
ABSTRACT
Mucopolysaccharidoses (MPS) are inherited metabolic diseases with strong neurological involvement. MPSs are caused by defects in lysosomal enzymes involved in the degradation of glycosaminoglycans (GAGs), which consequently accumulate into the lysosomes as primary storage. Macroautophagy/autophagy impairment is well known to drive neurodegeneration in MPSs, however, mechanisms underlying such dysfunction are still poorly understood. Recently, by studying a mouse model for MPS-III (Sanfilippo syndrome) we have shown that the progressive aggregation of amyloid proteins in neuronal cell bodies occurs downstream of the GAG storage and, in turn, impairs the autophagy pathway by affecting lysosomal-dependent autophagosome clearance
In MPSs, multiple organs and systems may be affected, yet progressive neurological deterioration is the most important clinical feature and, therefore, it is the major therapeutic target for these disorders. Importantly, while GAGs accumulate right after birth, neurodegenerative processes begin at later stages of disease progression in MPSs. In the last ten years a lot of effort has been put into trying to dissect mechanisms underlying neurodegenerative processes in MPSs.
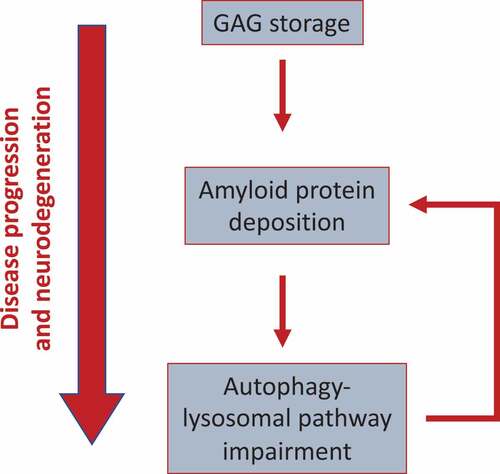
Figure 1. Model showing the functional link between GAGs, amyloid aggregation, and ALP dysfunction in MPS-III. GAG storage initially drives protein aggregation (likely, because of the capability of GAGs to provide a scaffold promoting amyloid aggregation). Perikaryal protein aggregation, in turns, triggers the block of the autophagy flux observed in MPS-III, likely by affecting lysosomal dynamics and trafficking. Then, because the ALP itself plays a key role in the clearance of protein aggregates, this may generate a vicious cycle, which boosts amyloid buildup and toxicity. This cascade of events drives disease progression and neurodegeneration
The autophagy-lysosomal pathway (ALP) is a major degradation process in the cell and is critical, particularly in neurons, to maintain cellular homeostasis. Accumulating evidence has established that impairment of the ALP plays a key role in driving neurodegenerative processes of several LSDs, including MPSs. In particular, by studying a mouse model of MPS-IIIA, one of the most common and severe forms of neurological MPSs, we have demonstrated that the autophagy flux is blocked downstream of autophagy initiation at the stage of lysosomal-mediated autophagosome maturation.
Additionally, deposition of insoluble protein aggregates composed of amyloidogenic proteins such as SNCA/α-synuclein, MAPT/tau protein and amyloid β-protein (Aβ) has been reported in several forms of neurological MPSs. These observations have been made in postmortem brains from patients (MPS-I, MPS-II and MPS-III) as well as in murine models of different neurological MPSs. Nevertheless, the neuropathogenic relevance of amyloid deposits in the context of MPSs has remained largely unexplored and, in most cases, such accumulation has been reported to occur mostly as an epiphenomenon.
In a very recent paper, we have provided novel insights linking GAG storage, protein aggregation and autophagy defects in the MPSs [Citation1]. We have shown that amyloid protein deposits localize mainly in the neuronal perikarya of MPS-IIIA mouse brains and contain primarily SNCA/α-synuclein together with MAPT/tau, Aβ and PRNP (prion protein). Moreover, such amyloid deposition occurs downstream of the primary GAG storage and increases progressively with age becoming massive at 5–6 months of age, when also ALP dysfunction and the first signs of neurological deterioration appear. The inhibition of protein aggregation in MPS-IIIA mice by treatment with CLR01, a small molecule capable of inhibiting efficiently and potently the abnormal self-assembly of multiple amyloidogenic proteins, results in a marked amelioration of neurodegenerative signs including neuroinflammation and cognitive dysfunction. Importantly, when we analyzed ALP in MPS-IIIA mouse brain, we found that CLR01 treatment reduces the enlargement of the lysosomal compartment (both the average size and the number of abnormal-sized lysosomes are reduced) and markedly decreases both the number of LC3-positive puncta and the protein levels of NBR1, an autophagy cargo receptor, thus indicating that lysosomal-mediated clearance of autophagosomes is efficiently restored by inhibiting amyloid deposition.
Overall, our data suggest additional important considerations that we would like to discuss here:
We have shown that brain deposition of multiple amyloid proteins occurs downstream the primary GAG storage and plays a gain of neurotoxic function in MPS-IIIA and likely in other MPSs by impairing the ALP. Therefore, our data indicate a pathological cascade of events initiated by primary GAG accumulation and involving, in succession, protein aggregation and autophagy defects. These processes take place during, and contribute to, disease progression and neurodegeneration in the MPSs (see model).
How amyloid protein deposition affects the ALP remains to be elucidated. In healthy neurons, autophagosomes are continuously generated in the axons and efficiently cleared upon fusion with lysosomes in the cell body and, therefore, are rarely found in neurites or cell bodies unless lysosome-dependent AV clearance is disrupted. In MPS-IIIA mouse brain the overall number of LC3-positive puncta is greatly increased compared to WT mouse brain. Interestingly, the majority of LC3 puntca are not within the soma of cells, but peripherally localized, a finding, which is consistent with a block of the autophagic flux, as previously reported. CLR01 treatment reduces the number of peripheral LC3-puncta in MPS-IIIA mouse brain. This suggests a model in which perikaryal aggregation of amyloidogenic proteins prevents lysosome-mediated clearance of autophagosomes, which consequently accumulate as unfused vesicles in the neuronal periphery. Of course, further studies will be required to support this model and understand how protein deposition has an impact on these trafficking processes.
Our data identify protein aggregation inhibition (by CLR01 or other molecules) as a a new approach for the treatment of brain lesions in MPS-IIIA and likely in other MPSs. The neuroprotective effect of amyloid inhibition in MPS-IIIA mice is mediated by the relief of the autophagy block. This finding brings up a consideration, which is relevant from a therapeutic point of view. Indeed, although promoting clearance of neurotoxic aggregates by reactivating autophagy flux in neurons is a potential powerful therapeutic strategy for MPSs, achieving this goal by simply inducing/accelerating autophagy (for example through repurposing of FDA-approved drugs) requires extreme caution in MPSs, where the autophagy flux is blocked at the stage of lysosomal-mediated autophagosome maturation. In fact, accelerating autophagy in such disease contexts could result in an increase in cargo delivery that exceeds the degradation capacity of the lysosomes, thus leading not only to ineffective treatment, but also potentially to detrimental effects. In this regard, a winning therapeutic strategy could be to develop drug-based protocols based on coupling the relief of the autophagy block with autophagy induction. In principle, the use of this combination of drugs, which act at different stages of the ALP, could result in a synergistic effect on autophagy flux.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
Reference
- Monaco A, Maffia V, Sorrentino NC, et al. The amyloid inhibitor CLR01 relieves autophagy and ameliorates neuropathology in a severe lysosomal storage disease. Mol Ther. 2020;28(4):1167–1176.