of new drugs. In this review, we summarize current knowledge on the role of ion channels and transporters in mammalian autophagy, and discuss their potential as drug targets in autophagy-related diseases.
The process of autophagy
Autophagy can be divided into three different forms: macroautophagy, microautophagy, and chaperone-mediated autophagy. Macroautophagy (hereafter referred to as autophagy) is the most studied form of autophagy in mammalian cells, and its distinguishing morphological hallmark is the formation of different cytoplasmic membrane vesicles in response to stress [Citation1,Citation2]. After sensing induction signals, autophagy begins with engulfing cytoplasmic materials by cup-shaped phagophores. Subsequently, the closure of phagophores results in the formation of double-membrane structures, namely autophagosomes. Finally, autophagosomes fuse with lysosomes to generate autolysosomes, where the cargo is degraded, and subsequently released back into the cytosol for reuse.
This dynamic membrane process is mediated by a number of ATG proteins and can be divided into several sequential steps: initiation, nucleation, expansion, fusion, degradation and recycling [Citation1,Citation2] (). The formation of a phagophore is initiated by the assembly and activation of the ULK complex, followed by phosphorylating components of the class III phosphatidylinositol 3-kinase (PtdIns3K) complex. The activation of ULK and the class III PtdIns3K complex produces phosphatidylinositol-3-phosphate (PtdIns3P) to support phagophore nucleation (mostly at the ER) by engaging PtdIns3P-binding ATG proteins and members of the WIPI (WD repeat domain, phosphoinositide interacting) proteins, coupled to the recruitment of ATG9-containing vesicles. This event further triggers the expansion of phagophores upon the incorporation of membrane from various sources, including the PM, ER, mitochondria, mitochondria-associated membranes (MAMs), GA, and recycling endosomes. The activation of ULK and class III PtdIns3K complexes is highly regulated by various kinases, especially MTOR (mechanistic target of rapamycin kinase) and AMP-activated protein kinase (AMPK).
Figure 1. The autophagy pathway. The autophagy process can be roughly divided into five phases: initiation, phagophore nucleation, phagophore expansion, autophagosome-lysosome fusion, and lysosomal substrate degradation.
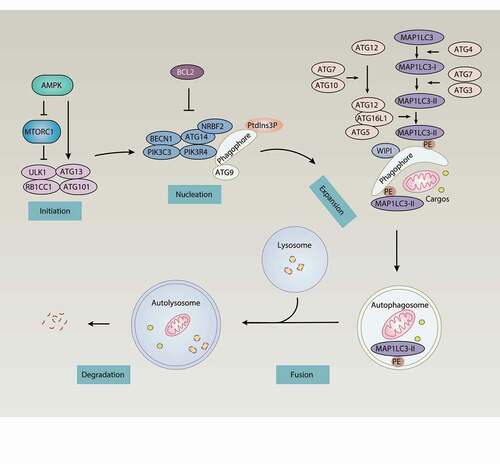
Phagophore expansion involves two ubiquitin-like conjugation systems. On the one hand, this involves the processing and activation of MAP1LC3 (microtubule associated protein 1 light chain 3; an ortholog of yeast Atg8) by the E1-like enzyme ATG7, the E2-like enzyme ATG3, and the cysteine protease ATG4, leading to the formation of MAP1LC3-I and the subsequent conjugation of MAP1LC3-I and phosphatidylethanolamine (PE), forming the membrane-bound MAP1LC3-II protein. This process is called MAP1LC3 lipidation, which is required for autophagosome formation and/or maturation. On the other hand, ATG7 and the E2-like enzyme ATG10 operate sequentially to catalyze the formation of the ATG12–ATG5-ATG16L1 (autophagy related 16 like 1) complex, which binds with WIPI and functions as an E3-like ligase to mediate lipidation of MAP1LC3 and its orthologs, such as GABARAP (GABA type A receptor-associated protein) and GABARAPL2/GATE-16 (GABA type A receptor associated protein like 2). Lipidated MAP1LC3 and its family members are required for expansion and closure of the phagophore membrane, and enable the phagophore with assembly sites and autophagic receptors (e.g., SQSTM1/p62 [sequestosome 1]) to mediate autophagy.
Upon closure, autophagosomes directly fuse with lysosomes to form autolysosomes. Alternatively, autophagosomes combine with endosomes to produce amphisomes, which in turn fuse with lysosomes to again form autolysosomes. Finally, lysosomes degrade autophagy cargoes through lumenal acidification and the activation of lysosomal hydrolases. The salvaged materials can be released back to the cytoplasm for reuse. Therefore, autophagy is a recycling system that balances the availability and demand of cellular components during stress.
Types of ion channels
Ligand-gated ion channels
Ligand-gated ion channels (LGICs) typically contain two different domains: the transmembrane domain (TMD) region encoding multiple TMDs forming ion pore, and an extracellular ligand-binding site that generates energy to open the gate after binding with ligands. By distinguishing between ligand types and protein structures, LGICs are grouped into several superfamilies: the pentameric ion channels (pLGICs, also called Cys-loop receptors), the ionotropic glutamate receptors (iGluRs), nucleotide-gated channels, proton-gated channels, calcium-activated channels, and other channels [Citation5].
pLGICs
The pLGICs, activated by several neurotransmitters, comprise nicotinic acetylcholine receptors (nAChRs), γ-aminobutyric acid type A/C receptors (GABAA/CRs), glycine receptors (GlyRs), 5-hydroxytryptamine type 3 receptors (5-HT3Rs), and zinc-activated ion channels (ZACs). The nAChRs and 5-HT3Rs have a similar structure that contains five subunits forming an ion-conducting pore, which can permeate cations (e.g., Na+, K+, and Ca2+). ZACs are comprised of four subunits, forming an intrinsic cation-selective channel that is permeable to Na+, K+, and Cs+. In contrast, GABAA/CRs and GlyRs are anion-selective channels, allowing the permeation of Cl−. These channels are widely-expressed in central and peripheral nervous systems and neuromuscular junctions, and are well-studied in neurotransmission. They are also expressed in non-excitable cells (e.g., epithelial or immune cells) and participate in several physiological processes.
iGluRs
The iGluRs are tetrameric LGICs that are activated by the neurotransmitter glutamate, comprising three main subtypes, namely GRIN/NMDAR (glutamate ionotropic receptor NMDA type), GRIA/AMPAR (glutamate ionotropic receptor AMPA type), and GRIK/kainate receptor (glutamate ionotropic receptor kainate type). The iGluRs are permeable to both K+ and Na+ to depolarize the postsynaptic membrane, and in some cases to divalent cations, such as Ca2+. They are expressed on pre- and postsynaptic cell membranes, primarily within the central nervous system (CNS). These enable iGluRs to control the strength of synaptic connections between neurons, which is crucial for advanced processes of brain function (e.g., learning and memory).
Nucleotide-gated channels
The nucleotide-gated channels contain two major groups, namely ATP-activated ion channels and cyclic nucleotide-activated ion channels. ATP-activated ion channels also contain P2RX/P2X receptor (purinergic receptor P2X) channels and CFTR (CF transmembrane conductance regulator). The P2RX channels are nonselective ion channels. In response to the binding of ATP, they can transport cations including Na+, K+, and Ca2+. The P2RX channels are expressed in peripheral and CNS structures (neurons and glia) and are involved in neurotransmission, pain signal generation, and neurotransmitter release [Citation8]. Among them, P2RX7 (purinergic receptor P2X7) mediates the influx of Ca2+ and Na+ and the efflux of K+ after activation. CFTR is expressed in epithelial cell membrane and is only opened after phosphorylation by PRKA/PKA (protein kinase cAMP-activated) to control the influx and efflux of Cl−. The deletion of mutant CFTR impairs the incorporation of CFTR into the PM as well as Cl− ion channel function, leading to cystic fibrosis and the dysregulation of epithelial fluid transportation in the lung, pancreas, and other organs. Cyclic nucleotide-activated ion channels consist of two structurally similar families: the cyclic nucleotide-gated (CNG) channels and the hyperpolarization-activated cyclic nucleotide-modulated (HCN) channels. CNG channels are nonselective cation channels formed as tetramers; they are activated by binding cAMP and cGMP to regulate membrane potential, but have little dependence on voltage. HCN channels serve as nonselective cation channels.
Proton-gated channels
Acid-sensing ion channels (ASICs) are pH-gated channels that are permeable to Na+, resulting in membrane depolarization. These channels are regulated by extracellular alkalosis, intracellular pH, and various other factors, and are expressed in postsynaptic membrane in peripheral and central neurons involved in pain sensing and synaptic plasticity. ASIC1 (acid sensing ion channel subunit 1) also shows Ca2+ permeability, and sends other messages.
Calcium-activated channels
Calcium-activated potassium channels contain calcium-activated chloride channels (CaCCs) and calcium-activated potassium channels (KCa). CaCCs are a type of Cl− channel, which is gated based on intracellular calcium concentration ([Ca2+]i), triggering either the influx or efflux of Cl− that is accompanied by water and Na+. Two proteins are identified as CaCC members: ANO1/TMEM16A (anoctamin 1) and ANO2/TMEM16B (anoctamin 2). The classical functions of ANO1 and ANO2 include roles in membrane excitability in the nervous system and cardiovascular system, whereas ANO1 has recently been found to have novel roles in the modulation of cancer, insulin secretion, and phospholipid scrambling [Citation9]. KCa is widely expressed in the PM of many types of cells and initiates the efflux of K+ triggered by [Ca2+]i and membrane potential. KCa has two subtypes: BK channels, which have a larger conductivity and are controlled by the synergy of membrane depolarization and Ca2+ concentration; and SK channels, which have less conductivity and are only controlled by Ca2+ concentration [Citation10].
KCNJ/Kir and SCNN1/ENaC
KCNJ/Kir (potassium inwardly rectifying channel) and SCNN1/ENaC (sodium channel epithelial 1) are also LGICs. KCNJ/Kir is a subset of K+ channels, comprising seven subfamilies in four functional groups: classical KCNJ/Kir channels, G protein-gated KCNJ/Kir channels, KCNJ/KATP (referred to as ATP-sensitive potassium channels) [Citation11], and KCNJ/K+-transport channels. All these channels require phosphatidylinositol(4,5)-bisphosphate (PtdIns [Citation4,Citation5]P2) for the activation and transportation of K+ in cells. KCNJ/Kir channels are widely distributed in the heart, brain, ventricular structures, and pancreas, located in cellular membrane net systems, and they stabilize resting membrane potential. KCNJ/Kir channels are important controllers of cell excitability, hormone release, and epithelial transport [Citation12]. SCNN1, having structural similarity with ASICs, are highly Na+-selective channels located on the PM of epithelial tissues.
Calcium channels
Ion channels, pumps, and organelles are required for the regulation of Ca2+ homeostasis. Generally, increased intracellular calcium is driven from two sources: influx from the extracellular space through the PM ion channels, and release from intracellular reservoirs, including the ER and lysosomes. PM Ca2+-permeable channels include most members of the TRP (transient receptor potential) family, Cav (voltage-gated Ca2+ channels), P2RX, and the calcium release-activated calcium channels, whereas candidate ER and lysosomal Ca2+-permeable channels include ITPR/InsP3R (inositol 1,4,5-trisphosphate receptor), RYR (ryanodine receptor), several members of the TRP family, and the TPCN/TPC (two pore segment channel) proteins [Citation13]. Conversely, ATP2B/PMCA (ATPase plasma membrane Ca2+ transporting)-mediated Ca2+ extrusion from the cytosol, ATP2A/SERCA (ATPase sarcoplasmic/endoplasmic reticulum Ca2+ transporting)-mediated calcium uptake from the cytosol to the sarcoplasmic/ER, and MCU (mitochondrial calcium uniporter)-mediated calcium uptake from the cytosol to mitochondria can reduce [Ca2+]i. It is likely that lysosomes uptake Ca2+ from ER. Although Ca2+/H+ exchanger (CAX) is required for lysosomal Ca2+ store filling in vertebrates [Citation14], no candidate channels or transporters have yet been identified in humans [Citation15] (). It was recently discovered that TMBIM6 (transmembrane BAX inhibitor motif containing 6/bax inhibitor 1)-mediated Ca2+ leak channels on ER [Citation16,Citation17] can enhance lysosomal Ca2+ levels through ER Ca2+ release [Citation18].
Figure 2. Calcium signaling in cells. Stimulation of the GPCR by specific antigens leads to the activation of PLC, the production of Ins(1,4,5)P3, and the release of Ca2+ from the ER to cytoplasm via ITPR channels. Ca2+ released by ITPR is also transferred into mitochondria through the VDAC and MCU at highly specialized membrane contact sites, termed MAMs, while SLC8B1 mediates mitochondrial Ca2+ extrusion. The source of Ca2+ uptake into endolysosomes is not clear, but the ER may contribute through ITPR. Other ER channels, including RYRs and several members of the nonselective TRPs, are involved in ER Ca2+ release, while candidate endolysosomal Ca2+ channels include TRPs and the TPCs. The decrease in Ca2+ levels within the ER lumen is sensed by low-affinity EF hands of STIMs. STIMs gain an extended conformation to trap and activate ORAI at the PM and induce SOCE. Other PM channels, including TRP channels, P2RX receptors, and Cav channels, are also involved in mediating Ca2+ signals. Cytoplasmic and ER Ca2+ homeostasis are mainly maintained by the actions of two transporters, including the ATP2B/PMCA that release Ca2+ from cytosol and the ATP2A/SERCA uptake of Ca2+ from cytosol. Abbreviations: ATP2A/SERCA, ATPase sarcoplasmic/endoplasmic Ca2+ transporting; ATP2B/PMCA, PM Ca2+ ATPase; Cav, voltage-activated Ca2+ channels; GPCR, G protein-coupled receptor; Ins(1,4,5)P3, inositol (1, 4, 5)-trisphosphate; ITPR, inositol 1,4,5-trisphosphate receptor; MCU, mitochondrial calcium uniporter; PLC, phospholipase C; RYRs, ryanodine receptors; SLC8B1, solute carrier family 8 member B1; STIM1, stromal interaction molecule 1; TPCs, two-pore channels; TRPs, transient receptor potential channels; VDACs, voltage-dependent anion channels.
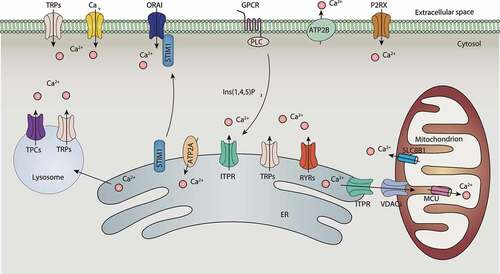
ORAI, ITPR/InsP3R, and RYR
The calcium release-activated calcium channels (CRACs), formed by ORAI1 (ORAI calcium release-activated calcium modulator 1), ORAI2 (ORAI calcium release-activated calcium modulator 2), and STIM1 (stromal interaction molecule 1) in the PM, are characterized by voltage independence, low conductance, and high Ca2+ selectivity. CRACs mediate the influx of Ca2+ from the extracellular space into the cell cytosol in response to the depletion of Ca2+ stored within the ER. This process is called store-operated calcium entry (SOCE) [Citation19]. In contrast, ITPR is an ER Ca2+ channel activated by inositol-(1,4,5)-trisphosphate (Ins [Citation1,Citation4,Citation5]P3), leading to the release of Ca2+ from the ER into the cytoplasm and mitochondria via the ER-mitochondria contact site. SOCE is typically activated by the engagement of G protein-coupled receptors or a tyrosine kinase cascade to activate PLC (phospholipase C) to cleave PtdIns(4,5)P2 and produce Ins(1,4,5)P3/InsP3. Ins(1,4,5)P3 further binds to ITPR, leading to the emptying of Ca2+ from the ER and the dissociation of ER lumenal Ca2+ from the EF-hand of STIM1 (a protein that homogeneously distributes in the ER). STIM1 subsequently forms oligomers and migrates to ER-PM junctions, which is followed by interaction with the proteins ORAI1 and ORAI2, leading to CRAC channel opening. Activated STIM1 can directly interact with voltage-operated calcium channels through its protein interaction domain, and strongly suppresses Ca2+ current and signaling [Citation20]. These findings indicate that STIM1 acts as a switch that selects ORAI1 and ORAI2 as Ca2+-activated signaling cascades rather than Cav, thereby activating different signaling pathways. Moreover, an ER membrane resident transmembrane protein known as SARAF/TMEM66 (store-operated calcium entry associated regulatory factor) is a binding protein of STIM1 and can be used as a negative regulator of SOCE to prevent excessive Ca2+ refilling [Citation21]. RYR proteins are Ca2+-release channels and are mainly expressed in the sarcoplasmic reticulum (SR) and ER in excitable cells, such as muscle cells and neurons. RYRs are also localized on mitochondria and regulate mitochondrial Ca2+ concentration by directly transporting Ca2+ to mitochondria.
TRPs and TPCs
In humans, TRP channels form a large superfamily, which contains six families (including TRPC [transient receptor potential cation channel subfamily C], TRPM [transient receptor potential cation channel subfamily M], TRPV [transient receptor potential cation channel subfamily V], TRPA1 [transient receptor potential cation channel subfamily A member 1], PKD/TRPP [polycystin, transient receptor potential cation channel], and MCOLN/TRPML [mucolipin]) based on amino acid homologies [Citation22]. These channels consist of six transmembrane segments with relatively nonselective permeability to cations, including Na+, K+, Ca2+, and Mg2+. Except for TRPM4 and TRPM5, most TRPs are permeable to Ca2+, and are located on the PM and intracellular membranes of various cell types. In addition to voltage, TRPs can also be gated by second messengers (e.g., cyclic ADP ribose, NAADP, and PtdIns [Citation4,Citation5]P2/PIP2), neurotransmitters, temperature, and mechanical force, making them suitable for function as environmental sensors. TPCs contain two members (TPCN1 [two pore segment channel 1] and TPCN2 [two pore segment channel 2]) in humans and are widely expressed in most human tissues, and located in endosomes and lysosomes. These channels are Na+- or H+-selective, whereas TPCN2 also conducts Ca2+ in sensing NAADP and releases Ca2+ from lysosomes. Through lysosomal localization, MCOLN1 and TPCN2 mediate lysosomal Ca2+ efflux.
Voltage-gated ion channels
In this review, we divide voltage-gated channels into two groups: the classical voltage-gated ion channels with a canonical voltage-sensing domain (VSD), and the nonclassical voltage-gated ion channels lacking a VSD.
Voltage-gated ion channels
Voltage-gated ion channels, including voltage-gated Na+ channels (Nav), voltage-gated K+ channels (Kv), Cav, and voltage-gated proton channels (Hv), are specific to Na+, K+, Ca2+, and H+, respectively [Citation23]. These channels are classic voltage-gated ion channels. Nav and Cav share similar functions in producing action potentials through mediating ion influx. Kv contributes to membrane repolarization by mediating ion efflux, and Hv balances the intracellular pH. Cav is involved in various cellular processes due to the regulation of [Ca2+]i. According to the activation threshold, Cav can be further divided into two subgroups: high-voltage-activated (including L-, P/Q-, N-, and R-types) and low-voltage-activated (including T-type) channels. Of note, the NALCN (sodium leak channel, nonselective) is structurally a member of the VGCCs family and is a voltage-insensitive channel that contains a nonselective pore domain. This domain allows NALCN to conduct Na+, Ca2+, K+, and Cs+, resulting in a current leak.
The CLCN/ClC family and KCNK/K2P
The CLCN/ClC (chloride voltage-gated channel) family and KCNK/K2P (potassium two pore domain channel subfamily K) are nonclassical voltage-gated ion channels that lack a VSD. CLCN family channels are voltage-gated Cl− channels expressed in brain, kidney, muscle, and other tissues. The CLCN family contains nine members and has the functions of maintaining resting membrane potential and ion homeostasis, mediating transepithelial transport, and regulating cell volume [Citation24]. The KCNK are voltage-activated K+ channels, expressed in many types of cells. KCNK-mediated background conductance not only stabilizes the resting membrane potential, but also contributes to repolarization [Citation25].
Other channels
VRACs and VDACs
The volume-regulated anion channels are composed of various combinations of LRRC8 (leucine rich repeat containing 8) proteins. These channels initiate passive efflux for Cl−, glutamate, and other neurotransmitters in response to cell swelling. They are ubiquitously expressed and play key roles in the regulation of cell volume, cell proliferation, and cell death [Citation26]. VRACs also mediate the efflux and influx of cyclic GMP-AMP (cGAMP, a small molecule ligand) in cells thereby expanding its role in drug uptake. VDACs/porins (voltage dependent anion channels) are ion channels located in the mitochondrial outer membrane. VDACs are permeable to small ions (e.g., Cl−, K+, Na+, and Ca2+), large anions (e.g., glutamate and ATP), and large cations (e.g., acetylcholine and dopamine), and produce a variety of cell survival and cell death signals [Citation27]. There are three isoforms of VDAC (VDAC1, VDAC2, and VDAC3) in mammalian cells, and the main type in cells is VDAC1, whose function is regulated by various ligands and binding proteins.
Gap junction channels
Gap junction channels (including GJ [gap junction protein]/connexins and PANXs [pannexins]) are a family of large-pore channel-forming proteins that allow the transport of ions, metabolites, small molecules, and second messengers (e.g., cGAMP) between adjacent cells in the PM without exposure to the extracellular environment [Citation28]. GJ proteins act as both classical gap junctions and hemichannels on cytoplasmic membranes, whereas PANXs mainly function on the cell surface.
TMEM175
TMEM175 (transmembrane protein 175) is a novel K+-selective ion channel expressed in both endosomes and lysosomes that regulates lysosomal pH stability, membrane potential, and organelle fusion [Citation29]. The pores of TMEM175-dependent channels can be blocked by zinc ion and 4-aminopyridine.
Ion transporters
Ion transporters in eukaryotes can be divided into two major subgroups: primary active transporters (containing ABC [ATP binding cassette] transporters, vacuolar-type H+-translocating ATPase [V-ATPase], and P-type ATPase [P-ATPase]), and secondary active transporters (e.g., SLC [solute carrier] family proteins) are typically driven by the Na+ gradients established by Na/K ATPases [Citation30].
Primary ion transporters
ABC transporters are classified into seven subfamilies labeled ABCA to ABCG, of which the ABCC transporters are the only ion transporters in the ABC transporter family. Most of the ABCCs export large hydrophobic anions, whereas CFTR (discussed above, also belongs to the ABCC subfamily) is only permeable to Cl−. The V-ATPase is an ATP-driven H+ pump that transports H+ across intracellular membranes and the PM in eukaryotic cells and is responsible for acidifying and maintaining the pH of intracellular organelles. Consequently, the V-ATPase plays multiple roles in various fundamental cellular processes, such as the targeting and posttranslational modification of proteins in the GA, the degradation of biological macromolecules in lysosomes, and the uptake and release of neurotransmitters in synaptic vesicles. Additionally, the V-ATPase has emerged as a critical regulator for MTOR, NOTCH, and WNT signaling [Citation31].
The ion transporters in P-ATPases can be classified as four types, namely Na+/K+ pumps, H+/K+ pump, Ca2+ pumps, and Cu2+ pumps. Na+/K+ pumps are ubiquitously expressed on the PM. For each ATP used, the Na+/K+ pumps transport three Na+ out of cells and two K+ into cells. This function is important for maintaining electrochemical gradients, leading to a resting membrane potential of cells and regulating cell volume. P-ATPases can also serve as scaffolding proteins in the signal transduction of a signal protein, such as PRKC/PKC (protein kinase C) and phosphoinositide 3-kinases (PI3Ks). Ca2+ pumps are distributed in all cells, comprising three members, namely ATP2A/SERCA, ATP2C/SPCA (ATPase secretory pathway Ca2+ transporting), and ATP2B/PMCA [Citation15]. ATP2A/SERCA is not only the main intracellular membrane-associated transporter for the uptake of Ca2+ from the cytosol into intracellular organelles, but also the only Ca2+ transporter on the SR/ER surface, and is usually related to the recovery phase after excitation. ATP2C/SPCA pumps Ca2+ and Mn2+ from the cytosol into the lumen of the GA, whereas ATP2B/PMCA is a cell surface pump for the efflux of Ca2+ from the cytosol [Citation32]. The H+/K+ pump acts as the H+ pump of the stomach by exchanging cytoplasmic H+ for K+ from the intestinal lumen, and its role is to acidify the stomach. In addition, Cu2+ pumps are involved in the efflux of Cu2+ and other heavy metals across the PM and cytoplasmic membranes.
Secondary ion transporters
To date, secondary ion transporters are known to include more than 400 transmembrane SLC (solute carrier) transporters in 65 families, which are localized on the cell surface or organelle membranes. The SLC family can transport a wide range of solutes, from simple inorganic ions to amino acids and sugars, to relatively complex organic molecules. There are two forms of SLC transporters: co-transporters (also known as symporters) and exchangers (also known as antiporters). In co-transporters, the direction of transport is the same for both the driving ion and driven ion/molecule. In contrast, in exchangers, the driving ion and driven ion/molecule move in opposite directions.
The role of ion channels and transporters in autophagy
Ion channels and transporters can induce or inhibit autophagy by controlling cytosolic ion concentration, membrane potential, or other signaling pathways, as described below.
Regulation of the MTORC1 pathway during initiation
Growth factors and the AKT-MTOR pathway
Growth factors induce autophagy by inhibiting the PI3K-AKT-TSC pathway and MTORC1 activity. The influx of Ca2+ from the extracellular space may increase [Ca2+]i and then modulate the AKT-MTOR pathway. For example, high-fat diet treatment increases [Ca2+]i and attenuates insulin signaling in HepG2 cells [Citation33]. This increased Ca2+ leads to the formation of Ca2+-PtdIns4P/PIP, thereby eliminating the interaction between the AKT-PH domain and PtdIns4P, and ultimately inhibiting AKT phosphorylation. The TRPV2 and TRPV4 channels also promote autophagy by inhibiting AKT via generating Ca2+ signals [Citation34,Citation35].
ER-to-cytosol Ca2+ efflux acts as a negative signal for the growth factor-MTORC1 pathway, thus inducing autophagy. RYR3 (ryanodine receptor 3)-mediated ER-to-cytosol Ca2+ efflux is increased following insulin withdrawal in hippocampal neural stem cells [Citation36]. The RYR antagonist dantrolene partially reduces autophagy and the expression of CALM (calmodulin) in the brain of neuronopathic Gaucher disease mice [Citation37]. The deletion of Fkbp1b (a molecular chaperone of RYR2 [ryanodine receptor 2]) leads to RYR2 channel leakage, activation of the AKT-MTOR pathway, and inhibition of autophagy in cardiomyocytes [Citation38]. ITPR-mediated Ca2+ release in the ER also induces autophagy in an MTOR-dependent manner in HeLa cells [Citation39,Citation40], nerve cells [Citation41] and cancer cells [Citation42].
Other regulators also affect Ca2+-related AKT activation in a context-dependent manner. KCl depolarization causes autophagy through the ionotropic glutamate receptor GRIN/NMDAR in hippocampal neurons [Citation43]. Interestingly, the blocking of GRIN leads to the destruction of autophagy flux and the accumulation of autophagosomes in vivo [Citation44], suggesting that GRIN may also contribute to the fusion stage of autophagy. In contrast, CRAC channels may be negative regulators of autophagy by activating the AKT-MTORC1 pathway in cancer cells [Citation45].
In addition to Ca2+ channels, two Cl− channels (CFTR and CLCN3 [chloride voltage-gated channel 3]) and one K+ channel, KCNH2/Kv11.1, regulate autophagy through the AKT-MTORC1 pathway. The overexpression of Cl− channel CFTR inhibits LPS-induced autophagy in lung cells via the activation of the AKT-MTORC1 pathway [Citation46], whereas defective CLCN3 channels induce autophagy in U251 or CNE-2Z cells by inhibiting AKT [Citation47,Citation48], indicating that chloride currents may function as a positive signal for the activation of the AKT-MTORC1 pathway. Pharmacological blockage of the potassium channel KCNH2 by macrolide antibiotics triggers autophagy by inhibiting AKT in acute leukemia cells [Citation49].
Energy stress and the MTOR pathway
The ER-mitochondria Ca2+ transfer comprises calcium release from the ER through ITPR and RYRs, across the outer mitochondrial membrane through VDACs, and across the inner mitochondrial membrane through the MCU. This process controls mitochondrial uptake of Ca2+ from the ER at ER-mitochondria contact sites, regulating mitochondrial fission, fusion, and function, as well as autophagy ().
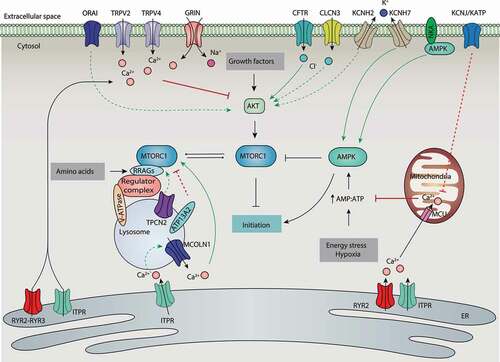
Figure 3. Regulation of MTORC1 activation by ion channels and ion transporters during autophagy initiation. Signals that activate the autophagic process (initiation) typically originate from various conditions of stress, such as growth factor stress, amino acid stress, energy stress, and hypoxia. The common target of these signaling pathways is MTORC1. Ion channels and ion transporters are involved in the regulation of several pathways upstream of MTORC1, including the AKT-dependent or AMPK-dependent pathways. Ion channels and transporters can also directly target MTORC1 activity at lysosomes. Abbreviations: AMPK, AMP-activated protein kinase; ATP13A2, ATPase cation transporting 13A2; CFTR, CF transmembrane conductance regulator; CLCN3, chloride voltage-gated channel 3; GRIN/NMDAR, glutamate ionotropic receptor NMDA type; ITPR, inositol 1,4,5-trisphosphate receptor; KCNJ/KATP, potassium inwardly rectifying channel subfamily J; Kv, voltage-gated K+ channels; NKA, sodium/potassium transporting ATPase; MCOLN1, mucolipin 1; MCU, mitochondrial calcium uniporter; MTORC1, MTOR complex 1; RRAGs/Rag GTPases, Ras related GTP binding; RYR2, ryanodine receptor 2; RYR3, ryanodine receptor 3; TPCN2, two pore segment channel 2; TRPV, transient receptor potential cation channel subfamily V; V-ATPase, vacuolar-type H+-translocating ATPase.
Ca2+ channels play a role as switches in the regulation of AMPK-dependent autophagy. Constitutive ITPR and MCU Ca2+ signaling are required for autophagy suppression in chicken DT40 B lymphocytes in normal media [Citation50], whereas the absence of ITPR or pharmacological inhibition of MCU results in a decrease of mitochondrial Ca2+ uptake and ATP production and a subsequent activation of AMPK and autophagy. MCUR1 (mitochondrial calcium uniporter regulator 1), an integral membrane protein that binds to MCU, contributes to MCU-dependent mitochondrial Ca2+ uptake during autophagy regulation [Citation51]. MCUR1 deficiency impairs ATP production and activates AMPK-dependent autophagy in various cells. The overexpression of MCU only partially reduces neuronal degeneration-induced autophagy in SH-SY5Y cells [Citation52], suggesting that other ion channels and transporters (e.g., RYR2) also play a role in mitochondrial Ca2+ uptake. The loss of RYR2 results in a reduction in mitochondrial Ca2+ and ATP production, thereby activating AMPK-dependent autophagy [Citation53].
Hypoxia and the MTOR pathway
During hypoxia, mitochondrial respiration is impaired, resulting in a low ATP:AMP ratio that activates AMPK and inhibits MTORC1 [Citation54] (). Hypoxia inhibits MTORC1 not only through the activation of AMPK, but also through the activation of TSC mediated by the induction of DDIT4 (DNA damage inducible transcript 4) [Citation55]. The plasma membrane KCNJ/KATP channel is activated to allow K+ influx into the cytoplasm during ATP-depleted metabolic states, such as hypoxia [Citation56]. The activation of KCNJ/KATP by epoxyeicosatrienoic acids protects against mitochondrial dysfunction and cellular death, in part by activating AMPK-dependent autophagy in starved cardiomyocytes [Citation57]. The KCNJ/KATP channel opener minoxidil can induce autophagy in PC12 cells, whereas blockers (e.g., quinine sulfate and tolazamide) slow the clearance of autophagy substrates [Citation58]. Based on how KCNJ/KATP activation reduces cytosolic Ca2+ overload [Citation59], KCNJ/KATP may protect mitochondrial function and maintain AMPK activity by indirectly reducing mitochondrial Ca2+ concentration.
In addition, a Na+/K+ pump, NKA (sodium/potassium transporting ATPase), interacts with and mediates the activation of AMPK, resulting in autophagy to protect against ischemic damage in N2a cells [Citation60]. However, NKA is dissociated from AMPK under hypoxia, indicating a feedback mechanism that controls autophagy activity. Furthermore, the pharmacological activation of KCNH7/Kv11.3 (using NS1643) mediates K+ efflux from the cytosol, and thereby induces AMPK-dependent autophagy in a melanoma cell line [Citation61]. Given that potassium currents result in the reduction of membrane depolarization and the induction of calcium influx [Citation62], it appears that potassium channels and transporters can regulate the activation of AMPK and subsequent autophagy by controlling the concentration of calcium.
Amino acid stress and the MTOR pathway
Unlike the other stimuli, MTORC1 sensing of amino acids from lysosomes is an “inside-out” mechanism that depends on the V‑ATPase [Citation63]. Upon amino acid signaling, MTORC1 translocates to the lysosomal membrane and interacts with the Ragulator complex, which serves as a scaffold for the V-ATPase and RRAG (Ras related GTP binding) proteins. This location change finally leads to RRAG-dependent MTOR activation in lysosomes. In contrast, the lysosome-localized amino acid transporter SLC36A1/PAT1 (solute carrier family 36 member 1), which exports amino acids out of the lysosome, inhibits MTORC1 activation.
Lysosomal pH, controlled by the V-ATPase, plays a significant role in the regulation of MTORC1 activity in osteoclasts [Citation64], linking the V-ATPase transporter activity and lysosomal pH to MTORC1 activation (). For example, ubiquinol regulates V-ATPase activity and lysosomal acidification by interacting with the V-ATPase, thereby increasing MTOR signaling in Drosophila and human neuronal cells [Citation65]. Cysteine 277 in the ATP6V1A (ATPase H+ transporting V1 subunit A) subunit of the lysosomal V-ATPase is crucial for RRAG activity, thereby regulating MTORC1 activity [Citation66]. Interestingly, follicular lymphoma-associated mutations in ATP6V1B2 (ATPase H+ transporting V1 subunit B2) activate MTOR and autophagic flux in lymphoma cell lines [Citation67]. These findings indicate that the redox and mutation status of V-ATPase affects autophagic activity.
In addition to V-ATPase, other lysosomal proteins also regulate MTORC1 activity and autophagy induction by controlling lysosomal function [Citation68] (). For example, mutations in a lysosomal P-ATPase, ATP13A2 (ATPase cation transporting 13A2), cause Parkinsonian syndrome or Kufor-Rakeb syndrome. The knockdown of ATP13A2 impairs autophagy induction in HeLa cells and neuronal cells by increasing the phosphorylation of MTORC1 [Citation69,Citation70], suggesting that ATP13A2 is a negative regulator of MTORC1. The physical interaction between MTOR and TPCN2 is further observed in skeletal muscle [Citation71]. In TPCN2 KO myotubes, the phosphorylation of MTOR is impaired in the absence of nutrients, indicating that TPCN2 is a repressor of autophagy by activating MTOR, while whether this process depends on the channel function of TPCN2 remains unknown.
Regulation of lysosomal MTOR activity
MCOLN1/TRPML1 (mucolipin 1) is a calcium-release channel on the membranes of late endosomes and lysosomes that affects MTORC1 activity. The knockdown of MCOLN1 prevents MTORC1 activity in HEK293T cells [Citation72], and thapsigargin can reverse this effect. Further evidence shows that ITPR-dependent Ca2+ signals maintain elevated MTORC1 activity and inhibit basal autophagy or autophagy caused by starvation [Citation42,Citation73]. Based on the further fact that a) the activity of lysosomal MTORC1 relies on lysosome function, which is tightly controlled by lysosomal Ca2+ release [Citation74], and b) ITPR is responsible for Ca2+ refilling of lysosomes from the ER [Citation75], we speculate that the storage and release of lysosomal Ca2+ in these channels regulate the phosphorylation of lysosomal MTORC1 and subsequent autophagy ().
Activation of the CAMKK2-AMPK pathway by calcium signaling during initiation
Increased [Ca2+]i activates CAMKK2 (calcium/calmodulin dependent protein kinase kinase 2), leading to the activation of AMPK [Citation76]. Thus, ion channels and transporters may control autophagy by regulation of the CAMKK2-AMPK pathway as discussed below.
The MTOR-dependent pathway
Two members in TRPC (TRPC4 and TRPC5) are positive regulators of autophagy in vascular endothelial cells [Citation77], glioblastoma cells [Citation78], or breast cancer cells [Citation79] by activating the Ca2+-CAMKK2-AMPK pathway, leading to MTOR inhibition (). CRAC is also involved in oxidized low-density lipoprotein-induced autophagy activation in bone marrow-derived endothelial progenitor cells [Citation80], partly through the activation of the CAMKK2-AMPK-MTOR pathway. The inhibition of ASIC1 (using drugs [e.g., PcTX1 and amiloride] or RNAi) blocks Ca2+-dependent activation of the CAMKK2-AMPK pathway, leading to autophagy resistance in chondrocytes and HSC-T6 cells in an acidic or high-glucose environment [Citation81]. However, pharmacological inhibition of ASICs enhances the autophagic degradation in rat pheochromocytoma PC12 cells [Citation82], indicating an anti-autophagy role of ASICs in some cases.
Figure 4. Regulation of the CAMKK2-AMPK pathway by ion channels and ion transporters during autophagy initiation. Ca2+ signaling activates CAMKK2, which further phosphorylates AMPK. The activated AMPK initiates autophagy by MTORC1-dependent or -independent pathways. Ion channels and ion transporters induce cytoplasmic Ca2+ signaling and activate the CAMKK2-AMPK pathway by mediating Ca2+ influx from the extracellular space or Ca2+ release from intracellular storage, thereby inducing AMPK-dependent autophagy. Abbreviations: AMPK, AMP-activated protein kinase; ASIC1, acid sensing ion channel subunit 1; ATP2A/SERCA, ATPase sarcoplasmic/endoplasmic reticulum Ca2+ transporting; CAMKK2, calcium/calmodulin dependent protein kinase kinase 2; GABAAR, γ-aminobutyric acid type A receptor; MCOLN1, mucolipin 1; MTORC1, MTOR complex 1; P2RX7, purinergic receptor P2X7; PtdIns3K, phosphatidylinositol 3-kinase; TPCN2, two pore segment channel 2; TRPC, transient receptor potential cation channel subfamily C; TRPM, transient receptor potential cation channel subfamily M; ULK1, unc-51 like autophagy activating kinase 1.
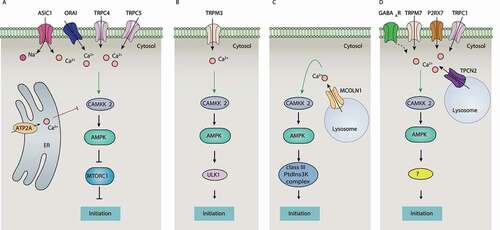
ATP2A is the main calcium transporter of intracellular organelles controlling intercellular Ca2+ concentration by the uptake of Ca2+ from the cytosol. In many cases, blocking ATP2A increases free cytosolic calcium and promotes autophagy induction via the activation of the CAMKK2-AMPK-MTOR pathway in various cell types (Table S1), suggesting that ATP2A-mediated cytosolic Ca2+ accumulation prevents autophagy induction ().
The MTOR-independent pathway
Two Ca2+-permeable channels (TRPM3 and MCOLN1) induce basal autophagy that bypasses MTOR by the activation of the CAMKK2-AMPK pathway (). Increased expression of TRPM3 in clear cell renal cell carcinoma leads to Ca2+ influx and activation of the CAMKK2-AMPK-ULK1 pathway [Citation83], thereby inducing the formation of phagophores. Similarly, MCOLN1 activates CAMKK2 and AMPK in ARPE-19 cells, leading to the formation of phagophores through the production of PtdIns3P and the activation of the class III PtdIns3K complex [Citation84,Citation85] ().
Other channels
Other ion channels and transporters related to Ca2+ signaling, such as TRPM7, TRPC1, P2RX7, and NAADP have also been reported as early autophagy promoters by activating Ca2+-CaMKK-AMPK pathway () [Citation86–88]. LRRK2 (leucine rich repeat kinase 2), whose mutation causes an autosomal dominant form of Parkinson disease, activates autophagy through a Ca2+-dependent pathway in HEK293T cells [Citation89]. This LRRK2-mediated effect can be prevented by using the NAADP antagonist NED-19 or TPCN2-negative mutants, indicating the presence of an NAADP-TPC-CAMKK2-AMPK axis in autophagy induction. The chloride channel GABAAR also participates in the induction of autophagy by activating the Ca2+-CAMKK2-AMPK pathway (). Exogenous GABA increases [Ca2+]i and activates AMPK-dependent autophagy in macrophages [Citation90].
Regulation of ROS-induced autophagy during initiation
Cytosolic Ca2+ signaling and ROS production
Given that calcium signaling contributes to ROS production in many conditions [Citation91], ion channels and transporters could also regulate autophagy by affecting ROS production. In thymocytes, the activation of TRPV1 triggers AMPK-dependent autophagy, which is associated with elevated ROS and increased [Ca2+]i [Citation92]. ROS stimulates AMPK activity via oxidative modification of its PRKAA subunit. Similarly, the activation of P2RX7 by the agonist BzATP triggers both ROS- and Ca2+-dependent AMPK activation, leading to autophagy in microglia and BMDMs [Citation93], whereas the P2RX7 antagonist A438079 eliminates these effects. In addition, the pharmacological inhibition of KCNJ2/Kir2.1 channels (using ML133) elevates [Ca2+]i, which leads to increased ROS and subsequent autophagy in endothelial progenitor cells [Citation94]. Together, these data suggest that TRPV1, P2RX7, and KCNJ2 on the PM may induce ROS-related autophagy in ways that are Ca2+ influx-dependent ().
Figure 5. Regulation of ROS-induced autophagy by ion channels and ion transporters. Under various environmental stimuli, ROS are produced and stimulate autophagy through various signaling pathways. Ion channels and transporters not only regulate the production of ROS, but also control the activity of AMPK in a manner dependent on Ca2+ signals. Abbreviations: AMPK, AMP-activated protein kinase; KCNJ/KATP, ATP-sensitive K+ channel; KCNJ/Kir, potassium inwardly rectifying channel; P2RX7, purinergic receptor P2X 7; ROS, reactive oxygen species; TRPC, transient receptor potential cation channel subfamily C; TRPV, transient receptor potential cation channel subfamily V; V-ATPase, vacuolar-type H+-ATPase.
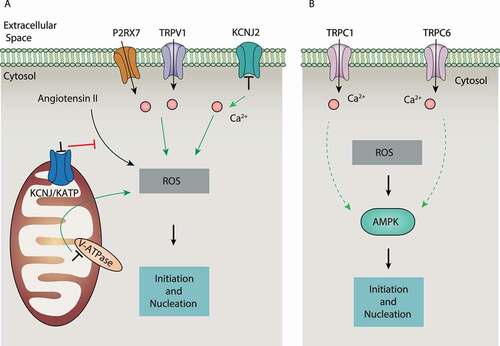
Mitochondrial ion homeostasis and ROS production
H+ pumps on mitochondria establish an H+ gradient across the inner mitochondrial membrane, which is crucial for Ca2+ flux across mitochondria and ROS production. The proton pump inhibitor esomeprazole, when targeting V-ATPase, can induce ROS and autophagy-dependent cell death in human melanoma cells by causing mitochondrial dysfunction and the activation of NADPH oxidase [Citation95], suggesting that acidic pH may accelerate ROS-dependent autophagy. Furthermore, the mitochondrial KCNJ/KATP channel inhibitor 5-hydroxydecanoate abolishes angiotensin II–induced autophagy caused by ROS production in vascular smooth muscle cells [Citation96]. These findings indicate that multiple mitochondrial channels regulate ROS production during autophagy ().
In addition to nonselective autophagy, alterations in mitochondrial Ca2+ signaling can lead to mitophagy. The mitochondria Ca2+ channel VDAC1 interacts with ITPR at ER-mitochondria contact sites, thereby potentiating the transfer of Ca2+ signals to mitochondria [Citation97]. VDAC1 also recruits PRKN (parkin RBR E3 ubiquitin protein ligase) to damaged mitochondria and promotes mitophagy [Citation98], although how Ca2+ and ROS signals synergistically affect VDAC1-dependent mitochondria remains unclear.
The ROS-AMPK pathway
ROS-mediated Ca2+ also triggers autophagy under hypoxia [Citation99]. This process can be controlled by TRPC6, a nonselective Ca2+-permeable cation channel widely expressed in the PM of kidney cells [Citation100]. For example, hypoxia and zinc oxide nanoparticles (nano-ZnO) induce TRPC6 expression by producing ROS, which finally leads to autophagy through increased Ca2+ influx in SH-SY5Y neuroblastoma cells [Citation101] or podocytes [Citation102]. Blocking TRPC6 (using SKF-96,365 or siRNA) decreases, whereas enhancement of TRPC6 activity (using flufenamic acid) increases, AMPK-dependent autophagy during hypoxia [Citation103]. Thus, TRPC6 plays an important role in linking ROS, Ca2+ influx, and AMPK during autophagy activation (). Unlike TRPC6, TRPC1-mediated Ca2+ influx may play a dual role autophagy depending on cell types [Citation104,Citation105].
ROS and TRPM2
TRPM2 serves as a Ca2+ permeable channel not only on the PM but also on lysosomes, activated by intracellular messengers, such as ADP-ribose (ADPR), nicotinamide adenine dinucleotide (NAD+) and cyclic ADPR (cADPR) [Citation106,Citation107]. trpm2−/- mice show reduced hippocampal autophagosome formation and protein aggregation, accompanied by learning and memory disorders [Citation108]. TRPM2 plays a crucial role in nano-ZnO- and EGF (epidermal growth factor)-activated ROS-induced autophagy in human perivascular cells [Citation109] and human corneal epithelial cells [Citation110], indicating a positive role of TRPM2 in ROS-induced autophagy. However, TRPM2 also has the ability to inhibit H2O2-induced autophagy through a Ca2+-dependent feedback mechanism [Citation111,Citation112], suggesting that targeting TRPM2 in autophagy is context-sensitive.
ROS and VRAC
The Cl− channel VRAC is a negative regulator of ROS-induced autophagy in glucose-exposed cardiomyocytes [Citation113], although VRAC also plays a pro-autophagic role in mediating the oxidative death of myocardial hypoxia-reoxygenation injury [Citation114]. V-ATPase-mediated pH formation regulates the expression and activity of VRAC during oxidative stress, thereby affecting autophagy induction in different cells [Citation115].
Regulation of the class III PtdIns3K complex during nucleation
Ion channels and transporters can control the nucleation of autophagy by modulating the activity of class III PtdIns3K complexes via BECN1. In particular, ITPR directly affects the activity of the class III PtdIns3K complex by binding to BECN1 (). The inhibition of ITPR induces autophagy independent of MTOR and Ca2+ signaling [Citation116]. In contrast, xestospongin B (an antagonist)-induced and nutrient starvation-induced autophagy are related to the disruption of the interaction between ITPR and BECN1 in HeLa cells [Citation117], whereas the autophagy repressor BCL2 may bridge the interaction of ITPR and BECN1. Interestingly, during starvation, BECN1 may enhance ITPR activity in Ca2+ release independent of BCL2, although ITPR impairs BECN1 activity when forming a BECN1-ITPR complex [Citation42]. The dynamic changes of the BECN1-ITPR complex may form a complex feedback mechanism to control autophagy induction as well as cell death. The formation of this complex depends on the ligand-binding domain of ITPR, and whether this process depends on the channel activity of ITPR is unclear. In addition, the overexpression of PKD2 (polycystin 2, transient receptor potential cation channel), a binding protein of ITPR [Citation118], induces autophagy by interacting with BECN1 through its channel activity [Citation119].
Figure 6. Regulation of BECN1 activity by ion channels and ion transporters. The phosphorylation of BECN1 is crucial for activating the class III PtdIns3K complex, leading it to produce PtdIns3P for phagophore nucleation. The activation of BECN1 is negatively regulated by BCL2 through protein-protein interaction. Ion channels and ion transporters play a dual role in the regulation of BECN1 in three ways: A) inhibition of BECN1 activity through enhancing the interplay between BECN1 and BCL2; B) inhibition of BECN1 activity through the CAMK2 pathway; and C) direct or indirect promotion of BECN1 activity. Abbreviations: BCL2, BCL2 apoptosis regulator; BECN1, beclin 1; CAMK2, calcium/calmodulin-dependent protein kinase II; CFTR, CF transmembrane conductance regulator; ITPR, inositol 1,4,5-trisphosphate receptor; NKA, sodium/potassium transporting ATPase; PIK3C3, phosphatidylinositol 3-kinase catalytic subunit type 3; PKD2, polycystin 2, transient receptor potential cation channel; TGM2, transglutaminase 2; TRPM, transient receptor potential cation channel subfamily M; VDAC2, voltage dependent anion channel 2.
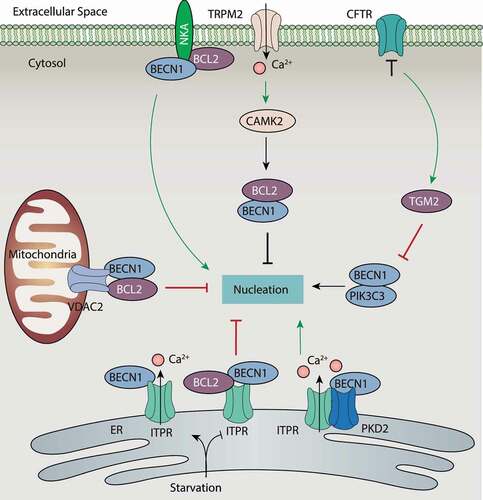
VDAC2-mediated autophagy suppression also depends on interactions with BECN1 or BCL2 to stabilize the BECN1-BCL2 complex [Citation120]. NKA interacts with BECN1 during autophagy caused by hypoxia, starvation, and other stresses, resulting in a type of autophagy-dependent cell death (namely autosis) [Citation121–123]. NKA can also bind BCL2 or ITPR in the ER (), indicating an NKA network integrated in the modulation of autophagy-dependent cell death. TGM2 (transglutaminase 2) is a bridging molecule at ER-mitochondria contact sites by assembling the ITPR-VDAC complex. The loss of the Cl− channel CFTR leads to ROS-mediated SUMOylation of TGM2, which inhibits autophagy induction in airway epithelial cells by binding and inhibiting BECN1 activity in the class III PtdIns3K complex [Citation124] (). Thus, the TGM2-CFTR pathway plays multiple roles in the regulation of BECN1 activity and expression, ultimately affecting autophagy induction. Whether the interaction with BECN1-BCL2 complex involves channel activity remains to be further evaluated.
The activity of the class III PtdIns3K complex is further directly regulated by Ca2+ sensors or channels. TRPM2-mediated Ca2+ influx and activation of CAMK2 (calcium/calmodulin-dependent protein kinase II) is required for autophagy inhibition by promoting BCL2 interaction with BECN1 and diminishing BECN1-mediated PtdIns3K complex activation [Citation112] (). In contrast, the functional deficiency of ATP2A promotes the binding of CALM to the PtdIns3K complex, resulting in increased contact between the ER and phagophores for the induction of autophagy [Citation125]. However, this process is independent of Ca2+ signaling. Thus, Ca2+ signaling plays a context-dependent role in the regulation of PtdIns3K complex activation.
Other Ca2+-independent channels and transporters also participate in the regulation of class III PtdIns3K complex activation through an integrated mechanism. For example, the gap junction channel GJA1/connexin 43, which is located on the PM, regulates PtdIns3K complex formation by acting as a scaffold protein, but this effect is not related to its channel function [Citation126].
Regulation of autophagy-related genes during expansion
Regulation of MAP1LC3 lipidation and cargo carriers
The VGCC family, responsible for extracellular Ca2+ influx, has been identified as a direct regulator of phagopore expansion. Among the family, L-type Cav not only transmits inward Ca2+ current, but also triggers Ca2+ release from the SR/ER by CAPN (calpain)-mediated cAMP production and subsequent Ins(1,4,5)P3/InsP3-mediated Ca2+ release, creating a Ca2+-positive feedback loop [Citation58,Citation127] (). Constitutively active CAPN can cleave ATG5 in unknown ways, thereby inhibiting autophagy [Citation128]. L-type Cav antagonist (e.g., verapamil, loperamide, and nimodipine)-induced autophagy depends on the activation of the Ca2+-CAPN pathway in neuroblastoma cells, whereas L-type Cav agonists (e.g., bay-K8644) have the opposite effect. In contrast, fluspirilene (an antagonist of P/Q-type Cav) induces autophagy in H4 cells by inhibiting channel activity or blocking CAPN-mediated ATG5 cleavage [Citation127]. Other VGCC blockers also have a similar effect on autophagy in different models (Table S1). These data indicate that the excessive activation of calcium channels (e.g., VGCC) may inhibit the nucleation of autophagy through promoting CAPN-mediated ATG5 cleavage in a Ca2+-dependent manner.
Figure 7. Regulation of autophagy by ion channels and ion transporters. Ion channels and transporters directly regulate ATG16L1 activity and indirectly regulate ATG5 activity. In addition, they induce transcript expression of ATG genes mainly through TFEB and miRNA. Abbreviations: AMPK, 5ʹ-AMP-activated protein kinase; CAPN, calpain; CaV, voltage-activated Ca2+ channel; CHRNA7/α7nAChR, cholinergic receptor nicotinic alpha 7 subunit; ITPR, inositol 1,4,5-trisphosphate receptor; MCOLN1, mucolipin 1; P2RX7, purinergic receptor P2X 7; PPP3, calcineurin; STIM1, stromal interaction molecule 1; TFEB, transcription factor EB; TPCN1, two pore segment channel 1; TRPM, transient receptor potential cation channel subfamily M; V-ATPase, vacuolar-type H+-ATPase.
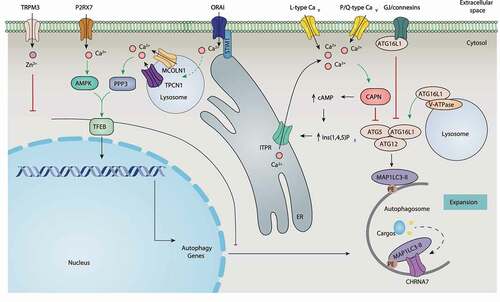
Ion channels and transporters can also regulate autophagy by interacting with ATG16L1 or MAP1LC3. Gap junction channels, including GJA1, GJA3, and GJB1/connexin 32, bind and inhibit ATG16L1 independent of their channel functions, thereby inhibiting basal autophagy in mouse embryonic fibroblasts (MEFs) [Citation126]. In contrast, V-ATPase-mediated recruitment of ATG16L1 initiates xenophagy after bacteria-induced vacuolar membrane damage in HeLa cells [Citation129]. The CHRNA7/α7nAChR (cholinergic receptor nicotinic alpha 7 subunit) can act as a cargo carrier to bind MAP1LC3 to activate autophagosome formation, thereby providing protection against various stresses [Citation130]. Overall, these data indicate that ion channels and transporters can target ATG family members to control phagophore expansion ().
Transcriptional regulation of autophagy-related genes
TFEB (transcription factor EB) is a nuclear regulator of autophagy by controlling the expression of ATG and other genes involved in autophagy [Citation131]. Lysosomal calcium is an important signal to activate TFEB-dependent autophagy. During starvation, MCOLN1-mediated lysosomal Ca2+ release activates PPP3/calcineurin, which binds and activates TFEB, leading to autophagy [Citation132]. Lysosomal damage leads to MAP1LC3 to interact with MCOLN1, facilitating Ca2+ efflux and TFEB activation [Citation133]. Oxidative stress further enhances this process by directly and specifically activating the MCOLN1-TFEB pathway, indicating the interaction of ROS and Ca2+ in the activation of TFEB-related autophagy [Citation134]. The Ca2+ channel-like protein TMBIM6 stimulates lysosome biogenesis and autophagy through TFEB activation [Citation135]. The activation of the MCOLN1-PPP3-TFEB pathway is also responsible for TMBIM6-mediated autophagy in HT1080 cells during starvation [Citation18]. In contrast, CTSB (cathepsin B; a lysosomal cysteine protease) inhibits autophagy by degrading MCOLN1, thereby limiting Ca2+ efflux and subsequent TFEB activity in infected BMDMs [Citation136]. These findings generally support the idea that lysosomal Ca2+ release contributes to TFEB activation and subsequent autophagy induction. In line with this, TPCN1-mediated lysosomal Ca2+ release triggers TFEB translocation to the nucleus in MEFs after sphingosine treatment [Citation137] ().
A change of [Ca2+]i is also involved in the modulation of TFEB activation. Caerulein triggers the interaction of STIM1 and CRAC channels in pancreatic tissues, thereby inducing the activation of SOCE and the entry of extracellular Ca2+, which further activates PPP3 and subsequent TFEB-induced autophagy [Citation138]. AMPK-dependent TFEB nuclear translocation mediates autophagy caused by the pharmacological activation of P2RX7 (using BzATP) in microglia [Citation93] (), indicating that the activation of TFEB is also affected by the energy state.
In addition to TFEB, ion channels can also control ATG expression through miRNA mechanisms. For example, the Zn2+ channel TRPM3 inhibits MAP1LC3 translational expression in clear cell renal cell carcinoma by downregulating MIR214 [Citation83] (). The overexpression of ORAI1 and SPCA impairs MIR519-induced autophagy in HeLa cells [Citation139]. A further understanding of the relationship between TFEB and miRNA may reveal the transcription mechanism of ion channel-mediated ATG expression.
Regulation of lysosomal function during fusion and degradation
Lysosomes provide a series of proteins as contact sites for autophagosomes, and serve as platforms of autophagosome-lysosome fusion. This lysosome-dependent process is regulated by ion channels and transporters, as described below.
Lysosomal Ca2+ channels and lysosomal fusion
The assembly of soluble N-ethylmaleimide-sensitive factor attachment protein receptors (SNAREs) complexes mediates lysosomal fusion events, which is enhanced by the release of Ca2+ from the lumen of lysosomes. The Ca2+ sensor SYT (synaptotagmin) acts as a bridge between the opposing bilayers and SNAREs, leading to membrane fusion in a Ca2+-dependent manner. Ca2+ homeostasis is also important for lysosomal acidification and lysosomal hydrolase activity [Citation140]. Unlike lysosomal Ca2+ that promotes autolysosome formation, high levels of cytosolic Ca2+ suppress fusion [Citation141], suggesting that the regulation of Ca2+ by ion channels and transporters during the fusion step of autophagy may be complex.
The MCOLN/TRPML channels consist of the MCOLN1, MCOLN2, and MCOLN3 channels. They play conserved roles as Ca2+-permeable channels and regulate lysosomal function, and are localized in the late endosomal and lysosomal compartments [Citation142]. MCOLN1 is the best-characterized lysosomal Ca2+-release channel and can be activated by several stimuli (e.g., starvation) during autophagy. Loss-of-function mutations of human MCOLN1 cause type IV mucolipidosis (ML-IV), which is a lysosomal storage disorder with symptoms of neurodegeneration and muscular dystrophy [Citation143], as well as abnormal lysosome and autolysosome function [Citation144]. MCOLN1 also interacts with the lysosomal chaperone complex and acts as a docking site for HSPA8 (heat shock protein family A (Hsp70) member 8) in the lysosome, allowing it to combine with substrates for chaperone-mediated autophagy in fibroblasts [Citation145]. In vivo, Mcoln1-deficient mice exhibit defective autophagy with an abnormal accumulation of autophagosomes in neurons [Citation146] and fibroblasts [Citation144]. These data further suggest that the activation of MCOLN1 is required for the efficient fusion of autophagosomes with lysosomes ().
Figure 8. Regulation of lysosomal function by ion channels and ion transporters during fusion and degradation steps of the autophagic process. To control autophagic fusion (A), ion channels and ion transporters produce calcium signaling by mediating the lysosomal release of Ca2+ or increasing the concentration of ER Ca2+. To control autophagic degradation (B), ion channels and ion transporters regulate lysosomal ion homeostasis of H+, K+, and Cl−, which are crucial for creating or maintaining a low pH environment. Abbreviations: ATP2A/SERCA, ATPase sarcoplasmic/endoplasmic Ca2+ transporting; Cav, voltage-activated Ca2+ channels; CFTR, CF transmembrane conductance regulator; CLCN7, chloride voltage-gated channel 7; HDAC6, histone deacetylase 6; ITPR, inositol 1,4,5-trisphosphate receptor; MCOLN1, mucolipin 1; MCOLN3, mucolipin 3; RYRs, ryanodine receptors; SNAREs, soluble N-ethylmaleimide sensitive factor attachment protein receptors; TMEM175, transmembrane protein 175; TPCN2, two pore segment channel 2; V-ATPase, vacuolar-type H+-ATPase.
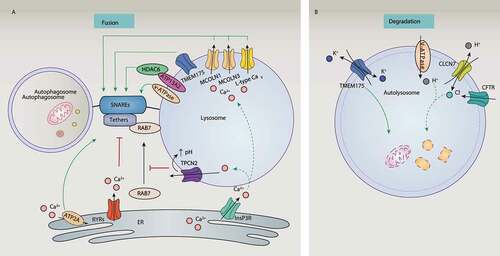
MCOLN3 is also involved in the fusion process of autophagy in some cells [Citation147]. MCOLN3 specifically interacts with the MAP1LC3 homolog GABARAPL2 to regulate autophagosome maturation in HeLa cells and HEK293 cells [Citation148], while more studies on its gain-of-function are needed. The function of MCOLN3 in autophagy may depend on its Ca2+ channel activity and palmitoylation [Citation149]. Interestingly, autophagy in HeLa cells induced by starvation does not seem to require MCOLN2, although it can bind to MCOLN1 to maintain lysosomal integrity [Citation150], indicating that the function of MCOLN family members in the formation of autolysosomes is not equal.
Other nonmucolipin channels also facilitate the fusion process of autophagy by regulating lysosomal Ca2+ release (). For example, the silencing of L-type Cav channels results in the accumulation of MAP1LC3-II and SQSTM1 in cardiomyocytes [Citation151] or primary cerebellar cultured neurons [Citation152]. P2RX4 (purinergic receptor P2X 4), a Ca2+ channel located on endolysosomal membranes, may also contribute to the fusion between lysosomes and autophagosomes in fibroblasts [Citation153].
However, the role of lysosomal TPCs in autophagy is controversial. TPCN2 appears to be a negative regulator of the fusion between basal autophagosomes and lysosomes in HeLa cells and breast cancer cells [Citation154] because the activation of TPCN2 by NAADP induces the efflux of lysosomal Ca2+, alkalizes the lysosomal pH, and prevents recruitment of RAB7 [Citation155], which is a critical step during autophagosome-lysosome fusion (). In addition, new lysosomal ATP-sensitive Na+ channels formed by TPCs control the pH stability of lysosomes, but have no effect on liver autophagy caused by starvation [Citation156]. Inversely, muscles derived from tpcn2−/- mice [Citation71] and the knockdown of TPCs in cardiomyocytes [Citation157] exhibit altered lysosomal pH, impaired lysosomal hydrolase activity, and an accumulation of autophagosomes under starvation conditions. These findings further indicate the selectivity and complexity of different TPC channels in autophagy.
ER Ca2+ channels and transporters and lysosomal fusion
Several ion channels and transporters on the ER regulate autophagy fusion by controlling Ca2+ signaling. The inhibition of RYRs in HEK293 cells, C2C12 myoblasts, and primary hippocampal neurons regulates autophagy flux by reducing ER Ca2+ release and increasing lysosomal turnover [Citation158]. The Ca2+ pump ATP2A on the ER also contributes to the fusion of autophagosomes with endosomes and lysosomes [Citation159]. Given that ER Ca2+ channels are responsible for Ca2+ refilling of lysosomes [Citation75], ER Ca2+ channels and transporters (ITPR, RYRs, and ATP2A) could control the transfer and refilling of Ca2+ from ER to lysosomes to regulate autophagic fusion ().
Lysosomal ATPase and lysosomal fusion
The lysosomal P-ATPase encoded by ATP13A2 is also involved in lysosomal function and autophagy. Mutations of ATP13A2 cause impairment of lysosomal acidification [Citation160], accompanied by SNCA/α-synuclein accumulation [Citation161]. ATP13A2 deficiency also impairs autophagosome-lysosome fusion in HEK293 cells and HeLa cells [Citation162]. Mechanistically, ATP13A2 enhances the fusion of autophagosomes and lysosomes by recruiting HDAC6 (histone deacetylase 6), resulting in the deacetylation of CTTN (cortactin) and assembly of the F-actin network (). Although it is unclear whether transporter activity is required for the fusion step, these findings provide an example of the interaction of lysosomes and cytoskeleton in the regulation of autophagy membrane dynamics.
Lysosomal ATPase and lysosomal degradation
Although lysosomal pH is crucial for lysosomal hydrolase activity during lysosomal Ca2+ homeostasis, lysosomal acidification is not a prerequisite for fusion [Citation159]. Because the acidic pH of the lysosome is mainly maintained by the V-ATPase embedded in the lysosomal membrane, V-ATPase family members can regulate the fusion or degradation process of autophagy. Several V-ATPase inhibitors, such as bafilomycin A1 and concanamycin A, limit autolysosome formation and cargo degradation [Citation163]. However, these inhibitors may have off-target effects independent of the V-ATPase. For example, bafilomycin A1 not only interferes with autophagy flux by inhibiting V-ATPase-dependent acidification, but also by blocking ATP2A-dependent autophagosome-lysosomal fusion [Citation164]. ZT-25, a new V-ATPase inhibitor, induces autophagy in HepG2 cells by blocking the MTOR pathway [Citation165]. Therefore, the effects of the V-ATPase on autophagy should be carefully explained when using these inhibitors.
The activity of the V-ATPase on autolysosome formation and cargo degradation is further controlled by its binding proteins. For example, ITM2A (a type II integral membrane protein) is involved in cell differentiation and inhibits the fusion process of autophagy by interacting with the components of the V-ATPase in HEK293 cells [Citation166]. UBQLNs (chaperone proteins of the ubiquitin-proteasome system) inhibit the activity of the V-ATPase by binding to its subunit ATP6V0A1 (ATPase H+ transporting V0 subunit a1), thereby impairing autophagy degradation in Drosophila and mammalian neuronal cells [Citation65]. LRRK2 blocks V-ATPase activity by binding to the ATP6V0A1 subunit of primary neurons, and plays a similar role in inhibiting autophagy [Citation167]. The binding protein not only inhibits V-ATPase activation in autophagy, but also enhances its function. For example, V-ATPase subunit ATP6V0D2 (ATPase H+ transporting V0 subunit d2) promotes autophagosome-lysosome fusion in macrophages by interacting with STX17 (syntaxin 17) and VAMP8 (vesicle associated membrane protein 8), thereby restricting bacterial infection [Citation168]. Although autolysosome formation requires the V-ATPase subunit ATP6AP2 (ATPase H+ transporting accessory protein 2) in liver cells [Citation169], its binding protein is still unknown. These data indicate that the V-ATPase may play a fundamental role in lysosomal degradation ().
Ion channels and lysosomal degradation
Cl− influx plays a major role in neutralizing the positive charge of the lumen produced by the V-ATPase, thereby maintaining the balance of acidic pH and membrane potential within the lysosome. Synthetic ion transporters can promote the transport of chloride ions, leading to an increase in the pH of the lysosome, which ultimately damages the lysosome and blocks autophagy [Citation170]. It seems that two lysosomal Cl− channels (CLCN7 [chloride voltage-gated channel 7] and CFTR) are required for lysosomal acidification via mediating Cl− influx to lysosomes [Citation171,Citation172] (). Functionally, knocking out clcn7 in mice leads to lysosomal dysfunction and increased levels of MAP1LC3-II, which is related to various pathological conditions [Citation173]. Alveolar macrophages from cftr−/- mice fail to kill internalized bacteria due to dysfunctional lysosomes [Citation174].
K+ channels (e.g., TMEM175 and KCNM/BK) also have the ability to maintain lysosomal pH and membrane potential by conducting K+ across lysosomal membranes [Citation29]. TMEM175 deficiency or KCNM/BK dysfunction leads to aberrant lysosomal pH and lysosomal hydrolase activity in cells (e.g., neurons and macrophages), thereby causing abnormal autophagosome-lysosome fusion and impairing clearance of autophagy [Citation175]. TMEM175 is also expressed in endosomes, indicating that the loss of its function may also affect the fusion of autophagosomes with endosomes (). Together, these data suggest that ion channels or transporters conduct H+, K+, and Cl− across lysosomes to control lysosomal pH and autophagic degradation.
Conclusions and perspectives
Over the past decade, autophagy has been recognized as the central regulator of cell homeostasis and quality control, although its molecular mechanisms and regulatory networks are context-dependent. Accordingly, research on the function of ion channels and transporters in autophagy has rapidly increased in various models. The increased ion signals (especially Ca2+, ROS, and pH signals) involve multiple stages of autophagy, from phagophore production to autophagosome maturation to autolysosome formation. This can regulate the expression and activity of autophagy through transcriptional (e.g., TFEB-mediated biogenesis), translational (e.g., miRNA-regulated ATG expression), and post-translational processes (e.g., phosphorylation-controlled activity of MTOR, AMPK, and BECN1), thereby forming an integrated mechanism that controls the autophagic membrane dynamic. According to research on gain- or loss-of-function, calcium channels (e.g., ITPR, RYRs, CACNA1A, TPCNs, MCOLN1, MCOLN3, and TRPM2), chloride channels (e.g., CFTR, CLCN3, and CLCN7), potassium channels (e.g., TMEM175), and transporters (e.g., P-type ATPase and SERCA) directly or indirectly affect autophagy by coupling with MTOR, AMPK, and ROS pathways. In addition to autophagy, ion channels and transporters also affect cell death [Citation176] and proliferation [Citation177]. In particular, Ca2+ and K+ channels mediate apoptosis and pyroptosis [Citation176]. Our understanding of this area of research is still incomplete, and many important questions remain to be answered.
How do different ion channels and transporters synergistically regulate autophagy?
A functionally defective ion channel or transporter may be quickly replaced and restored by the same type of subclass in response to autophagic stress. Certain ion channels and transport proteins may also directly affect the transport of amino acids, which requires the use of stable tracing techniques for confirmation. Ion channels and transporters can also regulate autophagy by directly affecting the formation of ATG complexes. However, distinguishing the ion-dependent and -independent roles of ion channels and transporters in autophagy remains a challenge, especially in vivo. The most-studied event in autophagy involving ions is probably Ca2+ signaling. Different sources of intracellular Ca2+ play different roles in autophagy, which is closely related to oxidative stress. It is still interesting to understand how non-Ca2+ channels or transporters affect ER stress and Ca2+ homeostasis. Because Ca2+ and ROS play a dual role in cell survival and cell death, autophagy-dependent survival and death may come from the activation of specific ion channels and transporters.
What is the role of autophagy in ion signals?
In contrast to extensive research on the role of ion signals in autophagy, the functional impact of autophagy in regulating ion production and signaling is poorly understood. The degradation of ion channels and transporters is multifaceted and occurs in various cell compartments, such as the PM and ER. The ubiquitin-proteasome system (UPS) has been extensively studied and regulates the stability of ion channels and transporters. As with the UPS, the degradation of ion channels and transporters mediated by autophagy may form a feedback mechanism to control the excessive activation of autophagy during stress. Although it is currently unclear, this autophagic degradation process may depend on specific autophagy receptors and signals, such as autophagic turnover of gap junction channels [Citation178,Citation179]. The interplay between ions, autophagy, and UPS signals changes dynamically, which is part of homeostasis.
How can we target ion signal-related autophagy in disease?
The use of drugs that target ion channels and transporters (Table S1) may be a strategy for treating autophagy-related diseases. Numerous preclinical studies are currently underway to evaluate the effects of ion channel agonists or antagonists on autophagic diseases. For example, amiloride inhibits ASICs to prevent Parkinson disease by promoting autophagic degradation of SNCA in vitro [Citation82] and in vivo [Citation131]. The calcium channel blocker verapamil is a clinically approved drug that enhances the fusion of autophagosomes and lysosomes in the liver, thereby preventing the accumulation of protein and lipid droplets and subsequent inflammation and insulin resistance [Citation141]. Moreover, the inhibition of TRPC5 can reduce basal autophagy and reverse the cell resistance of temozolomide and doxorubicin, which are first-line chemotherapy drugs for many cancers [Citation78,Citation79].
In summary, the involvement of ion channels and transporters in autophagy is complicated, depending on cell types or environmental conditions. Further research is needed to define the location and function of ion channels and transporters during autophagy, which depends on employing novel technologies, methods, drugs, and models.
Supplemental Material
Download MS Word (570.6 KB)Supplementary Material
Supplemental data for this article can be accessed here.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- Yang Z, Klionsky DJ. Eaten alive: a history of macroautophagy. Nat Cell Biol. 2010;12(9):814–822
- Dikic I, Elazar Z. Mechanism and medical implications of mammalian autophagy. Nat Rev Mol Cell Biol. 2018;19(6):349–364.
- Levine B, Kroemer G. Biological Functions of Autophagy Genes: A Disease Perspective. Cell. 2019;176:11–42.
- Ramirez A, Garcia-Quiroz J, Aguilar-Eslava L, et al. Novel Therapeutic Approaches of Ion Channels and Transporters in Cancer. Rev Physiol Biochem Pharmacol. 2020. doi:https://doi.org/10.1007/112_2020_28
- Goldschen-Ohm MP, Chanda B. SnapShot: Channel Gating Mechanisms. Cell. 2017;170(3):594–594
- Gadsby DC. Ion channels versus ion pumps: the principal difference, in principle. Nat Rev Mol Cell Biol. 2009;10(5):344–352.
- Kondratskyi A, Kondratska K, Skryma R, et al. Ion channels in the regulation of autophagy. Autophagy. 2018;14(1):3–21.
- Khakh BS, Alan North R. P2X receptors as cell-surface ATP sensors in health and disease. Nature. 2006;442(7102):527–532.
- Whitlock JM, Hartzell HC. Anoctamins/TMEM16 Proteins: chloride Channels Flirting with Lipids andExtracellular Vesicles. Annu Rev Physiol. 2017;79(1):119–143.
- Krabbendam IE, Honrath B, Culmsee C, et al. Mitochondrial Ca(2+)-activated K(+) channels and their role in cell life and death pathways. Cell Calcium. 2018;69:101–111.
- Paggio A, Checchetto V, Campo A, et al. Identification of an ATP-sensitive potassium channel in mitochondria. Nature. 2019;572(7771):609–613.
- Hibino H, Inanobe A, Furutani K, et al. Inwardly rectifying potassium channels: their structure, function, and physiological roles. Physiol Rev. 2010;90(1):291–366.
- Berridge MJ. The Inositol Trisphosphate/Calcium Signaling Pathway in Health and Disease. Physiol Rev. 2016;96(4):1261–1296.
- Lloyd-Evans E. On the move, lysosomal CAX drives Ca2+ transport and motility. J Cell Biol. 2016;212(7):755–757.
- Lloyd-Evans E, Waller-Evans H. Lysosomal Ca(2+) Homeostasis and Signaling in Health and Disease. Cold Spring Harb Perspect Biol. 2020;6:12. DOI:https://doi.org/10.1101/cshperspect.a035311
- Bultynck G, Kiviluoto S, Henke N, et al. The C terminus of Bax inhibitor-1 forms a Ca2+-permeable channel pore. J Biol Chem. 2012;287(4):2544–2557.
- Bultynck G, Kiviluoto S, Methner A. Bax inhibitor-1 is likely a pH-sensitive calcium leak channel, not a H+/Ca2+ exchanger. Sci Signal. 2014;7(343):pe22.
- Kim HK, Lee GH, Bhattarai KR, et al. TMBIM6 (transmembrane BAX inhibitor motif containing 6) enhances autophagy through regulation of lysosomal calcium. Autophagy. 2020. 1–18. doi:https://doi.org/10.1080/15548627.2020.1732161
- Putney JW. Origins of the concept of store-operated calcium entry. Front Biosci (Schol Ed). 2011;3(3):980–984.
- Park CY, Shcheglovitov A, Dolmetsch R. The CRAC Channel Activator STIM1 Binds and Inhibits L-Type Voltage-Gated Calcium Channels. Science. 2010;330(6000):101–105.
- Palty R, Raveh A, Kaminsky I, et al. SARAF Inactivates the Store Operated Calcium Entry Machinery to Prevent Excess Calcium Refilling. Cell. 2012;149(2):425–438.
- Zhang X, Hu M, Yang Y, et al. Organellar TRP channels. Nat Struct Mol Biol. 2018;25(11):1009–1018.
- de Lera Ruiz M, Kraus RL. Voltage-Gated Sodium Channels: Structure, Function, Pharmacology, and Clinical Indications. J Med Chem. 2015;58(18):7093–7118
- Jentsch TJ, Pusch M. CLC Chloride Channels and Transporters: structure, Function, Physiology, and Disease. Physiol Rev. 2018;98(3):1493–1590.
- Goldstein SA, Bockenhauer D, O’Kelly I, et al. Potassium leak channels and the KCNK family of two-P-domain subunits. Nat Rev Neurosci. 2001;2(3):175–184.
- Jentsch TJ. VRACs and other ion channels and transporters in the regulation of cell volume and beyond. Nat Rev Mol Cell Biol. 2016;17(5):293–307.
- Shoshan-Barmatz V, De Pinto V, Zweckstetter M, et al. VDAC, a multi-functional mitochondrial protein regulating cell life and death. Mol Aspects Med. 2010;31:227–285.
- Esseltine JL, Laird DW. Next-Generation Connexin and Pannexin Cell Biology. Trends Cell Biol. 2016;26(12):944–955.
- Cang C, Aranda K, Seo Y-J, et al. TMEM175 Is an Organelle K + Channel Regulating Lysosomal Function. Cell. 2015;162:(5):1101–1112.
- Wu ZS, Cheng H, Jiang Y, et al. Ion channels gated by acetylcholine and serotonin: structures, biology, and drug discovery. Acta Pharmacol Sin. 2015;36(8):895–907.
- Sun-Wada G-H WY. Role of vacuolar-type proton ATPase in signal transduction. Biochim Biophys Acta Bioenerg. 2015;1847(10):1166–1172.
- Stafford N, Wilson C, Oceandy D, et al. The Plasma Membrane Calcium ATPase and Their Role as Major New Players in Human Disease. Physiol Rev. 2017;97(3):1089–1125.
- Kang JK, Kim OH, Hur J, et al. Increased intracellular 2+ concentrations prevent membrane localization of PH domains through the formation of Ca 2+-phosphoinositides. Proc Natl Acad Sci U S A. 2017;114(45):11926–11931.
- Nabissi M, Morelli MB, Amantini C, et al. Cannabidiol stimulates Aml-1a-dependent glial differentiation and inhibits gliomastem-like cells proliferation by inducing autophagy in a TRPV2-dependent manner. Int J Cancer. 2015;137(8):1855–1869.
- Zhan L, Yang Y, Ma -T-T, et al. Transient receptor potential vanilloid 4 inhibits rat HSC-T6 apoptosis throughinduction of autophagy. Mol Cell Biochem. 2015;402(1–2):9–22.
- Chung KM, Jeong E-J, Park H, et al. Mediation of Autophagic Cell Death by Type 3 Ryanodine Receptor (RyR3) in AdultHippocampal Neural Stem Cells. Front Cell Neurosci. 2016;10:116.
- Liou B, Peng Y, Li R, et al. Modulating ryanodine receptors with dantrolene attenuates neuronopathic phenotypein Gaucher disease mice. Hum Mol Genet. 2016;25:5126–5141.
- Yuan Q, Chen Z, Santulli G, et al. Functional role of Calstabin2 in age-related cardiac alterations. Sci Rep. 2014;4(1):7425.
- Decuypere JP, Kindt D, Luyten T, et al. mTOR-Controlled Autophagy Requires Intracellular Ca(2+) Signaling. PLoS One. 2013;8(4):e61020.
- Luyten T, Welkenhuyzen K, Roest G, et al. Resveratrol-induced autophagy is dependent on IP3Rs and on cytosolic Ca(2). Biochim Biophys Acta Mol Cell Res. 2017;1864(6):947–956.
- Ren G, Zhou Y, Liang G, et al. General Anesthetics Regulate Autophagy via Modulating the Inositol1,4,5-Trisphosphate Receptor: implications for Dual Effects of Cytoprotection andCytotoxicity. Sci Rep. 2017;7(1):12378.
- Decuypere J-P, Welkenhuyzen K, Luyten T, et al. Ins(1,4,5)P3 receptor-mediated Ca2+ signaling and autophagy induction are interrelated. Autophagy. 2011;7(12):1472–1489.
- Shehata M, Matsumura H, Okubo-Suzuki R, et al. Neuronal Stimulation Induces Autophagy in Hippocampal Neurons That Is Involved in AMPA Receptor Degradation after Chemical Long-Term Depression. J Neurosci. 2012;32(30):10413–10422.
- Roux C, Aligny C, Lesueur C, et al. NMDA receptor blockade in the developing cortex induces autophagy-mediated death of immature cortical GABAergic interneurons: an ex vivo and in vivo study in Gad67-GFP mice. Exp Neurol. 2015;267:177–193.
- Chen Y-W, Chen Y-F, Chen Y-T, et al. The STIM1-Orai1 pathway of store-operated Ca2+ entry controls the checkpoint incell cycle G1/S transition. Sci Rep. 2016;6(1):22142.
- Sui H, Luo M, Miao Y, et al. Cystic fibrosis transmembrane conductance regulator ameliorates lipopolysaccharide-induced acute lung injury by inhibiting autophagy through PI3K/AKT/mTOR pathway in mice. Respir Physiol Neurobiol. 2020;273:103338.
- Su J, Xu Y, Zhou L et al. Suppression of chloride channel 3 expression facilitates sensitivity of humanglioma U251 cells to cisplatin through concomitant inhibition of Akt andautophagy. Anatomical record:Hoboken, NJ. 2007 2013. Vol. 296. 595–603
- Zheng Y, Chen Z, Gu Z, et al. Starvation-induced autophagy is up-regulated via ROS-mediated ClC-3 chloride channel activation in the nasopharyngeal carcinoma cell line CNE-2Z. Biochem J. 2019;476(9):1323–1333.
- Pillozzi S, Masselli M, Gasparoli L, et al. Macrolide antibiotics exert antileukemic effects by modulating the autophagic flux through inhibition of hERG1 potassium channels. Blood Cancer J. 2016;6(5):e423.
- Cardenas C, Miller RA, Smith I, et al. Essential regulation of cell bioenergetics by constitutive InsP3 receptor Ca2+ transfer to mitochondria. Cell. 2010;142(2):270–283.
- Tomar D, Dong Z, Shanmughapriya S, et al. MCUR1 Is a Scaffold Factor for the MCU Complex Function and PromotesMitochondrial Bioenergetics. Cell Rep. 2016;15(8):1673–1685.
- Zhao M, Chen J, Mao K, et al. Mitochondrial calcium dysfunction contributes to autophagic cell death induced byMPP(+) via AMPK pathway. Biochem Biophys Res Commun. 2019;509(2):390–394.
- Bround MJ, Wambolt R, Luciani DS, et al. Cardiomyocyte ATP production, metabolic flexibility, and survival require calciumflux through cardiac ryanodine receptors in vivo. J Biol Chem. 2013;288(26):18975–18986.
- Papandreou I, Lim AL, Laderoute K, et al. Hypoxia signals autophagy in tumor cells via AMPK activity, independent of HIF-1,BNIP3, and BNIP3L. Cell Death Differ. 2008;15(10):1572–1581.
- Cam H, Easton JB, High A, et al. mTORC1 signaling under hypoxic conditions is controlled by ATM-dependentphosphorylation of HIF-1alpha. Mol Cell. 2010;40(4):509–520.
- Yao Z, Cavero I, Gross GJ. Activation of cardiac KATP channels: an endogenous protective mechanism during repetitive ischemia. A J Physiol. 1993;264(2 Pt 2):H495–504.
- Samokhvalov V, Alsaleh N, El-Sikhry HE, et al. Epoxyeicosatrienoic acids protect cardiac cells during starvation by modulatingan autophagic response. Cell Death Dis. 2013;4(10):e885.
- Williams A, Sarkar S, Cuddon P, et al. Novel targets for Huntington’s disease in an mTOR-independent autophagy pathway. Nat Chem Biol. 2008;4(5):295–305.
- Baczkó I, Giles WR, Light PE. Pharmacological activation of plasma-membrane KATP channels reduces reoxygenation-induced Ca2+ overload in cardiac myocytes via modulation of the diastolic membrane potential. Br J Pharmacol. 2004;141(6):1059–1067.
- Zhu M, Cao L, Xiong S, et al. Na+/K+-ATPase-dependent autophagy protects brain against ischemic injury. Signal Transduct Target Ther. 2020;1:5. DOI:https://doi.org/10.1038/s41392-020-0153-7
- Perez-Neut M, Haar L, Rao V, et al. Activation of hERG3 channel stimulates autophagy and promotes cellular senescencein melanoma. Oncotarget. 2016;7(16):21991–22004.
- Enyedi P, Czirják G. molecular background of leak k+ currents: two-pore domain potassium channels. Physiol Rev. 2010;90(2):559–605.
- Zoncu R, Bar-Peled L, Efeyan A, et al. mTORC1 senses lysosomal amino acids through an inside-out mechanism that requiresthe vacuolar H(+)-ATPase. Science (New York, NY). 2011;334(6056):678–683.
- Hu Y, Carraro-Lacroix LR, Wang A, et al. Lysosomal pH plays a key role in regulation of mTOR activity in Osteoclasts. J Cell Biochem. 2016;117(2):413–425.
- Şentürk M, Lin G, Zuo Z, et al. Ubiquilins regulate autophagic flux through mTOR signalling and lysosomal acidification. Nat Cell Biol. 2019;21(3):384–396.
- Chung CY-S, Shin HR, Berdan CA, et al. Covalent targeting of the vacuolar H+-ATPase activates autophagy via mTORC1 inhibition. Nat Chem Biol. 2019;15(8):776–785.
- Wang F, Gatica D, Ying ZX, et al. Follicular lymphoma–associated mutations in vacuolar ATPase ATP6V1B2 activate autophagic flux and mTOR. J Clin Investig. 2019;129(4):1626–1640.
- Narita M, Young AR, Arakawa S, et al. Spatial coupling of mTOR and autophagy augments secretory phenotypes. Science. 2011;332(6032):966–970.
- Gusdon AM, Zhu J, Van Houten B, et al. ATP13A2 regulates mitochondrial bioenergetics through macroautophagy. Neurobiol Dis. 2012;45(3):962–972.
- Bento CF, Ashkenazi A, Jimenez-Sanchez M, et al. The Parkinson’s disease-associated genes ATP13A2 and SYT11 regulate autophagy via a common pathway. Nat Commun. 2016;7:7.
- Lin P-H, Duann P, Komazaki S, et al. Lysosomal two-pore channel subtype 2 (TPC2) regulates skeletal muscle Autophagic Signaling. J Biol Chem. 2015;290(6):3377–3389.
- Li R-J, Xu J, Fu C, et al. Regulation of mTORC1 by lysosomal calcium and calmodulin. eLife. 2016. 5. doi:https://doi.org/10.7554/eLife.19360
- Khan MT, Joseph SK. Role of inositol trisphosphate receptors in autophagy in DT40 cells. J Biol Chem. 2010;285(22):16912–16920.
- Raffaello A, Mammucari C, Gherardi G, et al. Calcium at the center of cell signaling: Interplay between endoplasmic reticulum, mitochondria, and lysosomes. Trends Biochem Sci. 2016;41(12):1035–1049.
- Garrity AG, Wang W, Collier CM, et al. The endoplasmic reticulum, not the pH gradient, drives calcium refilling oflysosomes. eLife. 2016;5. DOI:https://doi.org/10.7554/eLife.15887
- Nakanishi A, Hatano N, Fujiwara Y, et al. AMP-activated protein kinase-mediated feedback phosphorylation controls theCa(2+)/calmodulin (CaM) dependence of Ca(2+)/CaM-dependent protein kinase kinase beta. J Biol Chem. 2017;292(48):19804–19813.
- Zhang L, Dai F, Cui L, et al. Novel role for TRPC4 in regulation of macroautophagy by a small molecule invascular endothelial cells. Biochim Biophys Acta. 2015;1853(2):377–387.
- Zou Y, Chen M, Zhang S, et al. TRPC5induced autophagy promotes the TMZresistance of glioma cells via theCAMMKbeta/AMPKalpha/mTOR pathway. Oncol Rep. 2019;41(6):3413–3423.
- Zhang P, Liu X, Li H, et al. TRPC5-induced autophagy promotes drug resistance in breast carcinoma viaCaMKKbeta/AMPKalpha/mTOR pathway. Sci Rep. 2017;7(1):3158.
- Yang J, Yu J, Li D, et al. Store-operated calcium entry-activated autophagy protects EPC proliferation viathe CAMKK2-MTOR pathway in ox-LDL exposure. Autophagy. 2017;13(1):82–98.
- Gao W-F, Xu -Y-Y, Ge J-F, et al. Inhibition of acidsensing ion channel 1a attenuates acidinduced activation ofautophagy via a calcium signaling pathway in articular chondrocytes. Int J Mol Med. 2019;43(4):1778–1788.
- Sun X, Cao Y-B, Hu L-F, et al. ASICs mediate the modulatory effect by paeoniflorin on alpha-synuclein autophagicdegradation. Brain Res. 2011;1396:77–87.
- Hall DP, Cost NG, Hegde S, et al. TRPM3 and miR-204 establish a regulatory circuit that controls oncogenicautophagy in clear cell renal cell carcinoma. Cancer Cell. 2014;26(5):738–753.
- Scotto Rosato A, Montefusco S, Soldati C, et al. TRPML1 links lysosomal calcium to autophagosome biogenesis through the activation of the CaMKKβ/VPS34 pathway. Nat Commun. 2019; (1):10. DOI:https://doi.org/10.1038/s41467-019-13572-w.
- Huang P, Xu M, Wu Y, et al. Multiple facets of TRPML1 in autophagy. Cell Calcium. 2020;88:102196.
- Oh HG, Chun YS, Park C-S, et al. Regulation of basal autophagy by transient receptor potential melastatin 7(TRPM7) channel. Biochem Biophys Res Commun. 2015;463(1–2):7–12.
- Sukumaran P, Sun Y, Antonson N, et al. Dopaminergic neurotoxins induce cell death by attenuating NF-kappaB-mediatedregulation of TRPC1 expression and autophagy. Faseb J. 2018;32(3):1640–1652.
- Guse AH, Lee HC. NAADP: a universal Ca2+ trigger. Sci Signal. 2008;1(44):re10.
- Gomez-Suaga P, Luzon-Toro B, Churamani D, et al. Leucine-rich repeat kinase 2 regulates autophagy through a calcium-dependent pathway involving NAADP. Hum Mol Genet. 2012;21(3):511–525.
- Kim JK, Kim YS, Lee H-M, et al. GABAergic signaling linked to autophagy enhances host protection againstintracellular bacterial infections. Nat Commun. 2018;9(1):4184.
- Yan Y, Wei CL, Zhang WR, et al. Cross-talk between calcium and reactive oxygen species signaling. Acta Pharmacol Sin. 2006;27(7):821–826.
- Farfariello V, Amantini C, Santoni G. Transient receptor potential vanilloid 1 activation induces autophagy inthymocytes through ROS-regulated AMPK and Atg4C pathways. J Leukoc Biol. 2012;92(3):421–431.
- Sekar P, Huang D-Y, Hsieh S-L, et al. AMPK-dependent and independent actions of P2X7 in regulation of mitochondrial andlysosomal functions in microglia. Cell Commun Signal. 2018;16(1):83.
- Zhang X, Cui X, Li X, et al. Inhibition of Kir2.1 channel-induced depolarization promotes cell biological activity and differentiation by modulating autophagy in late endothelial progenitor cells. J Mol Cell Cardiol. 2019;127:57–66.
- Marino ML, Fais S, Djavaheri-Mergny M, et al. Proton pump inhibition induces autophagy as a survival mechanism following oxidative stress in human melanoma cells. Cell Death Dis. 2010;1(10):e87–e.
- Yu K-Y, Wang Y-P, Wang L-H, et al. Mitochondrial KATP channel involvement in angiotensin II-induced autophagy invascular smooth muscle cells. Basic Res Cardiol. 2014;109(4):416.
- De Stefani D, Bononi A, Romagnoli A, et al. VDAC1 selectively transfers apoptotic Ca2+ signals to mitochondria. Cell Death Differ. 2012;19(2):267–273.
- Geisler S, Holmstrom KM, Skujat D, et al. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol. 2010;12(2):119–131.
- Mungai PT, Waypa GB, Jairaman A, et al. Hypoxia triggers AMPK activation through reactive oxygen species-mediated activation of calcium release-activated calcium channels. Mol Cell Biol. 2011;31(17):3531–3545.
- Reiser J, Polu KR, Moller CC, et al. TRPC6 is a glomerular slit diaphragm-associated channel required for normal renalfunction. Nat Genet. 2005;37(7):739–744.
- Liu Z, Du Z, Li K, et al. TRPC6-Mediated Ca(2+) entry essential for the regulation of Nano-ZnO Induced Autophagy in SH-SY5Y cells. Neurochem Res. 2020;45(7):1602–1613
- Zhao B, Yang H, Zhang R, et al. The role of TRPC6 in oxidative stress-induced podocyte ischemic injury. Biochem Biophys Res Commun. 2015;461(2):413–420.
- Ji T, Zhang C, Ma L, et al. TRPC6-Mediated Ca2+ signaling is required for hypoxia-induced autophagy in human podocytes. Cellular Physiology and Biochemistry: International Journal of Experimentalcellular Physiology, Biochemistry, and Pharmacology. 2018;48(4):1782–1792.
- Sukumaran P, Sun Y, Vyas M, et al. TRPC1-mediated Ca(2)(+) entry is essential for the regulation of hypoxia andnutrient depletion-dependent autophagy. Cell Death Dis. 2015;6(3):e1674.
- Hou X, Xiao H, Zhang Y, et al. Transient receptor potential channel 6 knockdown prevents apoptosis of renal tubular epithelial cells upon oxidative stress via autophagy activation. Cell Death Dis. 2018;9(10):1015
- Fliegert R, Bauche A, Wolf Perez AM, et al. 2ʹ-Deoxyadenosine 5ʹ-diphosphoribose is an endogenous TRPM2 superagonist. Nat Chem Biol. 2017;13(9):1036–1044.
- Massullo P, Sumoza-Toledo A, Bhagat H, et al. TRPM channels, calcium and redox sensors during innate immune responses. Semin Cell Dev Biol. 2006;17(6):654–666.
- Jang Y, Lee B, Kim H, et al. Trpm2 ablation accelerates protein aggregation by impaired ADPR and autophagicClearance in the Brain. Mol Neurobiol. 2019;56(5):3819–3832.
- Jiang Q, Gao Y, Wang C, et al. Nitration of TRPM2 as a molecular switch induces autophagy during brain pericyte injury. Antioxid Redox Signal. 2017;27(16):1297–1316.
- Huo Y, Chen W, Zheng X, et al. The protective effect of EGF-activated ROS in human corneal epithelial cells byinducing mitochondrial autophagy via activation TRPM2. J Cell Physiol. 2020(10). doi:https://doi.org/10.1002/jcp.29597.
- Wyrsch P, Blenn C, Bader J, et al. Cell death and autophagy under oxidative stress: roles of poly(ADP-Ribose)polymerases and Ca(2+). Mol Cell Biol. 2012;32(17):3541–3553.
- Wang Q, Huang L, Yue J. Oxidative stress activates the TRPM2-Ca(2+)-CaMKII-ROS signaling loop to inducecell death in cancer cells. Biochim Biophys Acta Mol Cell Res. 2017;1864(6):957–967.
- Wang L, Shen M, Guo X, et al. Volume-sensitive outwardly rectifying chloride channel blockers protect againsthigh glucose-induced apoptosis of cardiomyocytes via autophagy activation. Sci Rep. 2017;7(1):44265.
- Xia Y, Liu Y, Xia T, et al. Activation of volume-sensitive Cl- channel mediates autophagy-related cell death in myocardial ischaemia/reperfusion injury. Oncotarget. 2016;7(26):39345–39362.
- Nilius B, Prenen J, Droogmans G. Modulation of volume-regulated anion channels by extra- and intracellular pH. Pflugers Arch. 1998;436(5):742–748.
- Sarkar S, Floto RA, Berger Z, et al. Lithium induces autophagy by inhibiting inositol monophosphatase. J Cell Biol. 2005;170(7):1101–1111.
- Vicencio JM, Ortiz C, Criollo A, et al. The inositol 1,4,5-trisphosphate receptor regulates autophagy through itsinteraction with Beclin 1. Cell Death Differ. 2009;16(7):1006–1017.
- Sammels E, Devogelaere B, Mekahli D, et al. Polycystin-2 activation by inositol 1,4,5-trisphosphate-induced Ca2+ release requires its direct association with the inositol 1,4,5-trisphosphate receptor in a signaling microdomain. J Biol Chem. 2010;285(24):18794–18805.
- Pena-Oyarzun D, Rodriguez-Pena M, Burgos-Bravo F, et al. PKD2/polycystin-2 induces autophagy by forming a complex with BECN1. Autophagy. 2020. 1–15. doi:https://doi.org/10.1080/15548627.2020.1782035
- Yuan J, Zhang Y, Sheng Y, et al. MYBL2 guides autophagy suppressor VDAC2 in the developing ovary to inhibitautophagy through a complex of VDAC2-BECN1-BCL2L1 in mammals. Autophagy. 2015;11(7):1081–1098.
- Liu Y, Shoji-Kawata S, Sumpter RM Jr., et al. Autosis is a Na+,K+-ATPase-regulated form of cell death triggered by autophagy-inducing peptides, starvation, and hypoxia-ischemia. Proc Natl Acad Sci U S A. 2013;110(51):20364–20371.
- Fernandez AF, Liu Y, Ginet V, et al. Interaction between the autophagy protein Beclin 1 and Na+,K+-ATPase during starvation, exercise, and ischemia. JCI Insight. 2020; (1):5. DOI:https://doi.org/10.1172/jci.insight.133282.
- Lauf PK, Alqahtani T, Flues K, et al. Interaction between Na-K-ATPase and Bcl-2 proteins BclXL and Bak. Am J Physiol Cell Physiol. 2015;308(1):C51–60.
- Luciani A, Villella VR, Esposito S, et al. Defective CFTR induces aggresome formation and lung inflammation in cysticfibrosis through ROS-mediated autophagy inhibition. Nat Cell Biol. 2010;12(9):863–875.
- Zhao YG, Chen Y, Miao G, et al. The ER-Localized transmembrane protein EPG-3/VMP1 regulates SERCA activity to control ER-isolation membrane contacts for autophagosome formation. Mol Cell. 2017;67(974–89):e6.
- Bejarano E, Yuste A, Patel B, et al. Connexins modulate autophagosome biogenesis. Nat Cell Biol. 2014;16(5):401–414.
- Xia H-G, Zhang L, Chen G, et al. Control of basal autophagy by calpain1 mediated cleavage of ATG5. Autophagy. 2010;6(1):61–66.
- Yousefi S, Perozzo R, Schmid I, et al. Calpain-mediated cleavage of Atg5 switches autophagy to apoptosis. Nat Cell Biol. 2006;8(10):1124–1132.
- Xu Y, Zhou P, Cheng S, et al. A bacterial effector reveals the V-ATPase-ATG16L1 axis that initiates xenophagy. Cell. 2019;178(552–66):e20.
- Lin M-W, Chen Y-H, Yang H-B, et al. Galantamine Inhibits Abeta1-42-Induced neurotoxicity by enhancing alpha7nachrexpression as a cargo carrier for LC3 binding and Abeta1-42 engulfment duringautophagic degradation. Neurotherapeutics. 2020;17(2):676–689
- David R. TFEB perfects multitasking. Nat Rev Mol Cell Biol. 2011;12(7):405.
- Medina DL, Di Paola S, Peluso I, et al. Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat Cell Biol. 2015;17(3):288–299.
- Nakamura S, Shigeyama S, Minami S, et al. LC3 lipidation is essential for TFEB activation during the lysosomal damage response to kidney injury. Nat Cell Biol. 2020;22(10):1252–1263.
- Zhang X, Cheng X, Yu L, et al. MCOLN1 is a ROS sensor in lysosomes that regulates autophagy. Nat Commun. 2016;7:12109
- Yadav RK, Lee GH, Lee HY, et al. TMBIM6 (transmembrane BAX inhibitor motif containing 6) enhances autophagy and reduces renal dysfunction in a cyclosporine A-induced nephrotoxicity model. Autophagy. 2015;11(10):1760–1774.
- Qi X, Man SM, Malireddi RKS, et al. Cathepsin B modulates lysosomal biogenesis and host defense against Francisellanovicida infection. J Exp Med. 2016;213:2081–2097.
- Höglinger D, Haberkant P, Aguilera-Romero A, et al. Intracellular sphingosine releases calcium from lysosomes. eLife. 2015;4:e10616.
- Zhu Z-D, Yu T, Liu H-J, et al. SOCE induced calcium overload regulates autophagy in acute pancreatitis viacalcineurin activation. Cell Death Dis. 2018;9(2):50.
- Abdelmohsen K, Srikantan S, Tominaga K, et al. Growth inhibition by miR-519 via multiple p21-inducing pathways. Mol Cell Biol. 2012;32(13):2530–2548.
- Mindell JA. Lysosomal acidification mechanisms. Annu Rev Physiol. 2012;74(1):69–86.
- Kondratskyi A, Yassine M, Slomianny C, et al. Identification of ML-9 as a lysosomotropic agent targeting autophagy and celldeath. Cell Death Dis. 2014;5(4):e1193.
- Cheng X, Shen D, Samie M, et al. Mucolipins: intracellular TRPML1-3 channels. FEBS Lett. 2010;584(10):2013–2021.
- Boudewyn LC, Walkley SU. Current concepts in the neuropathogenesis of mucolipidosis type IV. J Neurochem. 2019;148(5):669–689.
- Vergarajauregui S, Connelly PS, Daniels MP, et al. Autophagic dysfunction in mucolipidosis type IV patients. Hum Mol Genet. 2008;17(17):2723–2737.
- Venugopal B, Mesires NT, Kennedy JC, et al. Chaperone-mediated autophagy is defective in mucolipidosis type IV. J Cell Physiol. 2009;219(2):344–353.
- Curcio-Morelli C, Charles FA, Micsenyi MC, et al. Macroautophagy is defective in mucolipin-1-deficient mouse neurons. Neurobiol Dis. 2010;40(2):370–377.
- Kim HJ, Soyombo AA, Tjon-Kon-Sang S, et al. The Ca2+ Channel TRPML3 Regulates Membrane Trafficking and Autophagy. Traffic. 2009;10(8):1157–1167.
- Choi S, Kim HJ. The Ca2+ channel TRPML3 specifically interacts with the mammalian ATG8 homologue GATE16 to regulate autophagy. Biochem Biophys Res Commun. 2014;443(1):56–61.
- Kim SW, Kim DH, Park KS, et al. Palmitoylation controls trafficking of the intracellular Ca2+ channel MCOLN3/TRPML3 to regulate autophagy. Autophagy. 2019;15(2):327–340.
- Zeevi DA, Lev S, Frumkin A, et al. Heteromultimeric TRPML channel assemblies play a crucial role in the regulationof cell viability models and starvation-induced autophagy. J Cell Sci. 2010;123(18):3112–3124.
- Pushparaj C, Das A, Purroy R, et al. Voltage-gated calcium channel blockers deregulate macroautophagy incardiomyocytes. Int J Biochem Cell Biol. 2015;68:166–175.
- Tian X, Gala U, Zhang Y, et al. A voltage-gated calcium channel regulates lysosomal fusion with endosomes andautophagosomes and is required for neuronal homeostasis. PLoS Biol. 2015;13(3):e1002103.
- Cao Q, Zhong XZ, Zou Y, et al. Calcium release through P2X4 activates calmodulin to promote endolysosomalmembrane fusion. J Cell Biol. 2015;209(6):879–894.
- Sun W, Yue J. TPC2 mediates autophagy progression and extracellular vesicle secretion in cancercells. Exp Cell Res. 2018;370(2):478–489.
- Lu Y, Hao B, Graeff R, et al. NAADP/TPC2/Ca2+ Signaling Inhibits Autophagy. Commun Integr Biol. 2013;6(6):e27595.
- Cang C, Zhou Y, Navarro B, et al. mTOR regulates lysosomal ATP-sensitive two-pore Na(+) channels to adapt tometabolic state. Cell. 2013;152(4):778–790.
- García-Rúa V, Feijóo-Bandín S, Rodríguez-Penas D, et al. Endolysosomal two-pore channels regulate autophagy in cardiomyocytes. J Physiol. 2016;594(11):3061–3077.
- Vervliet T, Pintelon I, Welkenhuyzen K, et al. Basal ryanodine receptor activity suppresses autophagic flux. Biochem Pharmacol. 2017;132:133–142.
- Mauvezin C, Nagy P, Juhasz G, et al. Autophagosome-lysosome fusion is independent of V-ATPase-mediated acidification. Nat Commun. 2015;6(1):7007.
- Dehay B, Ramirez A, Martinez-Vicente M, et al. Loss of P-type ATPase ATP13A2/PARK9 function induces general lysosomal deficiency and leads to Parkinson disease neurodegeneration. Proc Natl Acad Sci U S A. 2012;109(24):9611–9616.
- Usenovic M, Tresse E, Mazzulli JR, et al. Deficiency of ATP13A2 leads to lysosomal dysfunction, alpha-synuclein accumulation, and neurotoxicity. J Neurosci. 2012;32(12):4240–4246.
- Wang R, Tan J, Chen T, et al. ATP13A2 facilitates HDAC6 recruitment to lysosome to promote autophagosome–lysosome fusion. J Cell Biol. 2019;218(1):267–284.
- Mousavi SA, Kjeken R, Berg TO, et al. Effects of inhibitors of the vacuolar proton pump on hepatic heterophagy andautophagy. Biochim Biophys Acta. 2001;1510(1–2):243–257.
- Mauvezin C, Neufeld TP. Bafilomycin A1 disrupts autophagic flux by inhibiting both V-ATPase-dependent acidification and Ca-P60A/SERCA-dependent autophagosome-lysosome fusion. Autophagy. 2015;11(8):1437–1438.
- Lu Y, Zhang R, Liu S, et al. ZT-25, a new vacuolar H(+)-ATPase inhibitor, induces apoptosis and protectiveautophagy through ROS generation in HepG2 cells. Eur J Pharmacol. 2016;771:130–138.
- Namkoong S, Lee KI, Lee JI, et al. The integral membrane protein ITM2A, a transcriptional target of PKA-CREB,regulates autophagic flux via interaction with the vacuolar ATPase. Autophagy. 2015;11(5):756–768.
- Wallings R, Connor-Robson N, Wade-Martins R. LRRK2 interacts with the vacuolar-type H+-ATPase pump a1 subunit to regulate lysosomal function. Hum Mol Genet. 2019;28(16):2696–2710.
- Xia Y, Liu N, Xie X, et al. The macrophage-specific V-ATPase subunit ATP6V0D2 restricts inflammasome activation and bacterial infection by facilitating autophagosome-lysosome fusion. Autophagy. 2019;15(6):960–975.
- Kissing S, Rudnik S, Damme M, et al. Disruption of the vacuolar-type H+ATPase complex in liver causes MTORC1-independent accumulation of autophagic vacuoles and lysosomes. Autophagy. 2017;13(4):670–685.
- Busschaert N, Park S-H, Baek K-H, et al. A synthetic ion transporter that disrupts autophagy and induces apoptosis by perturbing cellular chloride concentrations. Nat Chem. 2017;9(7):667–675.
- Graves AR, Curran PK, Smith CL, et al. The Cl-/H+ antiporter ClC-7 is the primary chloride permeation pathway inlysosomes. Nature. 2008;453(7196):788–792.
- Di A, Brown ME, Deriy LV, et al. CFTR regulates phagosome acidification in macrophages and alters bactericidalactivity. Nat Cell Biol. 2006;8(9):933–944.
- Wartosch L, Stauber T. A role for chloride transport in lysosomal protein degradation. Autophagy. 2010;6(1):158–159.
- Busschaert N, Park SH, Baek KH, et al. A synthetic ion transporter that disrupts autophagy and induces apoptosis by perturbing cellular chloride concentrations. Nat Chem. 2017;9(7):667–675.
- Jinn S, Drolet RE, Cramer PE, et al. TMEM175 deficiency impairs lysosomal and mitochondrial function and increases α-synuclein aggregation. Proc Nat Acad Sci. 2017;114(9):2389–2394.
- Kunzelmann K. Ion channels in regulated cell death. Cell Mol Life Sci. 2016;73(11–12):2387–2403.
- Prevarskaya N, Skryma R, Shuba Y. Ion channels in cancer: Are cancer hallmarks oncochannelopathies? Physiol Rev. 2018;98(2):559–621.
- Lichtenstein A, Minogue PJ, Beyer EC, et al. Autophagy: a pathway that contributes to connexin degradation. J Cell Sci. 2011;124(6):910–920.
- Iyyathurai J, Decuypere JP, Leybaert L, et al. Connexins: substrates and regulators of autophagy. BMC Cell Biol. 2016;17(Suppl S1):20.