
ABSTRACT
ATG16L1 is a critical mediator of macroautophagy/autophagy required for LC3 lipidation and autophagosome formation. However, ATG16L1 has a C-terminal domain including 7 WD40-type repetitions (WD40 domain, WDD) that is unnecessary for the conventional autophagic pathway. Instead, this domain mediates unconventional activities where LC3 is lipidated in atypical subcellular localizations unrelated to canonical double-membrane autophagosomes. The WDD provides a docking surface for molecules including a specific amino acid motif, thus engaging the LC3 lipidation capabilities of ATG16L1 in single-membrane structures. The physiological implications of such atypical activities are poorly characterized. In a recent report we described the improvement of the WDD-binding motif and the identification of transmembrane molecules that harbor this element in their intracellular region. One of them, IL10RB (interleukin 10 receptor subunit beta), binds the WDD after IL10 activation to facilitate endocytosis, early trafficking and signaling of IL10-IL10R complexes without influencing their degradation rate. These results reveal a novel unconventional role of ATG16L1 in cytokine signaling that does not entail a degradative purpose, thus contributing to catalog the physiological roles played by unconventional activities of the autophagic machinery.
While the functions of most ATG (autophagy related) proteins have been relatively well established in the conventional autophagic route, a number of them have also been implicated in atypical activities unrelated to canonical autophagy. ATG16L1 is an interesting example of such dual functionality because its canonical and unconventional activities reside in different domains of the molecule. Thus, the role of ATG16L1 in canonical autophagy only requires its N-terminal region, whereas the C-terminal WDD provides a binding module for molecules including a tyrosine-based amino acid motif, an interaction that allows ATG16L1 engagement in atypical subcellular localizations. Therefore, identification of motif-containing, WDD-binding effectors could help in the discovery of novel cellular functions regulated by unconventional activities of ATG16L1. However, the original WDD-binding motif ([YWF]-X3-[ED]-X4-[YWF]-X2-L) is too degenerate for protein identification. In a recent article we described a systematic mutagenesis strategy directed to identify all permissive residues for WDD binding at every position of the interacting peptide [Citation1]. These studies produced a refined motif version that we used bioinformatically to find transmembrane molecules including this element in their intracellular domain. Through this approach we identified a wide collection of functionally diverse proteins, suggesting that the unconventional activities of ATG16L1 may control an unanticipated variety of cellular processes.
Intriguingly, the collection includes cytokine receptors, raising the idea that WDD-mediated recruitment of ATG16L1 to endosomes containing activated ILRs could promote their decoration with LC3 to regulate their trafficking and signaling properties. We tested this notion for IL10RB, one of the motif-containing receptors, and found that optimal IL10 signaling requires the WDD due to a role of ATG16L1 in endocytosis and early trafficking of IL10-IL10R complexes. Thus, cells lacking the WDD exhibit delayed accumulation of cytoplasmic IL10R-positive vesicles after IL10 treatment, revealing defective endocytosis. Also, when comparing time points with similar numbers of IL10R-positive endosomes, the vesicles generated in WDD-deficient cells have reduced colocalization with the early endosome marker EEA1, indicating an additional role of the WDD in IL10R trafficking to early endosomes. Surprisingly, IL10R colocalization with late endosomal/lysosomal markers (RAB7, LAMP1) is not affected in WDD-deficient cells, suggesting unaltered degradation. Consistently, an “endocytic flux” experiment done by treating cells with IL10 and bafilomycin A1 indicated that lysosome inhibition does not normalize the different number of IL10R endosomes found in both cellular systems (full-length- or ΔWDD-ATG16L1), arguing that such difference is not caused by dissimilar degradation rates. These results raise the idea that the unconventional role of ATG16L1 in IL10 signaling originates from the control of endocytosis and early trafficking of IL10-IL10R complexes. Increased endocytosis without changes in degradation would result in more endosomes carrying activated IL10Rs on their way to the lysosome. Because endosomes constitute an important signaling platform, this scenario would cause increased signaling. However, because the WDD also improves conversion of IL10R endosomes into EEA1-positive vesicles, we cannot exclude the possibility that access to an early compartment expressing adjuvant components may contribute to improve IL10R signaling.
While the general picture is reasonably clear, exactly how the WDD controls IL10R endocytosis and trafficking, and the possible role of LC3, remain to be established. On the one hand, we found that IL10RB overexpression induces WDD-dependent labeling of IL10R-positive vesicles with GFP-LC3, indicating an intrinsic ability of IL10RB to induce the atypical pathway. On the other hand, IL10 causes a very low level of IL10RB-LC3 colocalization in wild-type cells that is not affected by absence of the WDD, suggesting that it is unrelated to the unconventional mechanism. Instead, this colocalization might reflect formation of amphisomes, hybrid vesicles generated through fusion between endosomes and canonical autophagosomes. Long exposure times to bafilomycin A1 have been suggested to prevent amphisome formation, and lysosomal inhibition caused by this drug would also slow down IL10RB trafficking, thus facilitating detection of brief/weak unconventional LC3 decoration events that may be difficult to observe. We found that treatment with bafilomycin A1 before IL10 exposure causes substantial IL10RB-LC3 colocalization in cells expressing full-length ATG16L1 but poorer labeling in those lacking the WDD domain. Assuming efficient amphisome inhibition, these results argue that IL10R endosomes may become LC3 positive through a WDD-dependent unconventional mechanism. It would be interesting to analyze the ultrastructure of the IL10R-LC3-positive vesicles to explore if they include cytoplasmic cargo suggestive of an autophagic component.
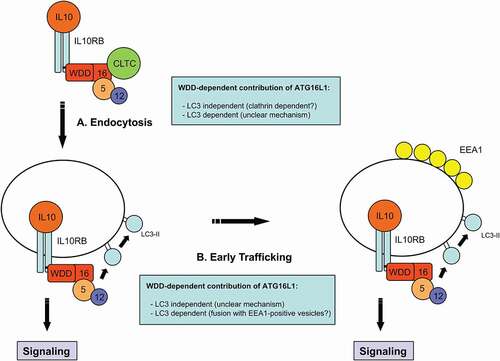
Because LC3 decoration generally favors vesicle targeting to the lysosomal compartment, the fact that the WDD does not promote IL10R endosome degradation argues against a relevant role of LC3 in any of the steps. However, LC3 also facilitates membrane fusion events, so transient decoration of IL10R-positive vesicles with LC3 could explain how they fuse with EEA1-positive compartments more efficiently in WDD-expressing cells. A potential role of LC3 in IL10R endocytosis is more difficult to anticipate. In this case ATG16L1 could function through a previously described interaction between its N-terminal domain and the clathrin heavy chain, leading to a dual model () where the WDD of ATG16L1 would favor IL10R endocytosis in a clathrin-dependent, LC3-independent manner, whereas its ability to transiently decorate IL10R endosomes with LC3 would promote their fusion with EEA1-positive endosomes and efficient early trafficking of IL10-IL10R complexes.
Figure 1. WDD-mediated unconventional functions of ATG16L1 in IL10R endocytosis and early trafficking in response to IL10. ATG16L1 contributes to IL10R endocytosis (A) by interacting with IL10RB through the WDD. Mechanistically, this effect could be independent of the LC3-lipidation capabilities of ATG16L1 and dependent on a previously described interaction between the N-terminal region of ATG16L1 and CLTC (clathrin heavy chain). Alternatively, LC3 lipidation promoted by ATG16L1 immediately after IL10R engagement could have a functional role in endocytosis, although the mechanistic details would be unclear in this case. The WDD also favors the early trafficking of IL10R endosomes to EEA1-positive vesicles (B). This function could be mediated by the transient unconventional labeling of IL10R endosomes with LC3 and its possible role in promoting membrane fusion events to improve their transition to EEA1-positive vesicles. Alternatively, ATG16L1 could have an LC3-independent function in this context, but the detailed mechanism would be unclear
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
Reference
- Serramito-Gómez I, Boada-Romero E, Villamuera R, et al. Regulation of cytokine signaling through direct interaction between cytokine receptors and the ATG16L1 WD40 domain. Nat Commun. 2020;11:5919.