
ABSTRACT
ATG7 drives macroautophagy, hereafter “autophagy”, by generating ATG12–ATG5 conjugates and lipidating Atg8 homologs including LC3. A pioneering body of work has defined the requirement of ATG7 for survival in mice and shown that neural-specific atg7 deletion causes neurodegeneration, but it has not been ascertained whether human life is compatible with ATG7 dysfunction. Recently, we defined the importance of ATG7 in human physiology by identifying twelve patients from five families harboring pathogenic, biallelic ATG7 variants causing a neurodevelopmental disorder. Patient fibroblasts show undetectable or severely diminished ATG7 protein levels, and biochemical assessment via autophagic flux and long-lived protein degradation assays demonstrated that attenuated autophagy underpins the pathology. Confirming the pathogenicity of patient variants, mouse cells expressing mutated ATG7 are unable to rescue LC3/Atg8 lipidation to wild-type levels. Our work defines mutated ATG7 as an important cause of human neurological disease and expands our understanding of autophagy in longevity and human health. We demonstrated that in certain circumstances, human survival with relatively mild phenotypes is possible even with undetectable levels of a nonredundant core autophagy protein.
Pioneering mouse genetics studies have demonstrated the vital nature of autophagy in mammalian development, where conditional deletion of core autophagy-related (ATG) genes causes perinatal lethality. In humans, accumulating evidence suggests that impairment of any stage of the autophagy pathway can lead to a number of diseases including adult neurodegeneration, cancer, infection and inflammation. Inherited impairments of autophagy have been identified but are rare.
ATG7 encodes a nonredundant core ATG protein with E1-like enzymatic activity. It is largely essential for autophagy, driving the conjugation of ATG12 to ATG5, and generating LC3-II via the lipidation of LC3-I with phosphatidylethanolamine. In the field, many laboratories utilize atg7-null cells for “autophagy deficient” studies. The physiological consequences of endogenous Atg7 inhibition in mice are profound. Tissue-specific atg7 ablation causes brain, liver and muscular dysfunction, and conditional whole-body atg7 deletion results in neonatal lethality.
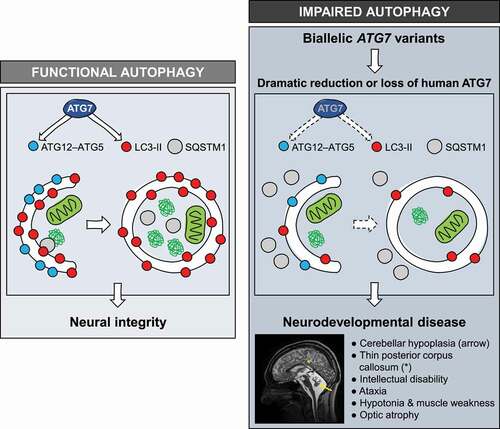
Using unbiased whole exome sequencing (WES), we first identified biallelic loss-of-function ATG7 variant alleles encoding splice site and nonsense changes in two siblings [Citation1]. This discovery prompted a collaboration with research groups in France, Switzerland, Germany and Saudi Arabia, via GeneMatcher (https://genematcher.org). Our colleagues identified another ten affected individuals from four families using similar WES pipelines, with each family harboring distinct, recessive ATG7 variants. Alleles encoded missense variants, with one leading to exclusion of exon 8 from ATG7 transcripts. Patients display ataxia, facial dysmorphism, hypotonia, muscle weakness, optic atrophy and intellectual disability, and the most severely affected individuals have seizures and spastic paraplegia. All patients present with neuroanatomical abnormalities comprising cerebellar hypoplasia and a thin posterior corpus callosum, providing defining features of ATG7 dysfunction. Clinical phenotypes appear to be static in adulthood, although one patient died early in childhood with diffuse brain atrophy and another has late-onset dementia.
First, we undertook formal histological and biochemical assessment of skeletal muscle biopsy from one patient with undetectable ATG7 protein and impaired LC3 lipidation. This patient (Patient 1) displayed muscle weakness, and their biopsy demonstrated mild myopathic changes, marked by inflammation, subsarcolemmal SQSTM1/p62 accumulation, and lipofuscin deposits. Supporting several ATG7 KO studies in mouse and human cells, basal autophagic structures were readily identified in skeletal muscle biopsy.
Dermal fibroblasts, available from at least one patient per family, demonstrated undetectable or severely diminished levels of ATG7 protein, accompanied by accumulation of basal SQSTM1 in most patient cell lines. Immunoblotting for LC3-II demonstrated that autophagic flux is impaired, and in the patient cell line with undetectable ATG7, LC3 lipidation is nearly absent even after concomitant autophagy induction and late-stage blockade. We further verified that these signatures reflected a bona fide impairment of nonselective flux using the classical long-lived protein degradation assay. Converging with previous data, autophagic sequestration of LDH (lactate dehydrogenase) is significantly diminished in primary patient fibroblasts.
Finally, to define the pathogenicity of individual missense variants, we re-expressed the human patient ATG7 mutants in atg7 knockout (KO) mouse embryonic fibroblasts (MEFs). Missense ATG7 variants fail to induce LC3 lipidation after concomitant autophagy induction and late-stage blockade to levels observed after introduction of wild-type ATG7, consolidating the causality of the ATG7 variants identified in these patients. These data are supported by studies using Saccharomyces cerevisiae expressing homologous atg7 variants or wild-type ATG7. In silico modeling suggests that missense variants interfere with ATG7 homodimerization, which is required for the transfer of activated LC3-I to ATG3. Hence, our converging data suggest that missense ATG7 variants interfere with ATG7 homodimerization, diminishing functional capacity which, combined with decreased steady-state levels of mutated ATG7, lead to impairment of autophagy.
Our work provides genetic and mechanistic demonstration that variant ATG7 is an important cause of autophagy dysfunction associated with human neurological disease (). Moreover, the identification of patients with relatively mild phenotypes and undetectable ATG7 demonstrates that, in some exceptional cases, humans can survive into adulthood despite the absence of a nonredundant core ATG protein. This is a remarkable finding given the perinatal lethality that characterizes autophagy-deficient mice, and the logical assumption that a human equivalent would also be unable to survive. Until now, this idea was supported by the identification of recessive, missense variants affecting ATG5 and WIPI2 that retain some functional activity. Null variants affecting WDR45 and WDR45B are associated with congenital autophagy disease, but evidence suggests that these homologs of yeast ATG18 encode functionally redundant proteins. Our discovery suggests that in some cases, additional degradative mechanisms may sustain human survival despite impaired autophagy, opening an exciting avenue for future research using patient-derived samples.
Figure 1. Functional autophagy in humans ensures neural integrity (left panel). Patients with bi-allelic ATG7 variants demonstrate loss or diminished levels of ATG7 protein, impairing autophagy (hallmarked by attenuated LC3 lipidation and SQSTM1 accumulation) and leading to complex neurodevelopmental disease (right panel)
Beyond autophagy, ATG7 is implicated in a diverse range of biological processes including innate immunity via LC3-associated phagocytosis, and modulating TP53/p53-mediated cell cycle arrest and apoptosis. Whether the dysfunction of these pathways, in combination with autophagy, contributes to human pathology resulting from variant ATG7 also remains unexplored. Surprisingly, the siblings displaying the most severe clinical presentation exhibited the mildest biochemical impairment of autophagy. Further interrogation of these patients’ exome and transcriptome datasets does not reveal strong candidate variants in other genes that may contribute to their severe phenotype. However, whereas rare heterozygous variants are usually benign in an otherwise healthy individual, it is reasonable to postulate that an individual with a background disease status (i.e., impaired autophagy) expressing such a variant might demonstrate a more severe clinical phenotype. Hence, we cannot rule out yet unidentified contributing genetic factors, and whole genome sequencing may provide more candidates. It is significant, however, that we observed variable clinical outcomes within the largest nuclear family. The future discovery of additional patients with deleterious variants affecting ATG7 or other core ATG genes will be essential to further define the phenotypic landscape of autophagy-deficient individuals. It will also be important to ascertain whether the same variants elicit consistent or heterogeneous manifestation of pathology between individuals, the latter being a characteristic hallmark of mitochondrial disease presentations.
Our discovery of a large patient cohort harboring recessive ATG7 variants consolidates the importance of autophagy in human health, building upon important work that identified pathogenic variants in ATG5, WIPI2, WDR45 and WDR45B. We identified patients surviving into adulthood with undetectable ATG7, and individuals in their seventh and eighth decades of life despite severely diminished ATG7 protein levels. It is tempting to speculate that autophagy-augmenting therapeutics or gene therapy could circumscribe the neurological phenotypes in patients with impaired autophagy, and the discovery of other autophagy-deficient cohorts and access to patient-derived biological material, including iPSC cell lines, promise to accelerate these investigations. As a consequence, this work could also have major implications for the many areas of unmet clinical need where aberrant autophagic activity is implicated.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
Reference
- Collier JJ, Guissart C, Oláhová M, et al. Developmental consequences of defective ATG7-mediated autophagy in humans. New England J Med. 2021;384:2406–2417.