
ABSTRACT
Macroautophagy is a highly conserved eukaryotic cellular pathway involving the engulfment of macromolecules, organelles, and invading microbes by a double-membrane compartment and subsequent lysosomal degradation. The mechanisms that regulate macroautophagy, and the interaction of its components with other cellular pathways, have remained unclear. Here, we performed proximity-dependent biotin identification (BioID) on 39 core human macroautophagy proteins, identifying over 700 unique high confidence proximity interactors with new putative connections between macroautophagic and essential cellular processes. Of note, we identify members of the OSBPL (oxysterol binding protein like) family as Atg8-family protein interactors. We subsequently conducted comprehensive screens of the OSBPL family for LC3B-binding and roles in xenophagy and aggrephagy. OSBPL7 and OSBPL11 emerged as novel lipid transfer proteins required for macroautophagy of selective cargo. Altogether, our proximity interaction map provides a valuable resource for the study of autophagy and highlights the critical role of membrane contact site proteins in the pathway.
Abbreviations
BioID: proximity-dependent biotin identification; GO: gene ontology; OSBPL: oxysterol binding protein like; VAPA: VAMP associated protein A; VAPB: VAMP associated protein B and C
Introduction
During macroautophagy, a double membrane compartment (the autophagosome) engulfs portions of cytoplasmic material for fusion with the lysosome, degrading sequestered macromolecules and damaged organelles. Macroautophagy is constitutively active in eukaryotic cells where it serves a critical role in cellular homeostasis, but can also be upregulated in response to changes in nutrient levels, environmental stresses or invading pathogens [Citation1]. Dysregulation of macroautophagy is associated with disease states such as cancer, infectious diseases, inflammatory bowel disease, and neurodegenerative disorders [Citation1], and will likely prove to be an important therapeutic target [Citation2]. Nonetheless, the mechanisms regulating the formation of autophagosomes remain unclear. Immuno-purification coupled with mass spectrometry has been a valuable approach for identifying key protein-protein interactions in the macroautophagy pathway [Citation3–6]. However, characterizing macroautophagy interactions that are transient, weak and/or highly dynamic, such as those found at membrane contact sites [Citation7–9], can be challenging using standard IP-based approaches [Citation10]. BioID has emerged as a powerful complementary technique, permitting the investigation of proximity interactions in compartments and pathways that are recalcitrant to standard approaches [Citation11–16]. The applicability of this technique to macroautophagy was recently demonstrated in plants (Nicotiana benthamiana), where ATG8-interacting proteins were identified [Citation17].
Here, we applied BioID to the study of macroautophagy in human cells, generating a proximity interaction map of 39 core macroautophagy proteins. The map provides important insights into how this critical process is organized into functional complexes, and how it interfaces with other cellular compartments and pathways. Our dataset also identifies several novel components of macroautophagy, including proteins with known links to human disease, and highlights connections to membrane contact sites and the importance of lipid transfer proteins in the pathway. Altogether, this dataset will be a valuable resource for future macroautophagy studies.
Results
Proximity interaction mapping of the macroautophagy pathway reveals new interactions between core macroautophagy proteins
We conducted BioID on 39 core macroautophagy proteins, encompassing the initiation, nucleation, and elongation steps, ATG9A, the Atg8-family proteins and ATG12 conjugation systems, and cargo receptors ( and Table S1). Stable HEK 293 cell pools expressing these proteins in a tetracycline-inducible manner were generated using the Flp-In T-REx site-specific recombination system [Citation18] (western blots and micrographs of two representative BirA*-tagged cell lines can be found in Figure S1). Following streptavidin purification and tandem mass spectrometry, 1670 high-confidence proximity interactions (Table S2) were identified between 39 baits and 773 unique proximity interactors (Table S3, Figure S2A).
Figure 1. BioID macroautophagy bait-bait interactome. (A) Schematic of the 39 BioID baits within the macroautophagy pathway, separated into their respective step in macroautophagy. (B) A bait-bait network was created based on a spring-embedded layout where edge-width is proportional to peptide counts. Bait-bait proximity interaction network based on a spring embedded layout where edge width is proportional to peptide counts. A total of 129 protein-protein interactions (PPI) between baits: 109 are previously reported (green arrows), 20 are novel uncharacterized PPI (red arrows), and one is a newly confirmed protein-protein interaction (yellow arrow). ATG8, Atg8-family proteins.
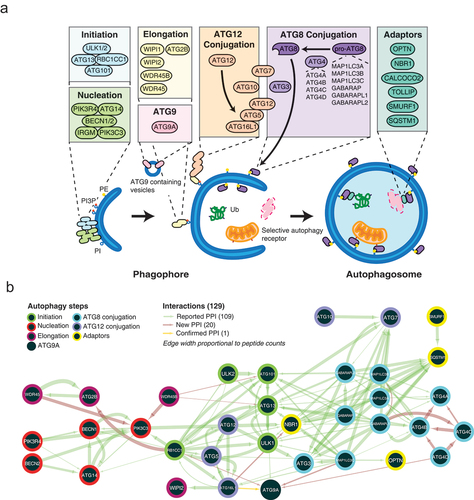
Amongst 129 proximity interactions detected between 39 bait proteins, 109 (84.5%) were previously reported (green arrows, ). These include known interactions between components of the initiation, nucleation and ATG12–ATG5-ATG16L1 complexes [Citation1,Citation19], between the Atg8-family proteins and ATG12 conjugation systems [Citation1] and Atg8-family protein members with the initiation complex [Citation20]. The bait-bait network also identified 20 previously uncharacterized proximity interactions (red arrows, ), one of which was validated here (yellow arrows, ). Co-immunoprecipitation validated an interaction between endogenous ATG9A and 3xMYC-ATG16L1 (Figure S2B), suggesting a point of contact for ATG9A-containing vesicles that dynamically interact with the developing phagophore [Citation21,Citation22]. The bait-bait proximity interactome thus identified known and novel protein-protein interactions between core macroautophagy proteins, highlighting the ability of BioID to capture bona fide interactions within the macroautophagy pathway.
The macroautophagy proximity interactome contains new fitness and disease-associated proteins
The bait-prey proximity interactome (Figure S2A, ) consists of 773 high-confidence proximity interactors, which contained 222 (28.7%) proteins with previously described roles in macroautophagy, including 48 proteins identified from high-throughput CRISPR or RNAi- screens (see Table S4 for references) to have functional links with macroautophagy. 123 (22.3%) proteins in the network not previously linked to macroautophagy are encoded by essential genes (, Table S3), including polypeptides linked to mitochondrial translation (MRPS; mitochondrial ribosomal proteins, tRNA synthetases), Golgi vesicular transport (coatomer protein complex, HAUS complex), microtubule cytoskeleton organization (CEP; centrosomal proteins, motor proteins) and cell cycle regulation. Dysregulation of macroautophagy is linked to multiple human diseases and our network contained 218 (28.2%) proteins encoded by human disease-related genes (, Table S3). Amongst proximity interactors with no previously reported functional role in macroautophagy, several are disease-associated proteins for conditions previously linked to macroautophagy, such as APBB2 in Alzheimer disease, DNAJC13 in Parkinson disease, DLG5 in inflammatory bowel disease and WDCP, GOLGA5, and HIP1 in various cancers (Table S3). Proteins encoded by disease-related genes not previously linked to macroautophagy were also identified, including ciliopathies (primary ciliary dyskinesia, short-rib thoracic dysplasia) and metabolic disorders (combined oxidative phosphorylation deficiency, congenital disorders of glycosylation) (Table S3). Together these data highlight extensive links between macroautophagy, essential and disease-associated cellular pathways.
Figure 2. Gene Ontology (GO) biological process enrichments in the macroautophagy BioID network. Top GO biological process enrichments of 420 high confidence proximity interactors with selected GO annotations. Bait proteins are organized based on steps of macroautophagy. Edge width of arrows proportional to peptide count. In the right panels, hits associated with each step of macroautophagy are depicted as black dots. Associated p- and FDR values can be found in Table S6. ATG8, Atg8-family proteins.
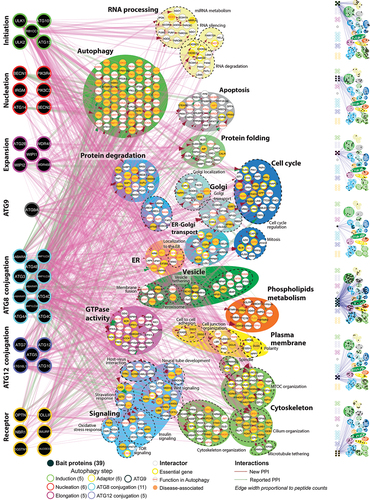
Figure 3. Analysis of bait-prey BioID interactions. (A) Summary of 773 high-confidence proximity interactors that are macroautophagy-associated, disease gene-encoded or essential gene-encoded. Of the 222 (28.7%) interactors with previously reported functional roles macroautophagy (“known macroautophagy proteins”), 48 proteins were identified in high-throughput CRISPR or RNAi-based screens (see Table S4 for references) to have functional effects on macroautophagy. Essential genes were determined based on cancer dependency data from 423 cell lines [Citation23] (see Table S3). (B) Prey proteins interacting with multiple baits in initiation, nucleation, Atg8-family protein conjugation, and ATG12 conjugation steps. Known and unknown macroautophagy interactors for each number of bait interactions is represented as a percentage of total prey proteins (“# of prey”). ATG8, Atg8-family proteins.
![Figure 3. Analysis of bait-prey BioID interactions. (A) Summary of 773 high-confidence proximity interactors that are macroautophagy-associated, disease gene-encoded or essential gene-encoded. Of the 222 (28.7%) interactors with previously reported functional roles macroautophagy (“known macroautophagy proteins”), 48 proteins were identified in high-throughput CRISPR or RNAi-based screens (see Table S4 for references) to have functional effects on macroautophagy. Essential genes were determined based on cancer dependency data from 423 cell lines [Citation23] (see Table S3). (B) Prey proteins interacting with multiple baits in initiation, nucleation, Atg8-family protein conjugation, and ATG12 conjugation steps. Known and unknown macroautophagy interactors for each number of bait interactions is represented as a percentage of total prey proteins (“# of prey”). ATG8, Atg8-family proteins.](/cms/asset/7a9ad28b-15e2-4430-83cb-95d33d051ef8/kaup_a_1965711_f0003_c.jpg)
GO enrichments and proximity interactions with proteins in membraneless organelles
The most highly enriched Gene Ontology (GO) categories in the proximity interactome include macroautophagy-related processes, ER-Golgi localization and transport, cytoskeletal related processes, and RNA processing (). When broken down into separate macroautophagy steps (, right panel), almost all prey proteins linked to ER-associated degradation interact with ATG9A, whereas prey proteins captured by initiation step bait proteins are primarily linked to miRNA metabolism, microtubule-organizing center organization and cilium organization and key components of stress granules and processing bodies (P-bodies), membraneless organelles that form biological condensates [Citation24–26]. Indeed, the macroautophagy proximity interactome contains 23 proteins previously linked to stress granules and P-bodies [Citation27], including proteins associated with the CCR4-NOT deadenylase complex, miRNA induced silencing, nonsense-mediated decay, and translational control (Figure S3). The connection of autophagy to these membraneless organelles is especially noteworthy as a recent study demonstrated that the phagophore assembly site in yeast is a liquid-like condensate of ATG proteins [Citation28].
About half of the prey proteins (331) are in proximity with two or more macroautophagy baits, and for 230 preys, these multiple bait proximity interactions belong to the same macroautophagy step (). Proteins with known functional roles in macroautophagy were in proximity with the highest number of baits in the initiation (ATG101, RB1CC1), nucleation (NRBF2), Atg8-family proteins (ATG4B, TBC1D5) and ATG12 conjugation (TECPR1) steps (, Table S3).
Members of the OSBPL family and the tethering protein VAPA are critical for macroautophagy
The identification of OSBPL family members as Atg8-family protein proximity interactors was intriguing (Table S2), as the mechanism of phospholipid synthesis and incorporation into the growing autophagosome has recently raised great interest. An emerging model suggests that phospholipids are synthesized in the ER then subsequently transferred in bulk via ATG2 to the nascent autophagosome [Citation29–33]. Despite the attractiveness of this model, it is clear that additional lipid transfer mechanisms must exist, as the theoretical phospholipid transfer rate of this mechanism is insufficient to explain observed rates of phagophore growth and fails to explain how the autophagosome obtains its unique phospholipid identity [Citation34].
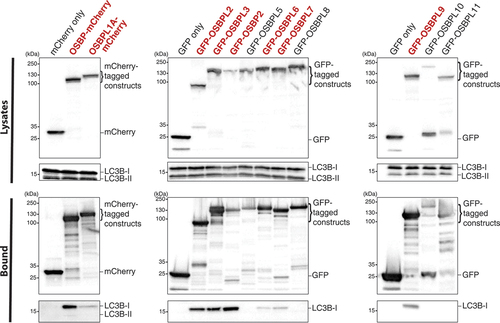
Given the important links between lipid transfer and membrane contact sites in macroautophagy, we conducted three macroautophagy screens focused on the OSBPL family. In the first, we conducted an unbiased screen of OSBPL family members to probe their ability to bind to the Atg8 homolog LC3B. The OSBPL family members, tagged with either mCherry or GFP, were used as bait in co-immunoprecipitation experiments and the bound protein fraction was probed for endogenous LC3B via western blotting (). In accordance with our BioID data, OSBPL3 and OSBPL6 both interacted with LC3B, but we also observed LC3B-binding to OSBP, OSBPL1A, OSBPL2, OSBP2, OSBPL7, and OSBPL9. Notably, the OSBPL proteins bind only the unlipidated (LC3B-I) form of the protein. Furthermore, the four OSBPL proteins that do not bind LC3B, OSBPL5, OSBPL8, OSBPL10, and OSBPL11 all lack an FFAT (two phenylalanines in an acidic tract) motif that binds the ER resident proteins VAPA (VAMP associated protein A) and VAPB (VAMP associated protein B and C) (, red labels) [Citation35]. Taken together, our data indicate that OSBPL family members can interact with Atg8 homologs.
Figure 4. OSBPL family members interact with LC3B. HEK293T cells were transfected with indicated mCherry- and GFP-tagged OSBPL constructs. Lysates were precipitated with RFP-Trap and GFP-Trap beads, and elutions were analyzed by western blot, probing for endogenous LC3BB. OSBPL family members that possess an FFAT (two phenylalanines in an acidic tract) motif involved in binding the ER-resident proteins VAPA and VAPB are highlighted in bold red text. Data are representative of three independent experiments.
In our second screen, we used MAP1LC3B (hereafter LC3B) recruitment to Salmonella as a model of selective targeting by components of the macroautophagy pathway [Citation36–39]. Following siRNA-mediated depletion of the indicated mRNAs (Figure S4), cells were infected with Salmonella for 1 h (Figure S5A). Under control conditions, ~30% of intracellular Salmonella are targeted by components of the macroautophagic machinery [Citation36], as evidenced by colocalization with LC3B. As expected, knockdown of ATG12, a key macroautophagy conjugation protein, was associated with a significant decrease in the number of LC3B+ bacteria (). We also examined a role for OSBPL binding partners VAPA and VAPB, proteins implicated in autophagosome biogenesis [Citation40] and which are critical for the formation of pathogen-host membrane-contact sites [Citation41]. A significant reduction in LC3B+ Salmonella was observed when OSBPL7, OSBPL11, or VAPA were knocked down, suggesting OSBPL-VAPA interactions are required for recruitment of LC3B to Salmonella (). Given the novelty of OSBPL7 and OSBPL11’s roles in autophagy, we validated the phenotype with additional siRNA against these two proteins (Figure S5B).
Figure 5. OSBPL7 and OSBPL11 are required for macroautophagy. (A) LC3B recruitment to Salmonella upon siRNA-mediated knockdown of OSBP, VAP, and OSBPL family members. Efficacy of siRNA-mediated knockdown was measured by RT-qPCR and depicted in Figure S4. The percentage of LC3B+ bacteria were counted in each siRNA treatment. Data represent three independent replicates. For quantification, bars represent the S.E.M. for three independent replicates and asterisks denote the following: p < 0.05 (*), p < 0.01 (**), p < 0.001 (***), p < 0.0001 (****). All 0 h treatments are not significant compared to the control siRNA-treated samples, unless otherwise indicated in the graph. (B) Cells treated with the indicated siRNA for 48 h were treated with puromycin to induce protein aggregate formation. After 4 h, the media was replaced with fresh media lacking puromycin to allow for protein aggregate clearance to begin. The clearance process proceeded for 8 h before the cells were fixed and stained for ubiquitin to identify protein aggregates. Representative images from the aggrephagy assay are shown. HeLa cells were stained for ubiquitin (green) to mark protein aggregates. Scale bar: 5 µm. (C) Aggrephagy assay results. The number of cells with over 4 ubiquitin-positive puncta were counted at 0, 4, and 12 h for three independent replicates. For quantification, bars represent the S.E.M. for three independent replicates and asterisks denote the following: p < 0.05 (*), p < 0.01 (**), p < 0.001 (***), p < 0.0001 (****). All 0 h treatments are not significant compared to the control siRNA-treated samples, unless otherwise indicated in the graph.
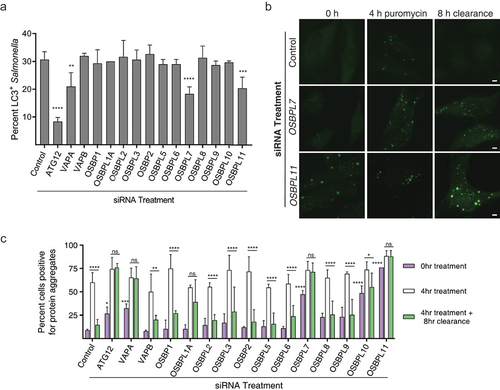
We conducted a third screen examining aggrephagy, the selective removal of protein aggregates by macroautophagy (). Puromycin treatment for 4 h induces the formation of ubiquitinated protein aggregates, and their subsequent clearance can be quantified after an 8 h chase period, as previously described [Citation42]. As with the LC3B recruitment to Salmonella assay, OSBPL7, OSBPL11, and VAPA were all found to be required for proper aggregate clearance (). OSBPL1A knock-down also impaired protein aggregate clearance, consistent with prior studies [Citation43], despite no apparent role in LC3B recruitment to Salmonella (). Notably, OSBPL7- and OSBPL11-siRNA treated cells also displayed an increase in protein aggregates under normal growth conditions (i.e. in the absence of puromycin), suggesting that they are important for basal macroautophagy of ubiquitinated proteins.
Discussion
Here we describe a proximity interactome map of human macroautophagy. Our dataset confirms many known interactions, validating its utility, and reports novel protein-protein proximity interactions, including those acting at membrane contact sites. Of note, while we performed BioID under normal growth conditions associated with “basal” (or “constitutive”) macroautophagy, we identified many factors required for macroautophagy responses to specific cellular stresses (i.e. “induced” macroautophagy). We also identified factors involved in different selective macroautophagy events. For example, TEX264 [Citation44,Citation45] and OSBPL1A [Citation43] were previously shown to be required for autophagy of cellular cargo. We propose that basal macroautophagy involves a diversity of cellular cargoes and specific recognition signals that operate to maintain cellular homeostasis and keep the macroautophagy system primed for large-scale induction. Further studies will be required to examine which factors in our dataset control different selective macroautophagy events.
We also identify OSBPL family members that are new regulators of LC3B recruitment to Salmonella, an important cellular defense mechanism against microbial infection and a powerful model to visualize selective autophagy. Prior observations have highlighted the importance of membrane contact sites in macroautophagy [Citation8,Citation40,Citation46]. Indeed, 3-dimensional electron microscopy reveals close proximity of the ER, mitochondria, vesicles, and lysosomes with the nascent phagophore [Citation47]. Membrane contact sites are known to be sites of lipid transport [Citation48–50] and in the case of macroautophagy, ATG2 was recently shown to promote phospholipid transport from the ER to the growing autophagosome [Citation29–32]. However, the role of other lipid transfer proteins in macroautophagy have not been defined. Here, we demonstrate that the OSBPL family of lipid transfer proteins are involved in autophagy as OSBPL3 and OSBPL6 are proximity interactors for all Atg8-family proteins and loss of OSBPL7 and OSBPL11 result in defective LC3 recruitment to bacteria and loss of aggrephagy. To date, only a single study has investigated OSBPL7 in detail, demonstrating that OSBPL7 regulates the degradation of GOSR1 (golgi SNAP receptor complex member 1), a protein important for ER-Golgi and intra-Golgi trafficking, by the proteasome [Citation51]. Likewise, little is known about OSBPL11, but it has been shown to act with OSBPL9 at the Golgi where together they are suggested to act as either lipid sensors or transporters important for maintaining proper trans-Golgi lipid content [Citation52]. However, for both OSBPL7 and OSBPL11, how these functions may be tied to autophagy is unclear.
In summary, we report a proximity interactome map of macroautophagy in human cells. We have identified numerous novel players with functional roles in autophagy, several of which have links to human disease. Furthermore, we identified a role for two lipid-transfer proteins, OSBPL7 and OSBPL11 in autophagy. These proteins likely complement the recently identified ATG2-mediated phospholipid transfer mechanism [Citation29–32]. Our identification of OSBPL7 and OSBPL11 as playing a role in autophagy sets the stage for future studies to unravel the importance of lipid transfer proteins in autophagy. The data presented here builds on our current understanding of autophagy interactions and will vastly improve our understanding of its mechanisms and connections with other biological processes.
Materials and methods
Oligonucleotides, plasmids and molecular biology
All primers used in this study and outsourced plasmids can be found in , respectively. OSBPL7 and OSBPL11 were N-terminally tagged with GFP using restriction cloning. OSBPL7 cDNA was graciously provided by Daoguang Yan (Jinan University, China). The PCR products were digested with HindIII and SalI (New England Biolabs, R0104 and R0138, respectively) and cloned into peGFP-C1 to construct pEGFP-C1-GFP-OSBPL7 and pEGFP-C1-GFP-OSBPL11. BioID bait constructs were cloned using AscI/NotI sites (New England Biolabs, R0558 and R0189, respectively) into the pcDNA5.1 FRT/TO FlagBirA* (N-terminal tag) vector. OSBPL9 and OSBPL10 were N-terminally tagged with GFP using Gibson Assembly (New England Biolabs) according to the manufacturer’s instructions. The OSBPL9 and OSBPL10 inserts were cloned from OSBPL9 and OSBPL10 cDNA derived from the MGC library at SPARC (SickKids, Toronto) and the backbone vector used for both constructs was pEGFP-C2 (Clontech, 6083-1). All constructs were verified by DNA sequencing (TCAG, Toronto, Canada). The UHRF1BP1 cDNA was derived from the MGC clone (SPARC, SickKids, Toronto). Macroautophagy bait CDSs have been amplified from MGC clones when available, or from HEK293 cDNAs (Genbank references are listed in Table S5). All unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Table 1. Primers used in this study
Table 2. Outsourced plasmids used in this study
Cell culture
HeLa and HEK293 cells (American Type Culture Collection, HeLa, CCL-2; HEK 293T, CRL-11268) were cultured in DMEM high glucose medium (Wisent, 319-005-CL) supplemented with 10% FBS (Wisent, 080-450) at 37°C with 5% CO2 and routinely passaged. As cell lines were obtained directly from suppliers, authentication was not performed. The cells were routinely tested for mycoplasma contamination using the e-Myco Mycoplasma test kit (Froggabio, 25235). For cellular transfection of DNA plasmids, GeneJuice (Sigma-Millipore, 709674) was used according to the manufacturer’s instructions. For siRNA transfections, Lipofectamine RNAiMax (Invitrogen, LS13778150) was used according to the manufacturer’s instructions. All siRNA strands () were transfected at a concentration of 100 nM for 48 h prior to fixation or subsequent treatment. Non-targeting siRNA and siRNA targeting ATG12 were used as negative and positive controls, respectively. Knockdown efficacy was monitored by RT-qPCR.
Table 3. siRNA sequences
Generation of stable cell lines
Stable cell line populations were created by transfecting HEK293 T-REx cells with the pcDNA5-FlagBirA*-FRT/TO constructs and the accessory plasmid pOG44 (Invitrogen, V600520) according to manufacturer’s instructions. Selection was performed using hygromycin B (Wisent, 450-141-XL) at a concentration of 200 μg/ml. For induction of the gene of interest, tetracycline was used at concentration of 1 μg/ml, and media was supplemented with biotin (50 μM; Bio Basic, BB0078). Biotinylation was generally performed for 20 h.
Bacteria strains and infections
Salmonella enterica serovar Typhimurium SL1344 and S. Typhimurium SL1344 expressing BFP from a pFPV25.1 vector under the rpsM promoter were used in this study. Late-log bacterial cultures were used for infecting HeLa cells during experiments. WT bacteria were grown for 16 h at 37°C with shaking and then sub-cultured (1:33) in LB without antibiotics for 3 h. Post subculture, bacteria were pelleted at 10,000 g for 2 min and resuspended in PBS, pH 7.2, with calcium and magnesium (Wisent, 311-420-CL). The inoculum was diluted 1:50 and added to HeLa cells at 37°C for 10 min. The cells were then washed at least 3 times with PBS with calcium and magnesium and fixed (if appropriate) at the time points indicated. Cells were maintained in DMEM with 10% FBS until 30 min, and this medium was supplemented with 100 μg/ml gentamicin (Wisent, 450-135-XL) until fixation.
Biotin-streptavidin affinity purification for mass spectrometry
Cell pellets were resuspended in 10 ml of Lysis Buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton X-100 [Bio-Rad, 1610407], 0.1% SDS, 1:500 protease inhibitor cocktail [Sigma-Aldrich, P3840], 1:1000 benzonase nuclease [Novagen, 707463]) and incubated on an end-over-end rotator at 4°C for 1 h, briefly sonicated to disrupt any visible aggregates, then centrifuged at 16,000 g for 30 min at 4°C. Supernatants were transferred to a fresh 15-ml conical tube, and 30 μl of packed, pre-equilibrated streptavidin sepharose beads (GE, 17-5113-01) were added. The mixtures were incubated for 3 h at 4°C with end-over-end rotation. Beads were pelleted by centrifugation at 1000 g for 2 min and transferred with 1 ml of Lysis Buffer to a fresh microcentrifuge tube. Beads were washed once with 1 ml Lysis Buffer and twice with 1 ml of 50 mM ammonium bicarbonate, pH 8.3. Beads were transferred in ammonium bicarbonate to a fresh centrifuge tube and washed two more times with 1 ml ammonium bicarbonate buffer. Tryptic digestion was performed by incubating the beads with 1 μg MS-grade TPCK trypsin (Promega, V5280) dissolved in 200 μl of 50 mM ammonium bicarbonate, pH 8.3, overnight at 37°C. The following morning, 0.5 μg MS-grade TPCK trypsin was added, and beads were incubated 2 additional hours at 37°C. Beads were pelleted by centrifugation at 2000 g for 2 min, and the supernatant was transferred to a fresh microcentrifuge tube. Beads were washed twice with 150 μl of 50 mM ammonium bicarbonate, and these washes were pooled with the first eluate. The sample was lyophilized and resuspended in Buffer A (0.1% formic acid). 1/5th of the sample was analyzed per MS run.
Mass spectrometry
Analytical columns (75-μm inner diameter) and pre-columns (150-μm inner diameter) were made in-house from fused silica capillary tubing from InnovaQuartz (FG75LCC) and packed with 100 Å C18–coated silica particles (Magic; Michrom Bioresources, 9996610000). Peptides were subjected to liquid chromatography (LC)-electrospray ionization-tandem mass spectrometry, using a 120 min reversed-phase (100% water–100% acetonitrile, 0.1% formic acid) buffer gradient running at 250 nl/min on a Proxeon EASY-nLC pump in-line with a hybrid LTQ-Orbitrap Velos mass spectrometer (Thermo Fisher Scientific, Waltham, MA). A parent ion scan was performed in the Orbitrap using a resolving power of 60,000, then up to the twenty most intense peaks were selected for MS/MS (minimum ion count of 1000 for activation), using standard collision induced dissociation fragmentation. Fragment ions were detected in the LTQ. Dynamic exclusion was activated such that MS/MS of the same m/z (within a range of 15 ppm; exclusion list size = 500) detected twice within 15 s were excluded from analysis for 30 s. For protein identification, Thermo .RAW files were converted to the .mzXML format using Proteowizard, then searched using iProphet against the human (Human RefSeq Version 45) database. X!Tandem search parameters were: 15 ppm parent mass error; 0.4 Da fragment mass error; complete modifications, none; cysteine modifications, none; potential modifications, +16@M and W, +32@M and W, +42@N-terminus, +1@N and Q. Each of the two biological replicates of FlagBirA*-Salmonella effector samples was analyzed using two technical replicates. Data were analyzed using the trans-proteomic pipeline (TPP), via the ProHits software suite. Proteins identified with a Protein Prophet cutoff of 0.9 and at least two unique peptides were analyzed with the SAINT express algorithm (v3.6.1). Eight control runs (consisting of Flag-BirA*only) prepared either following the BioID protocol (for the BioID analysis) were collapsed to the two highest spectral counts for each prey, and the SAINT score cutoff value was set to a BFDR < 0.01 (1% FDR). Network of high confidence proximity interactors was assembled using Cytoscape (3.6.0), and the high confidence proximity interactors detected by BioID are presented in Table S2 (with detailed control peptide counts).
Immunofluorescence
A table detailing all antibodies used in this study, including concentrations and suppliers, is available in . Cells were plated onto washed coverslips (#1 coverslips from Thermo Fisher) and left overnight to adhere. The cells were fixed with either 4% paraformaldehyde (Electron Microscope Sciences, 15710) or with −20°C methanol (Caledon Laboratories, 6700-1-40) for 20 min. The cells were blocked for 1 h using 10% FBS in PBS (Wisent, 311-010-CL) supplemented with 0.02% saponin (Calbiochem, 558255-100GM). The cells were incubated with primary and secondary antibodies for 1 h each at room temperature, with the exception of samples stained with LC3B, which were incubated overnight at 4°C. To detect specific primary antibodies, Alexa Fluor 488-, Alexa Fluor 594-, or Alexa Fluor 647-conjugated IgGs () were used as secondary antibodies at a dilution of 1:500. Cell nuclei were stained with 2 µg/mL DAPI (Invitrogen, D1306) in PBS for 10 min. Cells were mounted with Prolong Diamond Antifade Mountant (Life Technologies, P36961) For fixed cell confocal microscopy, the samples were imaged using a Quorum spinning disc confocal microscope (Leica DMI6000B inverted fluorescence microscope, Hamamatsu ORCA Flash 4 sCMOS and color camera, 63x/1.4 N oil immersion objectives, and processed using Perkin Elmer’s Volocity 6 software).
Table 4. Antibodies used in this study
Western blot
Total cell lysates were collected using Lysis Buffer (1% Triton X-100, 50 mM Tris pH 7.4, 150 mM NaCl, and 1 mM EDTA) supplemented with protease inhibitors (10 μg/ml aprotinin [BioShop, APR600.100], 10 μg/ml leupeptin [BioShop, LEU001.25], 1 μM pepstatin A [BioShop, PEP605.100], 1 mM PMSF [BioShop, PMS444.100]) and 1 mM DTT (BioShop, DTT002.50) and cell debris pelleted by spinning for 30 min at 16,000 g. Protein concentrations were determined using a BCA protein assay kit (Pierce, 23228). Protein lysate containing ~30 µg of total protein was loaded onto 12% Tris-Glycine gels (Invitrogen, XP00120BOX). Proteins were wet transferred to PVDF membrane (Bio-Rad, 1620177) for 2 h at 100 V and subsequently blocked with 5% skim milk in Tris-buffered saline with 0.1% Tween 20 (Fisher, BP337500; TBST) for 1 h. Membranes were subsequently incubated with specific antibodies () overnight at 4°C. Secondary antibodies conjugated with HRP () were used at a 1:2000 dilution. Blots were developed using the Clarity Western ECL (Bio-Rad, 1705060). Densitometry was performed on immunoblot images using ImageJ/Fiji (NIH).
Real-time quantitative polymerase chain reaction (RT-qPCR)
RNA was extracted from cells using the RNeasy kit (Qiagen, 74104) and cDNA was synthesized using the BioRad iScript cDNA Synthesis kit with 1 µg of RNA as the template. All quantitative PCR was performed using a QuantStudio 3 Real-Time PCR machine (Thermo Fisher). Data were analyzed using the QuantStudio 3 software (Thermo Fisher). The relative expression of the target genes was normalized to GAPDH transcript levels for each condition and then relative to expression in the non-targeting siRNA-treated sample. Primer sequences can be found in . No-template and no-reverse transcriptase controls were run for each primer pair to confirm the lack of primer–dimer formation/DNA contamination and genomic DNA contamination, respectively. At least three biological replicates were run per condition. All kits were conducted as per the manufacturer’s protocol.
Co-immunoprecipitations
HEK 293T cells were seeded into 10-cm diameter tissue culture dishes at approximately 25% confluency and grown at 37°C in 5% CO2. After 24 h cells were transfected with the appropriate DNA plasmid construct (6 μg DNA) using X-tremeGene 9 (Roche, 6365787001) transfection reagent as per manufacturer’s protocol for 24 h. Prior to cell lysis, where indicated, cells were starved in DMEM without amino acids and treated with 100 nM bafilomycin A1 (Cayman Chemicals, 11038-500) for 2 h. Cells were washed in PBS and lysed in Lysis Buffer (50 mM Tris- HCl, pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100) supplemented with 1 mM PMSF, 5 mM NaF, 5 mM NaVO4, 10 μg/ml aprotinin 10 mg/ml, 10 μg/ml leupeptin, and 1 μM pepstatin A. Cell lysates were incubated at 4°C for 1 h with GFP- or RFP-Trap resin (ChromoTek, gta-20 and rta-20, respectively; 15 μl packed volume), washed three times in Lysis Buffer, and eluted with 50 μl of 2x SDS-PAGE loading buffer. Samples were boiled for 10 min.
Protein aggregate clearance assay
HeLa cells were seeded in 24-well plates onto glass coverslips at a density of at 1 × 104 cells/coverslip. The following day, the cells were treated with the appropriate siRNA, as described above, for 48 h, followed by treatment with 5 µg/mL puromycin. After 4 h, the cells were washed three times with PBS and allowed to recover in regular media for 8 h. Cells were fixed at 0, 4, and 8 h time points and immunostained for polyubiquitinated proteins (FK2). At least 100 cells were counted for the presence or absence of at least 4 large ubiquitin-positive aggregates, at each time point. This protocol was adapted from Zheng et al. [Citation60].
Starvation, puromycin, rapamycin, and bafilomycin A1 treatment
HeLa cells were washed 3 times with PBS prior to treatment. For rapamycin treatment, cells with incubated with 25 µg/mL rapamycin (LKT Technologies, R0161-10MG) for 2 h and then fixed. For bafilomycin A1 treatment cells were incubated with 100 nM bafilomycin A1 for 2 h prior to fixation or harvesting for western blotting. For puromycin treatment, cells were incubated with 5 μg/ml puromycin for 2 h prior to fixation.
Statistical analysis
Statistical analyses were conducted using GraphPad Prism v6.0. The averages and S.E.M. are shown in the figures and p values were calculated using either a one-way or two-way ANOVA with Dunnett’s test as a post hoc analysis, where appropriate. For all figures, the following conventions were used: ns (p > 0.05), * (p ≤ 0.05), ** (p ≤ 0.01), *** (p ≤ 0.001), **** (p ≤ 0.0001).
Supplemental Material
Download Zip (33.1 MB)Acknowledgments
J.H.B. holds the Pitblado Chair in Cell Biology. Infrastructure for the Brumell Laboratory was provided by a John Evans Leadership Fund grant from the Canadian Foundation for Innovation and the Ontario Innovation Trust. N.M. was supported by a fellowship from the Research Training Centre at the Hospital for Sick Children. This work was supported by operating grants the Canadian Institutes of Health Research (MOP#97756; PJT#148668; FDN154329).
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The high confidence proximity interactors detected by BioID are presented in Table S2. The data has been deposited in the Mass Spectrometry Interactive Virtual Environment (MassIVE Database; https://massive.ucsd.edu/ProteoSAFe/static/massive.jsp) under accession number MSV000086586.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
Related Research Data
References
- Dikic I, Elazar Z. Mechanism and medical implications of mammalian autophagy. Nat Rev Mol Cell Biol. 2018;19:349–364.
- Rubinsztein DC, Codogno P, Levine B. Autophagy modulation as a potential therapeutic target for diverse diseases. Nat Rev Drug Discov. 2012;11:709–730.
- Maiuri MC, Criollo A, Kroemer G. Crosstalk between apoptosis and autophagy within the Beclin 1 interactome. EMBO J. 2010;29:515–516.
- Behrends C, Sowa ME, Gygi SP, et al. Network organization of the human autophagy system. Nature. 2010;466:68–76.
- Wild P, McEwan DG, Dikic I. The LC3 interactome at a glance. J Cell Sci. 2014;127:3–9.
- Jamilloux Y, Lagrange B, Di Micco A, et al. A proximity-dependent biotinylation (BioID) approach flags the p62/sequestosome-1 protein as a caspase-1 substrate. J Biol Chem. 2018;293:12563–12575.
- Nascimbeni AC, Giordano F, Dupont N, et al. ER –plasma membrane contact sites contribute to autophagosome biogenesis by regulation of local PI 3P synthesis. EMBO J. 2017;36:2018–2033.
- Hamasaki M, Furuta N, Matsuda A, et al. Autophagosomes form at ER-mitochondria contact sites. Nature. 2013;495:389–393.
- Tabara LC, Escalante R. VMP1 establishes ER-microdomains that regulate membrane contact sites and autophagy. PLoS One. 2016;11:1–20.
- Gingras AC, Abe KT, Raught B. Getting to know the neighborhood: using proximity-dependent biotinylation to characterize protein complexes and map organelles. Curr Opin Chem Biol. 2019;48:44–54.
- Gupta GD, Coyaud É, Gonçalves J, et al. A dynamic protein interaction landscape of the human centrosome-cilium interface. Cell. 2015;163:1484–1499.
- Lu Y, Zheng Y, Coyaud É, et al. Palmitoylation of NOD1 and NOD2 is required for bacterial sensing. Sci (80-). 2019;366:460–467.
- D’Costa VM, Coyaud E, Boddy KC, et al. BioID screen of Salmonella type 3 secreted effectors reveals host factors involved in vacuole positioning and stability during infection. Nat Microbiol. 2019;4:2511–2522.
- Coyaud E, Ranadheera C, Cheng D, et al. Global interactomics uncovers extensive organellar targeting by Zika Virus. Mol Cell Proteomics. 2018;17:2242–2255.
- Hua R, Cheng D, Coyaud É, et al. VAPs and ACBD5 tether peroxisomes to the ER for peroxisome maintenance and lipid homeostasis. J Cell Biol. 2017;216:367–377.
- Frendo-Cumbo S, Jaldin-Fincati JR, Coyaud E, et al. Deficiency of the autophagy gene ATG16L1 induces insulin resistance through KLHL9/KLHL13/CUL3-mediated IRS1 degradation. J Biol Chem. 2019;294:16172–16185.
- Macharia MW, Tan WYZ, Das PP, et al. Proximity-dependent biotinylation screening identifies NbHYPK as a novel interacting partner of ATG8 in plants. BMC Plant Biol. 2019;19:1–11.
- Ward RJ, Alvarez-Curto E, Milligan G. Using the Flp-InTM T-RexTM system to regulate GPCR expression. Methods Mol Biol. 2011;746:21–37.
- Yu L, Chen Y, Tooze SA. Autophagy pathway: cellular and molecular mechanisms. Autophagy. 2018;14:207–215.
- Alemu EA, Lamark T, Torgersen KM, et al. ATG8 family proteins act as scaffolds for assembly of the ULK complex: sequence requirements for LC3-interacting region (LIR) motifs. J Biol Chem. 2012;287:39275–39290.
- Yamamoto H, Kakuta S, Watanabe TM, et al. Atg9 vesicles are an important membrane source during early steps of autophagosome formation. J Cell Biol. 2012;198:219–233.
- Imai K, Hao F, Fujita N, et al. Atg9A trafficking through the recycling endosomes is required for autophagosome formation. J Cell Sci. 2016;129:3781–3791.
- Shimada K, Muhlich JL, Mitchison TJ. A tool for browsing the Cancer Dependency Map reveals functional connections between genes and helps predict the efficacy and selectivity of candidate cancer drugs. bioRxiv. 2019.
- Kato M, Han TW, Xie S, et al. Cell-free formation of RNA granules: low complexity sequence domains form dynamic fibers within hydrogels. Cell. 2012;149:753–767.
- Han TW, Kato M, Xie S, et al. Cell-free formation of RNA granules: bound RNAs identify features and components of cellular assemblies. Cell. 2012;149:768–779.
- Gomes E, Shorter J. The molecular language of membraneless organelles. J Biol Chem. 2019;294:7115–7127.
- Youn JY, Dunham WH, Hong SJ, et al. High-density proximity mapping reveals the subcellular organization of mRNA-associated granules and bodies. Mol Cell. 2018;69:517–532.e11.
- Fujioka Y, Alam JM, Noshiro D, et al. Phase separation organizes the site of autophagosome formation. Nature. 2020;578:301–305.
- Osawa T, Noda NN. Atg2: a novel phospholipid transfer protein that mediates de novo autophagosome biogenesis. Protein Sci. 2019;28:1005–1012.
- Valverde DP, Yu S, Boggavarapu V, et al. ATG2 transports lipids to promote autophagosome biogenesis. J Cell Biol. 2019;218:1787–1798.
- Osawa T, Kotani T, Kawaoka T, et al. Atg2 mediates direct lipid transfer between membranes for autophagosome formation. Nat Struct Mol Biol. 2019;26:281–288.
- Maeda S, Otomo C, Otomo T. The autophagic membrane tether ATG2A transfers lipids between membranes. Elife. 2019 Jul 4;8:e45777.
- Schütter M, Giavalisco P, Brodesser S, et al. Local fatty acid channeling into phospholipid synthesis drives phagophore expansion during autophagy. Cell. 2020;180:135–149.e14.
- Graef M. Recent advances in the understanding of autophagosome biogenesis. F1000Res. 2020;9:212.
- Murphy SE, Levine TP. VAP, a versatile access point for the endoplasmic reticulum: review and analysis of FFAT-like motifs in the VAPome. Biochim Biophys Acta - Mol Cell Biol Lipids. 2016;1861:952–961.
- Birmingham CL, Smith AC, Bakowski MA, et al. Autophagy controls Salmonella infection in response to damage to the Salmonella-containing vacuole. J Biol Chem. 2006;281:11374–11383.
- Huett A, Heath RJ, Begun J, et al. The LRR and RING domain protein LRSAM1 is an E3 ligase crucial for ubiquitin-dependent autophagy of intracellular salmonella typhimurium. Cell Host Microbe. 2012;12:778–790.
- Wild P, Farhan H, McEwan DG, et al. Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Sci (80-). 2011;333:228–233.
- Thurston TLM. The TBK1 adaptor and autophagy receptor NDP52 restricts the proliferation of ubiquitin-coated bacteria. Nat Immunol. 2009;10:1215–1221.
- Zhao YG, Liu N, Miao G, et al. The ER Contact Proteins VAPA/B Interact with Multiple Autophagy Proteins to Modulate Autophagosome Biogenesis. Curr Biol. 2018;28:1234–1245.e4.
- Stanhope R, Derré I. Making contact: VAP targeting by intracellular pathogens. Contact. 2018;1:251525641877551.
- Lystad AH, Simonsen A. Assays to monitor aggrephagy. Methods. 2015;75:112–119.
- Wijdeven RH, Janssen H, Nahidiazar L, et al. Cholesterol and ORP1L-mediated ER contact sites control autophagosome transport and fusion with the endocytic pathway. Nat Commun. 2016;7:1–14.
- An H, Ordureau A, Paulo JA, et al. TEX264 is an endoplasmic reticulum-resident ATG8-interacting protein critical for ER remodeling during nutrient stress. Mol Cell. 2019;74:891–908.e10.
- Chino H, Hatta T, Natsume T, et al. Intrinsically disordered protein TEX264 mediates ER-phagy. Mol Cell. 2019;74:909–921.
- Biazik J, Ylä-Anttila P, Vihinen H, et al. Ultrastructural relationship of the phagophore with surrounding organelles. Autophagy. 2015;11:439–451.
- Zachari M, Gudmundsson SR, Li Z, et al. Selective autophagy of mitochondria on a ubiquitin-endoplasmic-reticulum platform. Dev Cell. 2019 Sep 9;50(5):627–643.e5
- Balla T, Kim YJ, Alvarez-Prats A, et al. Lipid dynamics at contact sites between the endoplasmic reticulum and other organelles. Annu Rev Cell Dev Biol. 2019;35:85–109.
- Hanada K. Lipid transfer proteins rectify inter-organelle flux and accurately deliver lipids at membrane contact sites. J Lipid Res. 2018;59:1341–1366.
- Kentala H, Weber-Boyvat M, Olkkonen VM. OSBP-related protein family: mediators of lipid transport and signaling at membrane contact sites. Int Rev Cell Mol Biol. 2016;321:299–340.
- Zhong W, Zhou Y, Li S, et al. OSBP-related protein 7 interacts with GATE-16 and negatively regulates GS28 protein stability. Exp Cell Res. 2011;317:2353–2363.
- Zhou Y, Li S, Mäyränpää MI, et al. OSBP-related protein 11 (ORP11) dimerizes with ORP9 and localizes at the Golgi-late endosome interface. Exp Cell Res. 2010;316:3304–3316.
- Kabeya Y, Mizushima N, Ueno T, et al. LC3, a mammalian homolog of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;21:5720–5728.
- Campellone KG, Webb NJ, Znameroski EA, et al. WHAMM is an Arp2/3 complex activator that binds microtubules and functions in ER to Golgi transport. Cell. 2008;134:148–161.
- Cadwell K, Liu JY, Brown SL, et al. A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature. 2008;456:259–263.
- Goto A, Charman M, Ridgway ND. Oxysterol-binding protein activation at endoplasmic reticulum-golgi contact sites reorganizes phosphatidylinositol 4-phosphate pools. J Biol Chem. 2016;291:1336–1347.
- Xu Y, Liu Y, Ridgway ND, et al. Novel members of the human oxysterol-binding protein family bind phospholipids and regulate vesicle transport. J Biol Chem. 2001;276:18407–18414.
- Weber-Boyvat M, Kentala H, Peränen J, et al. Ligand-dependent localization and function of ORP-VAP complexes at membrane contact sites. Cell Mol Life Sci. 2015;72:1967–1987.
- Charman M, Colbourne TR, Pietrangelo A, et al. Oxysterol-binding protein (OSBP)-related protein 4 (ORP4) is essential for cell proliferation and survival. J Biol Chem. 2014;289:15705–15717.
- Zheng YT, Shahnazari S, Brech A, et al. The adaptor protein p62/SQSTM1 targets invading bacteria to the autophagy pathway. J Immunol. 2009;183:5909–5916.