
ABSTRACT
Excessive macroautophagy/autophagy is one of the causes of cardiomyocyte death induced by cardiovascular diseases or cancer therapy, yet the underlying mechanism remains unknown. We and other groups previously reported that autophagy might contribute to cardiomyocyte death caused by sunitinib, a tumor angiogenesis inhibitor that is widely used in clinic, which may help to understand the mechanism of autophagy-induced cardiomyocyte death. Here, we found that sunitinib-induced autophagy leads to apoptosis of cardiomyocyte and cardiac dysfunction as the cardiomyocyte-specific Atg7−/+ heterozygous mice are resistant to sunitinib. Sunitinib-induced maladaptive autophagy selectively degrades the cardiomyocyte survival mediator CCN2 (cellular communication network factor 2) through the TOLLIP (toll interacting protein)-mediated endosome-related pathway and cardiomyocyte-specific knockdown of Ccn2 through adeno-associated virus serotype 9 (AAV9) mimics sunitinib-induced cardiac dysfunction in vivo, suggesting that the autophagic degradation of CCN2 is one of the causes of sunitinib-induced cardiotoxicity and death of cardiomyocytes. Remarkably, deletion of Hmgb1 (high mobility group box 1) inhibited sunitinib-induced cardiomyocyte autophagy and apoptosis, and the HMGB1-specific inhibitor glycyrrhizic acid (GA) significantly mitigated sunitinib-induced autophagy, cardiomyocyte death and cardiotoxicity. Our study reveals a novel target protein of autophagic degradation in the regulation of cardiomyocyte death and highlights the pharmacological inhibitor of HMGB1 as an attractive approach for improving the safety of sunitinib-based cancer therapy.
Introduction
Cardiomyocyte death is the major pathophysiological cause of cardiovascular disease [Citation1–3] and cardiotoxicity [Citation4–6]. The suppression of cardiomyocyte death is the logical therapeutic strategy, which requires an understanding of the responsible signaling pathway. Excessive autophagy is one of the known causes of cardiomyocyte death as cardiomyocyte death is attenuated by inhibiting the activation of autophagy in mouse models of ischemia/reperfusion [Citation7], pressure overload [Citation8], and doxorubicin-induced cardiomyopathy [Citation9]. However, other studies have also supported the notion that autophagy is protective in the heart at baseline and in response to stress [Citation10–12]. Thus, the activity of autophagy must be precisely regulated to maintain cardiac homeostasis. In general, cardiomyocyte death occurs when autophagy is activated in excess [Citation13]. Autophagic degradation of critical molecules involved in cell survival, such as Bruce, in flies [Citation14] and the antioxidant catalase in fibroblasts [Citation15], is the leading cause of cell death. Therefore, the identification of crucial inducers of cardiomyocyte autophagy and key protein(s) that undergo the autophagic degradation is essential for the deep understanding of the pathophysiological mechanism of cardiovascular diseases and drug-induced cardiotoxicity.
Clinical data showed that patients who received sunitinib, a small molecular multikinase inhibitor, used as the first-line therapy for various types of cancers in clinic [Citation16,Citation17], developed heart failure (HF) and this treatment resulted in substantial morbidity and, in some cases, mortality through longitudinal cardiac surveillance including serial assessment of left ventricular (LV) ejection fraction (EF), plasma creatine kinase myocardial band (CK-MB) and TNNT2/CTNT (troponin T2, cardiac) level. It was not completely reversible in most patients, even after termination with sunitinib, which limited its clinical application [Citation18–21]. We and other groups previously reported that both autophagy activation and inhibition could contribute to the death of rat H9c2 cardiomyocytes or pericytes upon treatment of sunitinib [Citation22,Citation23]. However, precise evaluation of cardiomyocyte autophagic flux upon sunitinib treatment is lacking. Endomyocardial biopsies obtained from patients treated with sunitinib who developed LV dysfunction also showed lysosomal precipitates by transmission electron microscopy [Citation18]. These findings prompted us to further identify the precise effect of sunitinib on autophagic flux and the role of autophagy in sunitinib-induced cardiotoxicity, which would make it possible to identify the potential causes of cardiomyocyte death by excessive autophagy.
HMGB1 (high mobility group box 1), a DNA-binding nuclear protein, is important for orchestrating autophagic responses [Citation24]. Increased cytoplasmic or nuclear expression of HMGB1 promotes autophagy progression via different pathways [Citation24,Citation25]. Moreover, we and other independent groups have identified that HMGB1 serves as a critical mediator in myocardial ischemia/reperfusion injury [Citation26], heart failure [Citation27] and anthracycline-induced cardiomyopathy [Citation9]. We previously found that sunitinib could induce HMGB1 nucleus-to-cytoplasm shift in cancer and noncancerous cells and promote the autophagic degradation of TP53/p53 [Citation28]. However, whether HMGB1 plays functions in bridging sunitinib-induced autophagy, cardiomyocyte death and cardiotoxicity is still unknown.
In the present study, we found that sunitinib promoted the left ventricular dysfunction and cardiac injury in mice. We provided evidence that sunitinib-induced cardiomyocyte autophagy was detrimental in vivo by blocking autophagy using cardiomyocyte specific heterozygous Atg7-deficient mice. We identified CCN2, a key protein in the heart, that underwent autophagic degradation in an endosome-dependent manner upon sunitinib treatment and knockdown of Ccn2 by adeno-associated virus serotype 9 (AAV9) significantly reduced the survival of cardiomyocyte and cardiac function, suggesting that the degradation of CCN2 is one of the causes of autophagy-triggered cardiomyocyte apoptosis. Notably, HMGB1 enrichment was associated with sunitinib-induced autophagy. In addition, inhibiting HMGB1 expression through deletion or use of the pharmacological inhibitor glycyrrhizic acid (GA) could significantly alleviate sunitinib-induced cardiomyocyte autophagy, death and cardiotoxicity, making it a potential therapeutic target against cardiotoxicity.
Results
Sunitinib causes the death of cardiomyocyte and cardiac injury
Cardiovascular toxicity is the most pressing issue related to the clinical application of sunitinib in anticancer treatment [Citation19,Citation23]. Our previous in vitro study showed that sunitinib treatment led to autophagy and subsequent death of cardiomyocyte. CCC-HEH-2, a human embryonic cardiac tissue-derived cell line, was applied to detect the death of cardiomyocyte. We also further confirmed these results using primary neonatal rat ventricular myocytes (NRVMs) and adult mouse cardiomyocytes (MCMs). Sunitinib decreased the survival fraction of CCC-HEH-2, MCMs, H9c2 and NRVMs (). Sunitinib significantly enhanced the expression of c-PARP (cleaved poly (ADP-ribose) polymerase family) and cleaved (c)-CASP3 (caspase 3), markers of apoptosis, as detected by western blot of CCC-HEH-2 and MCMs cells (, C), S1(A, B)). Next, DAPI staining of the nuclei showed nuclear condensation in sunitinib-treated MCMs and CCC-HEH-2 cells ( and S1C), indicating the occurrence of apoptosis. Propidium iodide (PI) staining with flow cytometry was applied to detect the apoptosis rate. Sunitinib treatment significantly promoted the apoptosis of CCC-HEH-2 cells in a time-dependent manner (). In addition, 1.25 and 2.5 µM sunitinib slightly increased the apoptosis rate of CCC-HEH-2 cells but without significance, and 5 or 10 µM sunitinib dramatically increased the apoptosis rate to approximately 80% (Figure S1D). The pancaspase inhibitor Z-VAD-FMK apparently rescued the death and apoptosis of CCC-HEH-2 cells caused by sunitinib ( and S1E). These results indicated that sunitinib promoted cardiomyocyte apoptosis, which might be the key reason for its cardiotoxicity.
Figure 1. Sunitinib induces cardiomyocyte apoptosis and cardiotoxicity. (A) The results from the SRB staining assay showed the effect of sunitinib on the survival of CCC-HEH-2, MCMs, H9c2 and NRVMs cells. Three independent experiments were performed. (B-F) Sunitinib induces apoptosis of myocardial cells. (B) CCC-HEH-2 cells were treated with sunitinib (5 µM) for 0, 12, 24, 36 or 48 h. Total cell lysates were used to detect the expression levels of c-PARP, and c-CASP3. Western blot was repeated for three times and densitometric analysis was carried out. (C) Representative immunoblots of apoptosis proteins in MCMs cells stimulated with sunitinib in a concentration-dependent manner. Western blot was repeated at least three times and densitometric analysis was carried out. (D) DAPI staining showed cardiomyocyte nuclear condensation after sunitinib treatment. Scale bars: 10 µm. (E) Apoptotic cells were quantified by flow cytometry with PI staining after 12, 24, 36, or 48 h of exposure to 5 µM sunitinib. Four independent experiments were performed. (F) CCC-HEH-2 cells were treated with 20 μM Z-VAD-FMK, 5 µM sunitinib or both for 24 h, and then cell images were obtained by microscopy. Scale bars: 100 µm. (G-H) Sunitinib induces cardiotoxicity in vivo. Nude mice transplanted with 786-O human xenografts were randomly divided into 2 groups (n = 6 per group, 3 males and 3 females) and treated with saline or sunitinib (40 mg/kg) for 32 days. Heart tissues were harvested after the mice were sacrificed, and total tissue lysates were prepared. (G) Heart sections were stained with H&E. Scale bars: 100 µm. (H) Immunofluorescence assay was performed to show the apoptosis of myocardium caused by sunitinib. The heart sections were stained with c-CASP3 (green), ACTN2 (red) and DAPI (blue). Scale bar: 50, 25 µm. The p value was calculated by one-way ANOVA (Dunnett’s multiple comparisons tests). NS: no significance, *: p< 0.05, **: p< 0.01 and ***: p < 0.001. Abbreviations: SUNI, sunitinib; MCMs, adult mouse cardiomyocyte; NRVMs, neonatal rat ventricular myocytes.
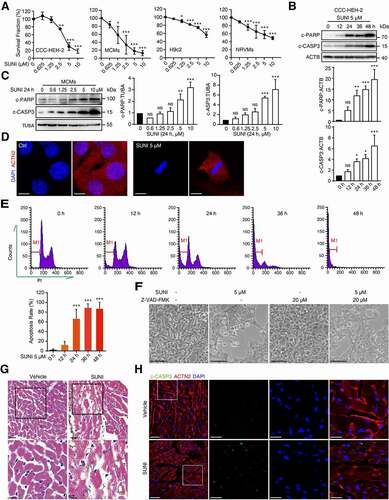
To gain clearer insights into cardiomyocyte death and toxicity, we assessed the relationship between the cardiotoxicity and anticancer activity of sunitinib in vivo to mimic the clinical occurrence. We developed a mouse xenograft model of renal cell carcinoma, which is one of the clinical indications of sunitinib. Consistent with reports from the clinic, we found that sunitinib inhibited tumor growth (Figure S1F), accompanied with increased level of serum CK-MB and TNNT2 (Figure S1G), indicating elevated cardiac injury. Histological analysis of sunitinib-treated mouse hearts by hematoxylin and eosin (H&E) staining identified the loss of cytoplasm (cytoplasmic vacuolization) and disorganization of the myofibrils, suggesting the existence of cardiomyocyte death, which was defined as myocardial injury (). To confirm cardiomyocyte apoptosis post sunitinib treatment in the heart, confocal microscopy after c-CASP3 and ACTN2/α-actinin (a marker of cardiomyocytes) costaining was performed and we found that hearts after sunitinib treatment showed more c-CASP3+ cells, which were also ACTN2 positive (), suggesting that sunitinib promoted apoptosis of cardiomyocyte in the heart. The mRNA expression of Nppa (natriuretic peptide type A), Myh6 (myosin, heavy polypeptide 6, cardiac muscle, alpha), and Myh7 (myosin, heavy polypeptide 7, cardiac muscle, beta) was slightly but not significantly increased by sunitinib (Figure S1H). However, the mRNA level of Nppb (natriuretic peptide type B) was significantly increased (Figure S1H), indicating that sunitinib caused slight cardiac remodeling. Wheat germ agglutinin staining revealed that sunitinib failed to cause cardiac hypertrophy (Figure S1I). We also found some collagen deposition in heart sections from sunitinib-treated mice using Sirius Red and Masson’s trichrome staining, indicating that sunitinib caused cardiac fibrosis (Figure S1(J, K)). Taken together, our in vivo study demonstrated that sunitinib treated mice showed slight cardiotoxicity when it exerted anti-cancer activity.
Maladaptive autophagy promotes the cardiomyocyte apoptosis and cardiotoxicity of sunitinib
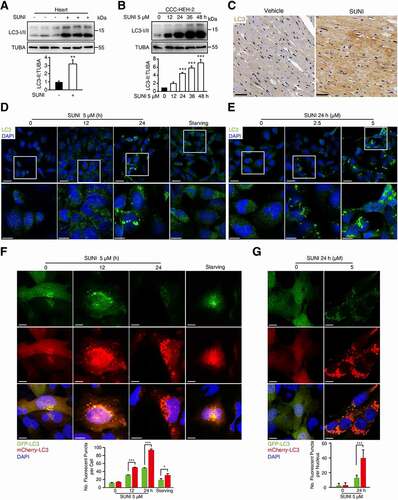
Since we and other groups have verified that autophagy promotes sunitinib-induced cardiomyocyte death in vitro [Citation22,Citation23], we next tested the activation of autophagy in the heart tissues of sunitinib-treated mice. The level of MAP1LC3/LC3-II, an acknowledged marker of autophagic activity [Citation29], was predominantly upregulated in response to sunitinib treatment in vivo (). Furthermore, we performed LC3-II expression analysis to detect autophagic activation over a time course and at different concentrations of sunitinib treatment in vitro. LC3-II expression in CCC-HEH-2 and MCMs cells was markedly increased in a time- and concentration-dependent manner after sunitinib treatment (, S2(A, B)). Immunohistochemistry and immunofluorescence microscopy also found increased accumulation of LC3 puncta in sunitinib-treated hearts or cardiomyocyte (). Notably, the accumulation of LC3-II is a marker for both the induction and inhibition of autophagy [Citation30]. Hence, we measured sunitinib-induced autophagic flux compared with a typical autophagy inducer, starvation, to titrate the autophagic response by infecting cardiomyocytes with Ad-mCherry-GFP-LC3 virus as described in our previous report [Citation31]. As shown in , G) and S2C, more red-fluorescent puncta (autolysosomes) were observed after sunitinib treatment than green-fluorescent puncta, implying autophagy induction. Under sunitinib stimulation, the percentage of red-fluorescent puncta was much higher than that under starvation, indicating the excessive activation of autophagy. Interestingly, we found that the protein level of SQSTM1/p62 (sequestosome 1), another marker of autophagic activity, increased upon sunitinib treatment (Figure S2(D-E)), which might be due to the feedback of increased mRNA (Figure S2F).
Figure 2. Sunitinib activates autophagy of cardiomyocyte. (A) Total heart tissue lysates from nude mice treated with saline or sunitinib (40 mg/kg, n = 3 per group) for 32 days were analyzed by western blots using an anti-LC3 antibody. Densitometric analysis was carried out. (B) CCC-HEH-2 cells were treated with 5 µM sunitinib for different periods of time, as indicated (between 0 and 48 h), and the expression of LC3-II was analyzed by western blot using TUBA as a loading control. Western blot was repeated for three times and densitometric analysis was carried out. (C) Heart sections were stained with LC3 antibody by immunohistochemistry. Scale bars: 25 µm. (D-E) CCC-HEH-2 cells were treated with sunitinib (0, 2.5 or 5 µM) for 24 h or sunitinib (5 µM) for 12 or 24 h or starving for 2 h. LC3 was detected by immunofluorescence staining. Scale bars: 10 µm (upper); 5 µm (lower). (F) CCC-HEH-2 cells expressing mCherry-GFP-LC3 were treated with 5 µM of sunitinib for different periods of time (0, 12 and 24 h). Autophagic flux assays was performed. (G) MCMs expressing mCherry-GFP-LC3 were treated with 5 µM sunitinib for 24 h and autophagic flux was detected. (F-G) Scale bars: 10 µm. Bottom: quantification of the percentage of mCherry-positive, GFP-positive puncta per nucleus (n = 100 cells pooled from three independent experiments). Data are presented as mean ± SD. The p value was calculated by Student’s t-test (unpaired, two-tailed, 2 groups). *: p < 0. 05, **: p < 0. 01 and ***: p < 0.001. Abbreviations: SUNI, sunitinib; MCMs, adult mouse cardiomyocyte.
Next, we evaluated the role of autophagy in sunitinib-induced cardiomyocyte apoptosis and cardiotoxicity. ATG5 (autophagy related 5) and ATG7 (autophagy related 7) are both essential for ATG conjugation systems and autophagosome formation [Citation32,Citation33]. Thus, we applied siRNA targeting ATG7 or ATG5 to silence the expression of ATG7 or ATG5 gene. We found that sunitinib-induced increased expression of c-PARP, c-CASP3 and LC3-II (, B)) and cell death (), was reversed in ATG7- or ATG5-silenced cells, suggesting that inhibition of autophagy led to the rescue of cardiomyocyte apoptosis, supporting the hypothesis that cardiomyocyte autophagy contributes to apoptosis.
Figure 3. Autophagy contributes to the cardiotoxicity of sunitinib. (A-C) Inhibition of autophagy rescues the apoptosis of cardiomyocyte. CCC-HEH-2 cells were transfected with nontargeting siRNA (NC) or siRNA targeting ATG7 or ATG5, and then treated with or without sunitinib for 24 h. (A-B) The expression of ATG7 or ATG5, LC3-I/II and c-CASP3 was detected by western blot, using TUBA as a loading control. Western blot was repeated three times and densitometric analysis was carried out. (C) Cell morphology was obtained by microscope. Scale bars: 100 µm. (D) C57BL/6 mice with heterozygous cardiac-specific Atg7 knockout (Atg7−/+) were generated by crossing Atg7-floxed C57BL/6 mice (Atg7flox/flox, Atg7f/f) with Myh6-MerCreMer transgenic C57BL/6 mice and then treated with 25 mg/kg tamoxifen for 3 days. 2 weeks after induction, Atg7f/+ and Atg7−/+ mice were treated with saline or sunitinib (40 mg/kg) for 42 days (n = 5 per group) by means of intragastric administration. Heart tissues were harvested after the mice were sacrificed, and total tissue lysates were prepared. (E) The deletion efficiency was determined by detecting the levels of ATG7 in total heart tissue lysates using GAPDH as a loading control. (F) The protein expression levels of ATG7 and LC3-I/II were analyzed in lysates from total heart tissues from 3 mice of each group. Densitometric analysis was carried out (G) Heart weight to body weight ratio. (H) Serum was analyzed for CK-MB and TNNT2 levels. (I-J) Cardiac function was examined by echocardiography, and the percentage of Ejection Fraction (EF) and Fractional Shortening (FS) was shown. n = 5. (K) Heart sections were stained with H&E. Scale bars: 100 µm. The p value was calculated by one-way ANOVA and the Sidak test. *: p < 0.05, **: p< 0.01, ***: p < 0.001, #: p < 0.05, ##: p < 0.01 and ###: p < 0.001. Abbreviations: SUNI, sunitinib; HW, heart weight; BW, body weight; CK-MB, creatine kinase myocardial band; TNNT2, troponin T2, cardiac.
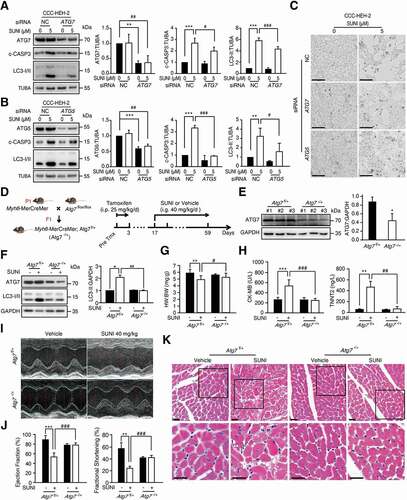
Then, we examined the role of autophagy in sunitinib-induced cardiotoxicity in vivo. Cardiomyocyte-specific Atg7-deficient mice were used to inhibit autophagy induction and progression. Considering that cardiomyocyte-specific knockout of Atg7 in mice could cause severe contractile dysfunction, myofibrillar disarray and vacuolar degeneration of cardiomyocyte [Citation34,Citation35], heterozygous Atg7-deficient (Atg7−/+) mice were applied as they showed normal cardiac function and intermediate autophagy activity. Atg7flox/flox (referred to as atg7f/f) mice were crossed with Myh6-MerCreMer mice to enable inducible knockdown of Atg7 in adult hearts (). After tamoxifen induction, western blot analysis confirmed the specificity of Atg7 knockdown in hearts compared to liver, lung, kidney or spleen from Atg7−/+ mice ( and S3A), which was further confirmed by immunofluorescence assay in heart sections (Figure S3B). As shown in , the sunitinib-induced increase in LC3-II was downregulated by Atg7 disruption, implying that the conversion from LC3-I to LC3-II was blocked, suggesting that sunitinib-induced autophagy was significantly reduced. Atg7 heterozygous disruption alleviated the sunitinib-induced decrease in the heart weight (HW):body weight (BW) ratio (). Meanwhile, we also found that inhibiting cardiomyocyte autophagy could reduce sunitinib-induced increases in serum CK-MB and TNNT2 levels (). Applying echocardiography to monitor cardiac function, we observed a trend toward a decreased Ejection Fraction (EF) and Fractional Shortening (FS) in Atg7f/+ mice treated with sunitinib (), which reflected LV dysfunction. As expected, sunitinib-induced impairment of LV function could be ameliorated by Atg7 heterozygous disruption (). Histological analysis revealed markedly reduced characteristics of heart injury in Atg7−/+ mice compared to Atg7f/+ mice under sunitinib treatment (). Thus, these data lend support for a detrimental role of maladaptive autophagy in sunitinib-induced cardiotoxicity in vivo.
The reduction in CCN2 contributes to autophagy-dependent cardiotoxicity
It has been reported that lethal autophagic death might be attributed to the excessive elimination of mitochondria or survival-related proteins [Citation36,Citation37]. Thus, we first detected whether mitochondrial damage occurred in sunitinib-induced cardiotoxicity. As shown in Figure S4A, the mitochondrial membrane potential (MMP) of cardiomyocyte was not affected by sunitinib treatment. We also noticed that sunitinib showed minimal effects on the levels of TOMM20 (translocase of outer mitochondrial membrane 20; a major mitochondrial import receptor [Citation38]) and HSPD1/HSP60 (heat shock protein family D (Hsp60) member 1; Figure S4B), which are implicated in mitochondrial protein import and macromolecular assembly [Citation39]. These data suggested that mitochondrial damage and degradation might not be involved in the autophagy-promoted cardiotoxicity of sunitinib. Therefore, we speculated that autophagy might evoke the degradation of the important intracellular proteins and, in turn, promote cardiomyocyte death.
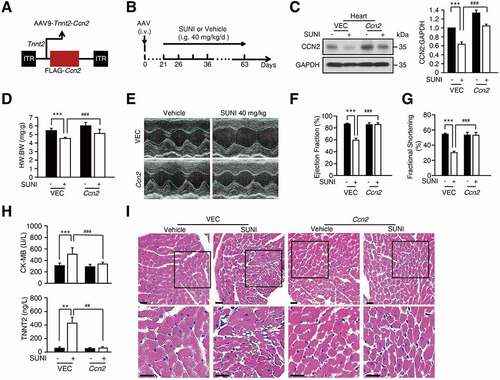
To identify the target proteins of autophagy, we used pepstatin A and E-64d to block lysosomal degradation and analyzed the enriched proteins in lysosomes using mass spectrometry (MS). We found that approximately 60 proteins were significantly upregulated by ≥ 2-fold in sunitinib-, pepstatin A-, and E-64d-treated cardiomyocytes compared with vehicle-treated cardiomyocytes (Table S1). Among the upregulated proteins, we observed that the autophagy cargo receptors SQSTM1, NBR1 (NBR1 autophagy cargo receptor) and TOLLIP (toll interacting protein) were enriched in sunitinib-, pepstatin A-, and E-64d-treated cells, indicating that the strategy is feasible. We noticed that the level of the cardiomyocyte survival factor CCN2 increased 5-fold in lysosomes from sunitinib-, pepstatin A-, and E-64d-treated cells compared with Ctrl cells (Table S1). These results implied that sunitinib might promote the aggregation of CCN2 in lysosomes for autophagic degradation. Thereafter, we validated this hypothesis by western blot and immunofluorescence assays and found that sunitinib treatment reduced CCN2 protein in CCC-HEH-2 cells ( and S4C), heart tissues (), H9c2 (Figure S4D), NRVMs (Figure S4E) and MCMs (Figure S4F-G). The reduction in CCN2 protein levels was positively related to the survival fraction of cardiomyocytes (), which is consistent with the implication of CCN2 in cell survival [Citation40,Citation41].
Figure 4. Maladaptive autophagy promotes the cardiotoxicity of sunitinib via degradation of CCN2. (A) CCC-HEH-2 cells were treated with 5 µM sunitinib for different periods of time, as indicated (between 0 and 48 h). (B) CCC-HEH-2 cells were treated with 24 h of sunitinib for different concentrations, as indicated (between 0 and 12.5 µM). (A-B) The expression of CCN2 was measured by western blot. Western blot was repeated for three times independently and densitometric analysis was carried out. (C) Total heart tissue lysates from saline and sunitinib treated mice were analyzed by western blot using an anti-CCN2 antibody. TUBA was used as a loading control. Heart tissues from 3 mice of each group were used and densitometric analysis was carried out. (D) Immunocytochemistry for CCN2 and nuclear staining by DAPI in CCC-HEH-2 cells. CCC-HEH-2 cells were treated with or without 5 µM sunitinib for 24 h. The expression of CCN2 was measured by immunofluorescence assay. Scale bar: 50 µm (upper) and 20 µm (lower). (E-F) CCC-HEH-2 cells were transfected with CCN2 shRNA (#1, #2) and vector (Vsh) lentivirus and then collected 72 h later. (E) Representative western blot showing the effect of CCN2 knockdown on the apoptosis of CCC-HEH-2 cells. (F) Cells were harvested and stained with ANXA5 and PI, apoptosis rates were analyzed by flow cytometry and representative images are shown. The experiments were performed four times independently, and data are expressed as the mean ± SD. (G-L) AAV9-Tnnt2-Ccn2-null or AAV9-Tnnt2-NC virus was injected into C57BL/6 mice through the tail vein to specifically knockdown the expression of Ccn2 in the heart (n = 9), and the hearts were collected 4 weeks later. (G) Immunofluorescence assays were performed to detect the knockdown efficiency of Ccn2 in heart tissue achieved by AAV. Scale bars: 50 µm. (H) Heart weight to body weight ratio was calculated. (I-J) Cardiac function was examined by echocardiography. (J) Quantification of EF, FS and the rates of heartbeats. (K) Heart tissues from the mice were collected, and were stained with H&E for histopathological analysis. Scale bars: 50 µm. (L) Sirius Red staining was performed to detect fibrosis in heart tissue. Scale bars: 100 µm. The p value was calculated by Student’s t-test (unpaired, two-tailed, 2 groups) or by one-way ANOVA and Dunnett’s multiple comparisons test (multiple groups). *: p < 0.05, **: p < 0.01 and ***: p < 0.001. Abbreviations: SUNI, sunitinib; HW, heart weight; BW, body weight.
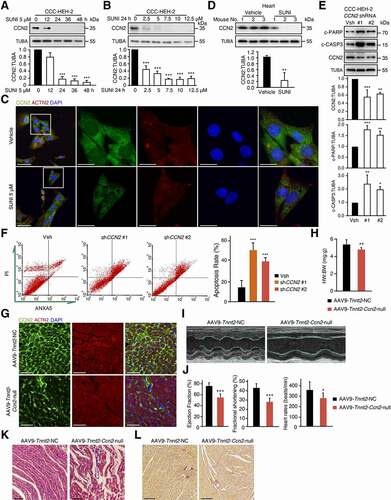
To demonstrate the relationship between the reduction in CCN2 and autophagy-dependent cardiotoxicity, we first confirmed the role of CCN2 reduction in cardiomyocyte death. Cardiomyocyte were infected with two shRNAs with different sequences targeting CCN2. Knockdown of CCN2 increased the protein levels of c-PARP and c-CASP3, and led to CCC-HEH-2 cell death ( and S4H), but failed to affect LC3 levels (Figure S4I), suggesting that the level of CCN2 was related to cell apoptosis and was not the cause of autophagy. Next, ANXA5/Annexin V-PI staining followed by flow cytometry was applied to detect the apoptosis rate. We found that knockdown of CCN2 significantly increased the apoptosis rate of CCC-HEH-2 cells (). Inhibition of the apoptosis by Z-VAD-FMK did not reverse the reduction in CCN2 (Figure S4J), further suggesting that decreased CCN2 is the cause of cardiomyocyte death.
Next, we evaluated the role of CCN2 reduction in sunitinib-induced cardiotoxicity by knocking down Ccn2 specifically in heart by applying adeno-associated virus serotype 9 (AAV9) vectors carrying the Tnnt2 promoter with siRNA targeting Ccn2 (Figure S4K). As shown in , Ccn2 expression in heart sections is almost gone in AAV9-Tnnt2-Ccn2-null-injected mice. Knockdown of Ccn2 led to a decrease in HW:BW ratio (). In addition, the EF, FS and heart rate were significantly reduced by knockdown of Ccn2 (), indicating LV dysfunction. H&E and Sirius Red staining showed the myofibril disorganization and slight fibrosis in AAV9-Tnnt2-Ccn2-null-injected mice () as seen in mice treated with sunitinib. We also found that knockdown of Ccn2 increased c-CASP3 levels in heart sections, as detected by immunofluorescence assay (Figure S4L). Taken together, our findings suggested that CCN2 is required for cardiac function and homeostasis.
We then validated this conclusion by overexpression of Ccn2 using AAV9-Tnnt2-Ccn2 virus (). Western blot assays of heart tissues suggested that overexpression of Ccn2 by the AAV9-Tnnt2-Ccn2 virus alleviated sunitinib-induced CCN2 reduction (). Overexpression of Ccn2 inhibited sunitinib-induced decreases in the HW:BW ratio (). In addition, the reduced EF and FS were rescued by overexpression of Ccn2 (), suggesting the alleviation of LV dysfunction. The increased serum level of CK-MB and TNNT2 induced by sunitinib was reversed by AAV9-Tnnt2-Ccn2 delivery (). Histological analysis revealed that heart injury were moderated in AAV9-Tnnt2-Ccn2-injected mice exposed to sunitinib compared with AAV9-vector-injected mice treated with sunitinib (). These results suggested that in this context, autophagy-promoted cardiotoxicity initiated by sunitinib mainly depended on the downregulation of CCN2.
Figure 5. Overexpression of Ccn2 rescues the cardiotoxicity caused by sunitinib. (A) Schematic representation of AAV9-Tnnt2-Ccn2 used in this study. (B) AAV9-Tnnt2-Ccn2 or AAV9-Tnnt2-VEC virus was injected into C57BL/6 mice through the tail vein to specifically reduce the expression of CCN2 in the heart, which was followed by saline or sunitinib (40 mg/kg) treatment for 42 days by means of intragastric administration (n = 5). (C) Representative western blots showing CCN2 expression in heart tissue lysates. GAPDH was used as a loading control. Total heart tissues from 3 mice of each group were used and densitometric analysis was carried out. (D) Heart weight to body weight ratio. (E-G) Cardiac function was examined by echocardiography. (H) Serum was analyzed for CK-MB and TNNT2 levels. (I) Heart sections were stained with H&E. Scale bars: 100 µm. The p value was calculated by one-way ANOVA (Dunnett’s multiple comparisons test). **: p < 0.01, ***: p < 0.001, ##: p < 0.01 and ###: p < 0.001. Abbreviations: AAV9, adeno-associated virus 9; SUNI, sunitinib; VEC, vector; HW, heart weight; BW, body weight; CK-MB, creatine kinase myocardial band; TNNT2, troponin T2, cardiac.
Sunitinib-induced autophagy promotes the degradation of CCN2
To define the mechanism of sunitinib-mediated downregulation of CCN2, we then examined whether sunitinib suppressed CCN2 transcriptional levels using quantitative real-time PCR (qPCR) analysis. No significant difference in the mRNA level of CCN2 was observed after sunitinib treatment (Figure S5A). Subsequently, we applied an inhibitor of protein synthesis, cycloheximide (CHX), to detect CCN2 protein turnover [Citation42]. The results showed that sunitinib treatment resulted in a shortening of the CCN2 half-life () confirming that the reduction in CCN2 protein levels was due to increased degradation.
Figure 6. Sunitinib promotes the degradation of CCN2 protein via the autophagy-lysosome pathway. (A) CCC-HEH-2 cells were treated with 10 µg/mL CHX and in the presence or absence of 5 µM sunitinib for different times, and CCN2 protein levels were measured by western blot. The half-life of CCN2 was detected. The CCN2 expression levels from biological triplicates were normalized with ACTB and quantified. (B) Representative western blots showing the role of autophagy-lysosome and proteasome on the degradation of CCN2 in CCC-HEH-2 cells. Cells were treated with 5 µM sunitinib for 24 h in the absence or presence of 10 µM CQ, or in the absence or presence of 20 µM MG132. The experiments were performed three times independently, and the CCN2 expression levels were normalized with TUBA and quantified (lower panel). (C) Representative western blots showed the effect of ATG5 knockdown on the sunitinib-induced degradation of CCN2 in CCC-HEH-2 cells. Western blot was repeated at least three times and densitometric analysis was carried out. (D) Levels of ATG7 and CCN2 in total heart tissue lysates were analyzed by western blots using GAPDH as a loading control. Total heart tissues from 3 mice of each group were used and densitometric analysis was carried out. (E) The colocalization of CCN2 with LC3 in untreated or sunitinib-treated MCMs cells. The cells were stained for CCN2 (green), LC3 (red) and DAPI (blue). Cells were treated with sunitinib for 24 h in the absence or presence of CQ for 2 h prior to the addition of sunitinib. Scale bars: 20 µm. (F) CCC-HEH-2 cells were transfected with NC or siRNA targeting TOLLIP. The expression levels of TOLLIP, CCN2 and GAPDH were determined after 24 h of exposure. Western blot was repeated at least three times and densitometric analysis was carried out. (G) CCC-HEH-2 cells were treated with sunitinib plus CQ. The cell lysates were subjected to immunoprecipitation using an antibody against CCN2, followed by western blots to detect CCN2 and TOLLIP expression. Western blot was repeated at least three times and densitometric analysis was carried out. (H) The cellular distribution of CD63 and CCN2 in CCC-HEH-2 cells was observed by immunofluorescence. The cells were stained for CCN2 (green), CD63 (red) and DAPI (blue). CCC-HEH-2 cells were treated with sunitinib for 24 h in the absence or presence of CQ for 2 h prior to the addition of sunitinib. Scale bars: 10 µm. The p value was calculated by one-way ANOVA (Dunnett’s multiple comparisons test or Sidak test). NS: no significance, **: p < 0.01, ***: p < 0.001 and #: p < 0.05. Abbreviations: SUNI, sunitinib; NC, nontargeting siRNA; WCL, whole cell lysate; CTRL, control; CQ, chloroquine; MCMs, adult mouse cardiomyocyte.
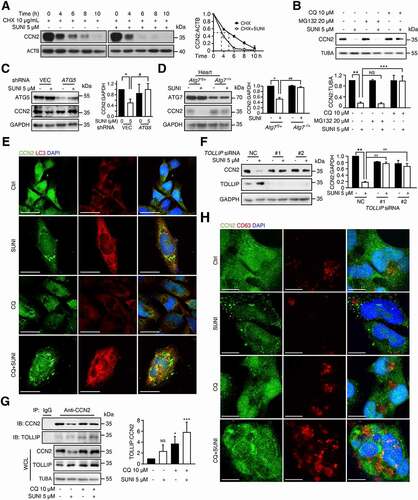
In addition to the autophagy-lysosome pathway, intracellular protein levels are also modulated by the ubiquitin-proteasome system [Citation43]. Therefore, we examined the roles of both systems in sunitinib-induced CCN2 degradation. Suppression of the proteasome degradation pathway by MG132, a specific proteasome inhibitor, failed to inhibit sunitinib-induced CCN2 degradation, whereas blockade of lysosomal function by chloroquine (CQ) completely reversed the sunitinib-induced decrease in CCN2 expression (). These results further indicated that lysosomes rather than proteasomes were required for sunitinib-induced CCN2 degradation. In addition, to further confirm the role of autophagy in the degradation of CCN2, ATG5 shRNA was applied to inhibit autophagy initiation and we found that knockdown of ATG5 reversed the reduction in CCN2 (). In addition, western blot analysis of heart tissues from sunitinib-treated Atg7f/+ and Atg7−/+ mice showed that Atg7 knockdown mitigated the downregulation of CCN2 (). Immunofluorescence assays showed that CCN2 colocalized with LC3 around the nuclei in CCC-HEH-2 cells upon sunitinib-plus-CQ treatment, suggesting that CCN2 was enclosed in autophagosomes ().
Emerging evidence has established that the binding of target proteins to autophagy cargo receptors is required for their selective degradation in lysosomes [Citation43]. Thus, we profiled autophagy cargo receptors with MS data to further confirm the autophagic degradation of CCN2. As mentioned before, the well-known autophagy cargo receptors SQSTM1, NBR1 and TOLLIP were enriched 5.0-, 4.0- and 2.2-fold in lysosomes from sunitinib-, pepstatin A-, and E-64d-treated treated cells compared with Ctrl cells, respectively (Table S1), suggesting that these proteins are candidates for autophagy cargo receptors for CCN2 degradation. However, siRNA-mediated SQSTM1 or NBR1 knockdown failed to inhibit sunitinib-induced CCN2 degradation (Figure S5(B, C)). Only TOLLIP knockdown notably inhibited sunitinib-induced CCN2 autophagic degradation (), suggesting that TOLLIP might be the autophagic cargo receptor for CCN2 degradation in lysosomes. To address this hypothesis, immunofluorescence assays were performed and showed that CCN2 could colocalize with TOLLIP around the nuclei in CCC-HEH-2 cells upon sunitinib- plus- CQ treatment (Figure S5D). Furthermore, we performed immunoprecipitation to test the interaction between endogenous CCN2 and TOLLIP, and found that sunitinib also enhanced the interaction between CCN2 and TOLLIP (), suggesting the involvement of TOLLIP in sunitinib-induced autophagic degradation of CCN2.
It has been reported that TOLLIP is essential for coordinating the endosomal compartment and mediating endocytic trafficking of ubiquitinated cargoes [Citation44–46], suggesting that CCN2 might be degraded by endosome-related autophagy. Therefore, we investigated whether endosomes were involved in sunitinib-induced CCN2 degradation. Immunofluorescence assays of CD63, a multivesicular body marker, found that sunitinib decreased the expression of CD63 and promoted the formation of large puncta of CD63 (Figure S5E), indicating that sunitinib might promote the clearance of late endosome. We also observed that the fraction of CCN2 and CD63 were colocalized (), further suggesting that CCN2 might be degraded by the TOLLIP-mediated endosome-related autophagy pathway, which led to cardiomyocyte apoptosis and cardiac dysfunction.
Accumulated nuclear HMGB1 is essential for sunitinib-activated autophagy and cardiotoxicity
According to the results above, we speculated that inhibition of autophagy initiation could be a possible strategy against sunitinib-induced cardiotoxicity. We previously reported that sunitinib could promote autophagy and enhance the level of cytoplasmic HMGB1 in cancerous and noncancerous cells [Citation28]. It has been comprehensively reported that HMGB1 plays critical roles in the regulation of autophagy and cardiovascular diseases [Citation47,Citation48]. Thus, we focused on the role of HMGB1 in the autophagy-dependent cardiotoxicity of sunitinib. Western blot analysis showed that HMGB1 protein levels were increased in cardiomyocyte treated with sunitinib (, S6(A, B)) and in heart tissues from sunitinib-treated mice (). Immunofluorescence assays revealed that sunitinib promoted the nuclear accumulation of HMGB1 in CCC-HEH-2 cells (Figure S6C) or cardiomyocyte (ACTN2 positive) in heart sections ().
Figure 7. Sunitinib-induced excessive HMGB1 induces maladaptive autophagy and subsequent apoptosis. (A) MCMs cells were treated with various concentrations of sunitinib as indicated (between 0 and 10 µM) for 24 h. Western blot was repeated at least three times and densitometric analysis was carried out. (B) Total heart tissue lysates from nude mice treated with saline or sunitinib (40 mg/kg, n = 5 per group) for 42 days. The HMGB1 expression levels from 3 hearts were normalized to GAPDH and densitometric analysis was carried out. (C) Immunofluorescence assays were performed to detect the expression of HMGB1 from heart tissues in the absence or presence of sunitinib. Scale bars: 20 µm, 10 µm. (D) CCC-HEH-2 cells were transfected with NC or siRNA targeting HMGB1, infected with mCherry-GFP-LC3 and then treated with or without sunitinib for 24 h. Autophagic flux assays showed the effect of HMGB1 silencing on sunitinib-induced autophagy. Scale bars: 20 µm, 10 µm. Right: quantification of the percentage of mCherry-positive or GFP-positive puncta per cell. n = 100 cells pooled from three independent experiments. (E) CCC-HEH-2 cells (WT and HMGB1 KO) were treated with sunitinib at a concentration of 2.5 or 5 µM, and then c-PARP and LC3-I/II were analyzed by western blot. The experiment was repeated three times and densitometric analysis is shown in the lower panels. (F) SRB assay showed the effect of HMGB1 knockout on sunitinib (5 µM)-induced inhibition in survival fraction of CCC-HEH-2 cells. Three independent experiments were performed. (G-L) C57BL/6 mice with cardiomyocyte-specific hmgb1 knockout (hmgb1 cKO) were generated by crossing Hmgb1-floxed C57BL/6 mice (Hmgb1flox/flox, Hmgb1f/f, refered to as WT) with Myh6-MerCreMer transgenic C57BL/6 mice and then treating the mice with 25 mg/kg tamoxifen for 3 days. 2 weeks after induction, WT and hmgb1 cKO mice were treated with saline or sunitinib (40 mg/kg) for 42 days (n = 7–11 per group) by means of intragastric administration. (G) Heart tissues from the mice were collected and stained with H&E for histopathological analysis. Scale bars: 50 µm. (H) Heart weight to body weight ratio was analyzed. (I-J) Cardiac function was examined by echocardiography. (K) Immunofluorescence assay was performed to show the apoptosis of myocardium caused by sunitinib. The heart sections were stained with c-CASP3 (green), ACTN2 (red) and DAPI (blue). Scale bars: 20 µm, 10 µm. (L) The CCN2 expression levels from 3 hearts of each group were normalized to GAPDH and densitometric analysis was carried out. The p value was calculated by Student’s t-test (unpaired, two-tailed, 2 groups) or by one-way ANOVA (Sidak test, multiple groups). NS: no significance, *: p< 0.05 **: p< 0.01, ***: p < 0.001, ##: p < 0.01 and ###: p < 0.001. Abbreviations: SUNI, sunitinib; HW, heart weight; BW, body weight; NC, nontargeting siRNA; WT, wild type; cKO, conditional knockout.
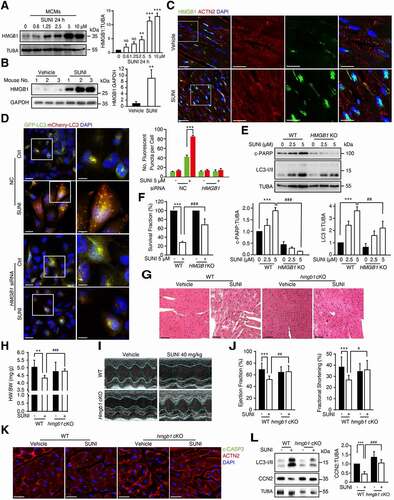
Notably, knockdown of HMGB1 by siRNA reduced sunitinib-enhanced autophagy flux (), suggesting that HMGB1 might be involved in cardiomyocyte autophagy in cardiotoxicity caused by sunitinib. To more precisely explore the role of HMGB1 in autophagy related cardiomyocyte death, we applied CRISPR-Cas9 with a 2 sgRNA pool to knock out HMGB1 expression in CCC-HEH-2 cells. HMGB1 KO cells were obtained from a single colony and the knockout efficiency was confirmed by qPCR (Figure S6D). In HMGB1 KO cells, sunitinib showed no apparent effects on LC3-I/II convention or PARP activation (). As a result of autophagy inhibition, sunitinib-induced CCN2 degradation was reversed in HMGB1 KO cells (Figure S6(E, F)). HMGB1 knockout significantly rescued cardiomyocyte death as detected by sulforhodamine B (SRB) assay (). In addition, PI staining followed by flow cytometry showed that knockout of HMGB1 slightly increased the apoptosis rate of CCC-HEH-2 cells (Figure S6G), which is consistent with the report that nuclear HMGB1 is required for inhibition of DNA damage [Citation49]. However, the apoptosis rate in 5 or 10 µM sunitinib-treated HMGB1 KO cells significantly decreased compared with that in WT cells (Figure S6G), indicating that HMGB1 knockout suppressed the apoptosis caused by sunitinib, which was consistent with the morphological changes detected by microscopy (Figure S6H). Collectively, our results revealed that HMGB1 is a potential master regulator of sunitinib-induced autophagy and apoptosis.
We next explored the in vivo role of HMGB1 in sunitinib-induced cardiotoxicity using cardiomyocyte-specific hmgb1 knockout mice. Hmgb1flox/flox (referred to as Hmgb1f/f and WT) mice were crossed with Myh6-MerCreMer mice to obtain hmgb1f/f; Myh6-MerCreMer (hmgb1 cKO) mice to enable inducible deletion of hmgb1 gene in cardiomyocytes. Immunofluorescence assays in heart sections showed the specificity of hmgb1 deletion in cardiomyocytes after tamoxifen induction (Figure S7A). Histological analysis revealed alleviation of heart injury (abnormal structure of cardiac myofibrils) in hmgb1 cKO mice compared to WT mice under sunitinib treatment (). As shown in , the sunitinib-induced decrease in the HW:BW ratio was rescued by hmgb1 deletion. Moreover, sunitinib-impaired LV function (EF and FS) could be alleviated by hmgb1 deletion as detected via echocardiography (, J)). Immunofluorescence assays in heart sections also showed reduced c-CASP3 levels in hmgb1 cKO mice upon sunitinib treatment (), indicating that cardiac specific hmgb1 deletion suppressed the apoptosis caused by sunitinib. Notably, the sunitinib-induced reduction in CCN2 and increase in LC3-II were reversed in hmgb1 cKO hearts (), suggesting that sunitinib-induced autophagy and CCN2 degradation were significantly reduced by hmgb1 deletion. Taken together, these data identified the master role of nuclear HMGB1 in autophagy-dependent cardiotoxicity of sunitinib in vivo.
The function of nuclear HMGB1 in autophagy is much less known than that of cytoplasmic and extracellular HMGB1. It has been reported that nuclear HMGB1 modulates the expression of HSPB1/HSP27 (heat shock protein family B (small) member 1), which is critical for autophagy and mitophagy [Citation50,Citation51]. Therefore, we further investigated the role of HSPB1 in sunitinib-induced HMGB1-mediated autophagy and apoptosis of cardiomyocyte. We found that overexpression of both WT and nuclear export signal (NES) mutant HMGB1 increased the expression of HSPB1, promoted activation of PARP and CASP3 and increased LC3-II levels (Figure S7B). In addition, silencing the expression of HSPB1 significantly reversed the increased expression of c-PARP, c-CASP3 and LC3-II caused by sunitinib (Figure S7C), suggesting that HMGB1 might mediate autophagy and apoptosis of cardiomyocyte through HSPB1 in sunitinib-induced cardiotoxicity.
Inhibition of HMGB1 confers functional protection from sunitinib-induced cardiotoxicity by blocking CCN2 degradation
Based on the above findings, it was reasonable to conclude that HMGB1 could be a reliable target for treating the cardiotoxicity of sunitinib. Glycyrrhizic acid (GA), a triterpene saponin glycoside, which is the primary bioactive component of the main extract of the root of the plant Glycyrrhiza glabra (licorice), a shrub from the Leguminosae family, has been effective against chronic hepatitis in China and Japan for more than 50 years [Citation52]. Previous studies reported that GA could inhibit HMGB1 expression [Citation53], and we confirmed that GA could downregulate the expression of HMGB1 in a concentration-dependent manner (). According to these reports, we hypothesized that GA could block the sunitinib-induced autophagic degradation of CCN2 by inhibiting HMGB1. First, GA obviously dampened the sunitinib-induced increase in LC3-II and CCN2 degradation (). Consistent with these findings, GA inhibited sunitinib-induced autophagic flux in MCMs and CCC-HEH-2 cells ( and S8A). As expected, we also found that GA significantly inhibited the increase in both c-PARP and c-CASP3 () and improved cardiomyocyte survival (, F)), suggesting that GA could play a protective role in the autophagy-dependent cardiotoxicity of sunitinib.
Figure 8. Inhibition of HMGB1 by GA confers functional protection against sunitinib-induced cardiotoxicity. (A) CCC-HEH-2 cells were treated with various concentrations of GA, as indicated (between 0 and 100 µM) after 24 h of exposure. Representative western blots showing HMGB1 expression upon GA treatment using TUBA as a loading control. (B) CCC-HEH-2 cells were exposed to GA (50 µM) with or without sunitinib (5 µM) for 24 h. LC3-I/II and CCN2 protein levels were measured by western blot analysis using TUBA as a loading control. Western blot was repeated at least three times and densitometric analysis was carried out. (C) Autophagic flux assays showed the effect of HMGB1 inhibition by GA on sunitinib-induced autophagy in MCMs cells. Scale bars: 10 µm. Right panel: quantification of the percentage of mCherry-positive, GFP-positive puncta per nucleus. n = 100 cells pooled from three independent experiments. (D) CCC-HEH-2 cells were exposed to GA (50 µM) with or without sunitinib (5 µM) for 24 h. c-PARP and c-CASP3 protein levels were measured by western blot analysis using TUBA as a loading control. Western blot was repeated at least three times and densitometric analysis was carried out. (E) Cells were treated with or without sunitinib, in the absence or presence of GA for 24 h, and cell morphology was obtained by microscopy. Scale bars: 100 µm. (F) CCC-HEH-2 cells were exposed to GA (50 µM) with or without sunitinib (5 µM) for 72 h. SRB assay showed the effect of GA on sunitinib-induced inhibition in the survival fraction of CCC-HEH-2 cells. Three independent experiments were performed. (G-J) C57BL/6 mice were treated with saline, sunitinib (40 mg/kg), GA (10 mg/kg) or GA plus sunitinib for 42 days by means of intragastric administration (n = 5 per group). (G) Heart weight to body weight ratio. (H) Serum was analyzed for CK-MB and TNNT2 levels. (I) Cardiac function was examined by echocardiography. (J) Heart tissues from the mice were collected, and stained with H&E for histopathological analysis. Scale bars: 100 µm. The p value was calculated by one-way ANOVA (Dunnett’s multiple comparisons test or Sidak test). **: p < 0.01, ***: p < 0.001, #: p < 0.05, ##: p < 0.01 and ###: p < 0.001. Abbreviations: SUNI, sunitinib; HW, heart weight; BW, body weight; CK-MB, creatine kinase myocardial band; TNNT2, troponin T2, cardiac; GA, glycyrrhizic acid; MCMs, adult mouse cardiomyocyte.
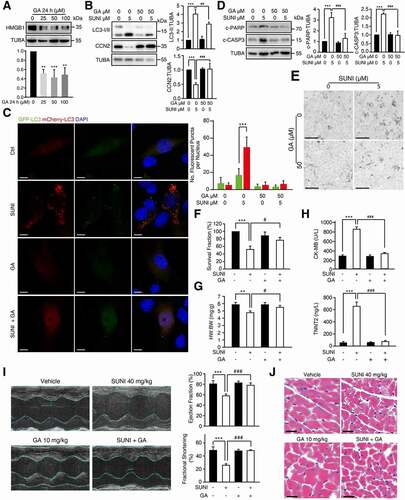
To further investigate the protective efficacy of GA on sunitinib-induced cardiotoxicity in vivo, we confirmed the reparatory effect of GA on the abnormal HW:BW ratio caused by sunitinib (). GA dramatically reduced the accumulation of LC3 caused by sunitinib as shown in Figure S8B, indicating that GA inhibited autophagy. GA also decreased the abnormally increased serum CK-MB and TNNT2 level caused by sunitinib (). In addition, we used an echocardiography-monitored strategy in a mouse model of sunitinib-induced cardiomyopathy. The decreased EF and FS values recovered to normal levels as the vehicle control with GA treatment (), indicating that GA could remedy sunitinib-stimulated cardiac dysfunction. H&E staining showed that GA rescued the cytoplasmic vacuolization caused by sunitinib (). We also checked the effects of GA on the anti-cancer activity of sunitinib. SRB results showed that GA synergized with sunitinib to reduce the survival fraction of HCT116 cells (Figure S8C). Taken together, these results demonstrated that GA played a protective role in sunitinib-induced cardiotoxicity by preventing HMGB1 mediated autophagic degradation of CCN2, implying that pharmacological inhibition of HMGB1 might be an attractive approach for improving the safety of sunitinib-based cancer therapy.
Discussion
In this study, we revealed that maladaptive autophagy induced by sunitinib, a clinical targeted anti-cancer drug associated with cardiac dysfunction, led to cardiomyocyte apoptosis and cardiotoxicity by degrading CCN2 via the cargo protein TOLLIP. We confirmed HMGB1 as an autophagy and cardiotoxicity master regulator and deletion or pharmacological inhibitor of HMGB1 as a therapeutic intervention to address the cardiotoxicity of sunitinib and even the autophagy related cardiac dysfunction.
Sunitinib malate, showed significant clinical benefits in the treatment of advanced or metastatic RCC. However, cardiovascular toxicity following sunitinib treatment is the major obstacle for its wide safe use in clinic. Several studies have revealed that sunitinib-treated patients showed a high incidence of hypertension, decreased LVEF and congestive heart failure [Citation13,Citation20,Citation21,Citation23]. Nevertheless, the definite pathophysiological changes in the hearts post sunitinib treatment are still unknown due to the difficulty of biopsy of human hearts. The myocardial injury caused by overload stress or cardiotoxic drugs promotes compensative remodeling, manifested by myocardial fibrosis (increased extracellular matrix and collagen deposition), hypertrophy and other morphological changes. Myocardial fibrosis is thought to be an early manifestation and the hallmark of hypertrophic cardiomyopathy [Citation54]. Here, we found that short-term treatment with sunitinib led to significant cardiomyocyte death, decreased heart weight, slight fibrosis and increased expression of cardiac remodeling gene Nppb, but showed no significant effects on cardiomyocyte size, indicating that short-term sunitinib treatment may cause early myocardial injury without the occurrence of cardiac hypertrophy. It is possible to speculate that sunitinib-induced cardiomyocyte death might contribute to the decreased heart weight. Furthermore, the long-term effects of sunitinib on cardiac pathophysiology remain to be investigated.
Despite the lack of definite pathophysiological changes in the hearts, the mechanism of the cardiotoxicity of sunitinib has been struggling explored. Reports have found that long-term treatment with high concentrations of sunitinib increases CASP9 activity in NRVMs or CASP3 activity in H9c2 cells, indicating that apoptosis might be involved in sunitinib-induced cardiotoxicity [Citation18,Citation55]. However, other groups also reported that short-term treatment with sunitinib could not induce apoptosis [Citation56] and no available strategy against sunitinib-induced cardiotoxicity based on inhibition of apoptosis was identified, indicating that the precise mechanism is yet to be discovered. Here, we confirmed that sunitinib is a strong inducer of excessive autophagy and subsequent apoptosis, which is consistent with our and others’ previous reports [Citation22,Citation23]. Notably, one study reported that sunitinib induced autophagic inhibition in pericytes [Citation57]. Cardiac pericytes and cardiomyocyte are two important cell components in the heart and both types of cells are essential for the maintenance of cardiac function and homeostasis. However, the effect of sunitinib on cardiomyocyte was lacking in this report. We speculated that the distinct gene expression and physiological functions of these two cell types led to the different effects of sunitinib on autophagy. In addition, the effects of sunitinib on autophagy of endothelial cells or fibroblasts may need to be identified, as these cells are also important for normal heart function.
Moreover, another group reported that sunitinib inhibits late autophagy in H9c2 cells [Citation58]. This conclusion lacks sufficient evidence to support. The article only showed data from western blot (LC3-II:LC3-I) assay to illustrate autophagy inhibition, and the increase in the ratio of LC3-II:LC3-I may be due to the activation of autophagy. Here, we proved that SUNI activates cardiomyocyte autophagy through various assays, including western blot, autophagy flux and immunohistochemistry, using various in vitro and in vivo models, which made our conclusions more convincing. Autophagy is a conserved and essential process that plays various functions under different physiological and pathological conditions [Citation59]. Whether autophagy is beneficial or detrimental to cells and tissue is dependent on the circumstances, which is different from apoptosis. Here, we provided evidence demonstrating that sunitinib-induced excessive autophagy is the cause of cardiomyocyte apoptosis and cardiotoxicity by silencing ATG5, ATG7 or cardiomyocyte-specific heterozygous deletion of Atg7 in mice (), which might at least partially clarify why sunitinib causes autophagy or apoptosis in different reports, and indicated that inhibition of autophagy is a potential therapeutic strategy against the cardiotoxicity of sunitinib rather than inhibition of apoptosis.
Detrimental autophagy promotes cell death through the excessive elimination of organelles or survival-related proteins [Citation36,Citation37]. Here, we identified CCN2 as a major substrate of autophagic degradation for the first time. In addition, we found that the autophagy cargo receptor TOLLIP was essential for sunitinib-induced CCN2 degradation, in addition to the classical receptor SQSTM1. It was recently reported that TOLLIP is important for cargo engulfment by endosomal membranes [Citation44] and late autophagosomes and late endosomes are involved in the formation of amphisomes, which also fuse later with lysosomes for degradation [Citation60–62]. These reports indicated that endosomes might be related to autophagic degradation of CCN2 by sunitinib. Indeed, we observed the clearance of endosomes by sunitinib, as the expression of CD63 was decreased upon sunitinib treatment. The colocalization of CCN2 and CD63 in cardiomyocytes cotreated with CQ and SUNI further confirmed the involvement of endosomes in the autophagic degradation of CCN2 by sunitinib. However, the precise role of TOLLIP in endosome-related autophagy has yet to be explored in the future.
CCN2 is highly expressed in the heart but its role in cardiac physiology and pathology is complicated and cell type specific. Heart-specific knockout or overexpression of Ccn2 by Myh7-Cre showed no effects on cardiac function or remodeling with aging or under stress [Citation63]. However, the expression of CCN2 is upregulated in the left ventricle of patients diagnosed with heart failure caused by dilated cardiomyopathy [Citation64]. The plasma CCN2 level is also upregulated in diastolic heart failure patients and is correlated with the severity of diastolic dysfunction and tissue fibrosis [Citation65]. Taken together, the role of CCN2 in the normal adult heart is still unclear. Here, we confirmed that a forced decrease in CCN2 levels by AAV9 vector with the Tnnt2 promoter could induce cardiomyocyte apoptosis and cardiac dysfunction, which were observed in sunitinib-treated mice. Cardiomyocyte-specific Ccn2 overexpression in mice using the AAV9 virus dramatically reduced the cardiotoxicity of sunitinib, which is consistent with other findings that transgenic overexpression of Ccn2 in mice showed a cardioprotective effect on cardiac hypertrophy and cardiac dysfunction and CCN2 showed antiapoptotic effects in cardiomyocyte [Citation66,Citation67]. Hence, these findings indicated that autophagic degradation of CCN2 is relevant to cardiomyocyte death caused by long-term treatment with sunitinib and cardiotoxicity induced by sunitinib, which might be caused by a distinct mechanism as heart failure caused by dilated cardiomyopathy. In addition, the distinct conclusions mentioned above may be caused by the strategy of Ccn2 knockout and the use of different cardiac specific promoters-driven cre. To clarify the function of CCN2 in normal adult hearts, transgenic mice with heart specific knockout of Ccn2 by Myh6-Cre or Tnnt2-Cre remain to be studied.
Notably, how CCN2 regulates cardiomyocyte survival/death and cardiac function needs to be further clarified to fully understand the cardiac dysfunction caused by anticancer drugs or diseases. Interestingly, while autophagy is detrimental and promotes cardiomyocyte death in myocardial ischemia/reperfusion [Citation7,Citation68,Citation69] and the cellular mechanisms by which autophagy is upregulated have been revealed [Citation70,Citation71], the mechanism by which autophagy promotes cell death is still unknown. Our findings suggested that CCN2 degradation might be the possible reason for ischemia/reperfusion injury, which deserves further investigation to clarify the role of CCN2 in autophagy-dependent cardiomyocyte death.
CCN2 is also essential for matrix metalloproteases and extracellular and cell- adhesion proteins in cardiac fibroblasts and upregulation of CCN2 contributes to TGFB (transforming growth factor beta)-SMAD signaling-mediated fibrosis in dilated cardiomyopathic hearts. Therefore, there is a concern that blocking CCN2 autophagic degradation may promote CCN2 secretion in fibroblasts, eventually resulting in cardiac fibrosis. Recent studies demonstrated that only activated fibroblast-derived CCN2 in an autocrine manner could induce cardiac fibrosis, and we found that sunitinib failed to activate fibroblasts. Moreover, recombinant CCN2 protein protected cardiomyocytes from several stresses. Thus, inhibiting CCN2 autophagic degradation might be feasible strategy against sunitinib-induced cardiotoxicity.
We previously found that sunitinib-induced autophagy selectively degraded the tumor suppressor TP53/p53 in cancer cells, which inhibited the anticancer efficiency of sunitinib. The expression of TP53 in adult cardiomyocytes is low, and upregulation of TP53 in cardiomyocyte leads to mitochondrial dysfunction, hypertrophy, inflammatory responses and cardiac dysfunction [Citation72–74]. Here, in cardiomyocyte, we did not observe the apparent accumulation of p53 in lysosomes from sunitinib-, pepstatin A-, and E-64d-treated cardiomyocyte, indicating that TP53 might not be involved in sunitinib-induced cardiotoxicity and autophagy and that the substrate for selective autophagy is cell type specific.
Research has shown that the target kinase selectivity of kinase inhibitors, including sunitinib, was not associated with the potential of cardiotoxicity [Citation56,Citation75], indicating that the cardiotoxicity of sunitinib might be caused by off-target effects. In attempting to elucidate the toxic process and explore potential targets for preventing this toxicity, we uncovered an essential role of HMGB1 in regulating sunitinib-stimulated cardiomyocyte autophagic death. Increased expression of HMGB1 promotes autophagy progression via different pathways depending on its subcellular localization and posttranslational modification [Citation24,Citation76–80]. Increased expression of HMGB1 is also one of the key factors in the pathogenesis of autoimmune myocarditis, myocardial infarction and cardiac fibrosis [Citation81–83]. Here, we found that sunitinib increased the protein level of HMGB1 in the nuclei of cardiomyocyte under sunitinib treatment, which led us to identify the role and mechanism of nuclear HMGB1 in autophagy and CCN2 degradation. We found that silencing the expression of HMGB1 or its downstream effector HSPB1 significantly reversed sunitinib-induced cardiomyocyte autophagy and apoptosis, suggesting the essential role of HMGB1/HSPB1 signaling in the autophagy and cardiotoxicity caused by sunitinib. Further study is still needed to clarify the deep mechanism by which HMGB1/HSPB1 regulate autophagy and endosome-dependent CCN2 degradation. In addition, it has been reported that a high dose of sunitinib inhibits AMP-activated protein kinase (AMPK) signaling by inhibiting ACAC/ACC in cardiomyocytes [Citation56]. AMPK is a promoter of autophagy via inhibition of MTOR (mechanistic target of rapamycin kinase) [Citation84,Citation85] and HMGB1 could also promote autophagy by regulating the AMPK-MTOR signaling pathway, indicating that more careful analysis of the involvement of AMPK-related autophagy in sunitinib-induced cardiotoxicity is needed.
GA, an HGMB1 inhibitor, is widely known as an anti-inflammatory compound and is used to protect organs from injury. GA can also inhibit fibrosis by reducing the CCN2 level in the liver of CCl4 treated rats [Citation86] and induce autophagy in the lungs of LPS-treated mice [Citation87]. As inflammation is involved in these processes, the target of GA is still unclear. GA showed cardioprotective effects by inhibiting the HMGB1-dependent phospho-JNK/Bax pathway, restoring the NRIP1/RIP140-NFKB/NF-kB pathway, inhibiting HMGB1 release or inhibiting calcium influx via L-type calcium channels, suggesting that HMGB1 might be a target of GA [Citation83,Citation88–90]. Importantly, our present study confirmed that GA reduced the autophagy-dependent cardiotoxicity caused by sunitinib. As we and other groups found that sunitinib showed little effect on cardiac fibrosis and inflammation [Citation18], the cardioprotective effect of GA might work through inhibiting HMGB1 expression. Although hundreds of chemicals can inhibit autophagy, few of these agents act as truly specific autophagy inhibitors in clinic, and most of them confer major toxic side effects. As a drug that has been used in Japan and China for more than 50 years, GA has a favorable safety profile and cardioprotective activity, which provides a security assurance for its clinical use to rescue sunitinib-induced cardiotoxicity [Citation52]. Remarkably, we previously demonstrated that inhibiting HMGB1 by shRNA knockdown could enhance the anticancer activity of sunitinib [Citation28]. Moreover, we found that GA enhanced anticancer activity of sunitinib in vitro. These data thereby provide a strong rationale for the clinical use of GA in combination with sunitinib-based cancer therapy.
In conclusion, the data presented in this study advances our understanding of autophagic cell death in drug-induced cardiac dysfunction and reveals that selective targeting of CCN2 for autophagic degradation in cardiomyocyte results in sunitinib-stimulated cardiomyocyte apoptosis and cardiotoxicity. In addition, our study has identified a potentially valuable therapeutic application by providing a mechanism-based rationale to combat sunitinib-induced cardiotoxicity.
Materials and methods
Animals
All animal-related experimental procedures and methodologies were approved by the Center for Drug Safety Evaluation and Research of Zhejiang University and performed according to the Institutional Animal Care and Use Committee (IACUC) protocol of Zhejiang University (IACUC-S18-15). Five- to six-week-old C57BL/6 mice or athymic nude mice (BALB/c-nude) were obtained from Beijing Vital River Laboratory Animal Technology Co., Ltd. The mice were housed in barrier facilities with a 12 h light/dark cycle, and food and water were available ad libitum. Approximately 1 week was provided before starting experiments to allow the animals to acclimate to the laboratory environment. Sunitinib malate (hereafter referred to as sunitinib; Selleck Chemicals, S1042) was dissolved in 0.9% NaCl, and the animals were treated with 40 mg/kg sunitinib through intragastric administration for 6 weeks (for BALB/c-nude mice, 32 days). Glycyrrhiza acid (GA; Sigma-Aldrich, 50,531) was dissolved in 0.9% NaCl, and the animals were treated with 10 mg/kg glycyrrhiza acid through intragastric administration for 42 days. Then, echocardiography was performed, creatine kinase myocardial band (CK-MB) and TNNT2/CTNT (troponin T2, cardiac type) were tested, and hearts were harvested for weight measurement and hematoxylin and eosin (H&E; Beyotime, C0105) staining. Experiments were performed using the number of animals as shown in the respective figure legends.
Generation of cardiomyocyte-specific Atg7−/+ or hmgb1 cKO mice
Atg7flox/flox mice (RBRC02759) were provided by RIKEN BRC through the National Bio-Resource Project of the MEXT, Japan [Citation32]. The details of the genetic background were as follows: B6.Cg-Atg7< tm1Tchi>. Atg7flox/flox mice were crossed with Myh6-MerCreMer transgenic C57BL/6 mice (Nanjing Biomedical Research Institute of Nanjing University) to produce Atg7flox/+; Myh6-MerCreMer mice.
Hmgb1-floxed mice were generated via the CRISPR-Cas9 system by Nanjing Biomedical Research Institute of Nanjing University, China. Hmgb1flox/flox mice were crossed with Myh6-MerCreMer transgenic C57BL/6 mice to produce Hmgb1flox/+; Myh6-MerCreMer. Then, Hmgb1flox/+; Myh6-MerCreMer mice were crossed with Hmgb1flox/flox mice to produce hmgb1flox/flox; Myh6-MerCreMer homozygous mice.
For tamoxifen-induced deletion, 5- to 7-week-old mice of the corresponding genotype were subsequently injected with 25 mg/kg tamoxifen (Sigma-Aldrich, T-5648) per day for 3 consecutive days to induce Cre recombinase expression. Two weeks after induction, mice were used for later experiments.
Cardiomyocyte-specific overexpression of Ccn2
Adeno-associated virus serotype 9 (AAV9) vectors carrying the Tnnt2 promoter and Ccn2 cDNA (AAV9-Tnnt2-Ccn2) were constructed and packaged by Vigene Biosciences (Shandong, China), and the titers of the AAV vectors (viral genomes/mL) were determined by quantitative real-time PCR (qPCR), as described previously [Citation91]. The vectors (1 × 1012 vg/mL) and AAV9-Tnnt2-Ccn2 (1 × 1013 vg/mL) were then injected into mice through the tail vein, with 5 × 1010 vg per C57BL/6 mouse. 3 weeks later, mice with cardiac-restricted overexpression of Ccn2 were treated with 40 mg/kg sunitinib for 6 weeks. Then, echocardiography was performed, CK-MB and TNNT2 levels were tested, and hearts were harvested for weight measurement and H&E staining. Experiments were performed using the number of animals as shown in the respective figure legends.
Cardiomyocyte-specific knockdown of Ccn2
AAV9 vectors carrying the Tnnt2 promoter and mir30-NC (AAV9-Tnnt2-NC) or mir30-Ccn2 siRNA (AAV9-Tnnt2-Ccn2-null) were constructed and packaged by Hanbio Biotechnology (Shanghai, China). The virus was purified and titers of the AAV vectors were measured by qPCR. AAV9-Tnnt2-NC (1.7 × 1012 vg/mL) and AAV9-Tnnt2-Ccn2-null (1.8 × 1012 vg/mL) were then injected into C57BL/6 mice through the tail vein (1.8 × 1011 vg per mouse). Four weeks after injection, mice were collected for testing as described above.
The siRNA sequences were as follows:
NC: 5ʹ- TTCTCCGAACGTGTCACGTAA -3ʹ;
Ccn2 siRNA: 5ʹ- CAGGAAGATGTACGGAGACAT -3ʹ.
In vivo xenograft tumor model
A xenograft mouse model bearing 786-O cells was established by subcutaneously inoculating tumor cells into nude mice. Then, 786-O cells (5 × 106) were inoculated subcutaneously into the right shoulders of the mice. When the tumors reached a mean size of 90 mm3, the mice were randomly divided into 2 groups and treated as indicated in the experiments. Sunitinib malate was dissolved in 0.9% NaCl, and the animals were treated with 40 mg/kg sunitinib through intragastric administration for 32 days. Heart samples were harvested and fixed. Tumor volumes were measured every four days and calculated as (length × width2)/2. The individual relative tumor volume (RTV) was calculated as previously reported [Citation92]. Then, CK-MB and TNNT2 were tested, and hearts were harvested for weight measurement and H&E staining. Experiments were performed using the number of animals as shown in the respective figure legends.
Echocardiography
Cardiac function was evaluated by detecting heart dimensions with 2D echocardiography in conscious mice with the Vevo 1100 Imaging System. The LV internal diameter in diastole (LVIDd) or systole (LVIDs) and the end-diastolic volume (EDV) or end-systolic volume (ESV) were measured and analyzed by VisualSonics software. The LVEF was calculated according to the following formula: EF (%) = [(EDV – ESV)/EDV] × 100. The LVFS was calculated according to the following formula: FS (%) = [(LVIDd – LVIDs)/LVIDd] × 100.
Biochemical analysis
On the last day of sunitinib treatment, serum was collected after centrifugation (2000 g for 10 min at 4°C) of the blood. The level of serum CK-MB was determined using a fully automatic biochemical detection machine (Cobas c 311, Roche Diagnostics GmbH, Germany) with specific detection kits (Roche Diagnostics GmbH, 07190808190). Serum TNNT2 was determined by the CARDIAC Troponin T Quantitative test (Roche Diagnostics GmbH, 04856627190).
Histopathological staining
Heart samples were fixed in 10% phosphate-buffered (composition: 137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.76 mM KH2PO4) formalin (pH 7.4; Sigma-Aldrich, F8775), embedded in paraffin, and sectioned at 3 µm intervals. After de-waxing and rehydration, the sections were stained with H&E, Sirius Red, Masson’s trichrome or wheat germ agglutinin (Alexa Fluor™ 488 Conjugate, 5 µg/ml; Thermo Scientific, W11261) for analysis and then examined by microscopy.
Cell culture and drug treatment
CCC-HEH-2 human embryonic cardiac tissue-derived cell lines (RRID: CVCL_VU29) were established from male human embryonic tissues at 11 weeks by the National Infrastructure of Cell Line Resource of China and H9c2 rat embryonic heart tissue-derived cell lines were purchased from Jennio Biological Technology (Guangzhou, China). The human colon cancer cell line HCT116 was obtained from the Institute of Biochemistry and Cell Biology (Shanghai, China). The cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Gibco, 10569010) supplemented with 10% fetal bovine serum (FBS; HyClone, SV30160.03), 100 U/mL penicillin and 100 µg/mL streptomycin (Gibco, 10378016) in a humidified atmosphere with 5% CO2 at 37°C. All cell lines routinely tested negative for mycoplasma contamination.
Throughout the experiments (if not stated otherwise), the cells were treated with 5 µM sunitinib for the indicated time points or 2.5, 5, 7.5, 10 or 12.5 µM sunitinib for 24 h for cell viability analysis and western blots. In selected samples, 10 μg/mL cycloheximide (CHX; MedChemExpress, HY-12,320) and 20 μM MG132 (Sigma-Aldrich, M8699) were used. In some samples, 10 µM chloroquine (CQ; Sigma-Aldrich, C6628) was added 2 h before sunitinib treatment.
Isolation of neonatal rat ventricular myocytes (NRVMs)
NRVMs cultures (DMEM supplemented with 10% FBS) were prepared. Briefly, hearts were removed from 1-day-old Sprague-Dawley rats, the atria and great vessels were trimmed off, and tissue was finely minced followed by sequential digestion with 0.5 mg/ml collagenase type I (Gibco, 17,018,029). Ventricular cardiomyocytes were separated from fibroblasts by differential plating and cultured in gelatin-coated 96-well tissue culture plates (0.1 hearts/well) in media containing DMEM, 10% FBS, 100 µM bromodeoxyuridine (Sigma-Aldrich, B5002), penicillin-streptomycin and L-glutamine (Invitrogen, 25,030–081).
Isolation of adult mouse cardiomyocyte (MCMs)
Isolation of MCMs was performed according to a previous study [Citation93]. Briefly, hearts were removed from adult mice and then modified Langendorff perfusion devices were applied. Type II collagenase (Gibco, 17,018,029) was used to separate myocytes from the isolated heart. Ventricular cardiomyocyte were separated from fibroblasts by differential plating and were cultured in different tissue culture plates in media containing DMEM, 10% FBS, 100 µM bromodeoxyuridine, penicillin-streptomycin and L-glutamine.
Cell survival assay
The cell survival assay was assessed by a sulforhodamine B (SRB; Sigma-Aldrich, S1402) colorimetric assay. The SRB colorimetric assay was performed as described previously. The absorbance at 510 nm was measured using a Multiscan Spectrum (Thermo Fisher Scientific, USA) until the absorbance values remained stable. Assays were performed in triplicate in three independent experiments.
Western blot analysis
Protein lysates were obtained from cells or tissues by IP buffer containing 0.3% NP-40 (Beyotime, ST366), 0.3% Triton X-100 (BioFROXX, 1139ML100), 0.25% leupeptin (Amresco, J580), 0.1% PMSF (Amresco, P8340) and 0.1% NaVO3 (Sigma-Aldrich, 450,243). Protein lysates (30–50 µg per sample) were separated on 8, 10 or 12% SDS-polyacrylamide gels and transferred to PVDF membranes (Merck Millipore, IPVH00010). Incubation with primary and secondary antibodies and signal detection followed the suppliers’ protocol using Western Lightning Plus-ECL Enhanced Chemiluminescence Substrate (PerkinElmer, NEL105001EA). Quantitative image analysis was performed using Quantity One software according to the manufacturer’s protocol.
Primary antibodies directed against ACTB/β-Actin (sc-1615), TUBA/α-tubulin (sc-53,646), NBR1 (sc-130,380), GAPDH (sc-25,778), HMGB1 (sc-26,351), HSPD1/HSP60 (sc-59,567), TOMM20 (sc-17,764), CCN2 (sc-14,939), TOLLIP (sc-292,758), SQSTM1 (sc-48,402) and HSPB1 (sc-13,132) were purchased from Santa Cruz Biotechnology. Antibodies directed against LC3 (2775s), SQSTM1 (5114s), c-PARP (5625s), c-CASP3 (9664s), ATG7 (2631s) and ATG5 (8540s) were purchased from Cell Signaling Technology. HRP-labeled secondary antibodies (GAR007, GAM007 and RAG007) were purchased from MultiSciences (Lianke) Biotech (Hangzhou, China).
Quantitative real-time PCR (qPCR)
Total RNA was extracted with TRIzol reagent according to the manufacturer’s protocol (Invitrogen, 15,596,026). cDNA was prepared using a cDNA reverse transcription kit (Transgene Biotech, AT311-03). qPCR was performed on a 7500 Fast System (Applied Biosystems, Singapore). The reactions were performed in triplicate. The reaction system (20 µL) was as follows: cDNA obtained from reverse transcription of 1 µg RNA, 1.25 µL of forward and reverse primers (10 µM), 10 µL of iTaq Universal SYBR Green Supermix (Bio-Rad, 1,725,125), and variable ddH2O to generate a volume of 20 µL. The samples underwent two-step amplification with an initial step at 95°C (3 min), followed by 95°C (3 sec) and 60°C (31 sec) for 39 cycles. The melting curve was analyzed. Fold changes in the expression of each gene were calculated by the comparative threshold cycle (Ct) method using the formula 2−(ΔΔCt).
The primer sequences were as follows:
Nppa forward: 5ʹ- GAGAGAAAGAAACCAGAGTG-3ʹ;
Nppa reverse: 5ʹ- GTCTAGCAGGTTCTTGAAATC-3ʹ;
Nppb forward: 5ʹ-AAGATGGTGCAAGGGTCTG-3ʹ;
Nppb reverse: 5ʹ-TGTGGAATCAGAAGCAGGTG-3ʹ;
Myh7 forward: 5ʹ- TTGGGAAATTCATCCGAATC-3ʹ;
Myh7 reverse: 5ʹ- CCAGAAGGTAGGTCTCTATG-3ʹ;
Myh6 forward: 5ʹ- AATCCTAATGCAAACAAGGG −3ʹ;
Myh6 reverse: 5ʹ- CAGAAGGTAGGTCTCTATGTC −3ʹ;
MAP1LC3 forward: 5ʹ- CCAGATCCCTGCACCATG- 3ʹ;
MAP1LC3 reverse: 5ʹ- CTGCTTCTCACCCTTGTATCG −3ʹ;
SQSTM1 forward: 5ʹ- AATCAGCTTCTGGTCCATCG −3ʹ;
SQSTM1 reverse: 5ʹ- TTCTTTTCCCTCCGTGCTC −3ʹ;
CCN2 forward: 5ʹ- ACCAATGACAACGCCTCC-3ʹ;
CCN2 reverse: 5ʹ- TTGGAGATTTTGGGAGTACGG-3ʹ;
HMGB1 forward: 5ʹ-GATATGGCAAAAGCGGACAAG-3ʹ;
HMGB1 reverse: 5ʹ-GGCGATACTCAGAGCAGAAG-3ʹ;
GAPDH forward: 5ʹ-CGGAGTCAACGGATTTGGTCGTAT-3ʹ;
GAPDH reverse: 5ʹ-ATCTTCTCCATGTCGTCCCAGTTG-3ʹ.
Transfection of siRNA oligonucleotides
Cells were seeded into 6-well plates at 1 × 105 cells per well and grown to 50%-60% confluence. Transfection was performed using Opti-MEM (Invitrogen, 31,985–062), siRNA and oligofectamine (Invitrogen, 12,252,001) according to the manufacturer’s recommendations. siRNA oligonucleotides were transfected at a final concentration of 12 nM using oligofectamine. The cell culture solution was changed to DMEM plus 10% FBS at 6 h after transfection. The cells were cultured for 24 h and placed in complete medium containing 5 µM sunitinib for the indicated times and treated or untreated cells were collected for further analysis. The following oligonucleotides were obtained from GenePharma (Shanghai, China) as siRNAs targeting the indicated genes:
TOLLIP-#1: 5ʹ- GCUGGAAUAAGGUCAUCCAdTdT -3ʹ;
TOLLIP-#2: 5ʹ- CGACUGAACAUCACGGUGGdTdT -3ʹ;
SQSTM1-#1: 5ʹ- GCAUUGAAGUUGAUAUCGAdTdT -3ʹ;
NBR1-#1: 5ʹ- CAGGAACCAAGUUUAUCAAdTdT -3ʹ;
NBR1-#2: 5ʹ- AGUUAAACAGGGAAACCAAdTdT -3ʹ;
HMGB1: 5ʹ- UUGGUGAUGUUGCGAAGAAACdTdT -3ʹ;
ATG5: 5ʹ- CAUCUGAGCUACCCGGAUAdTdT -3ʹ;
Negative Control: 5ʹ-UUCUCCGAACGUGUCACGUdTdT-3ʹ.
Knockdown of ATG5 or CCN2 by shRNA lentivirus
The pLKO.1 vectors (Addgene, 8453; deposited by Bob Weinberg Lab, Massachusetts Institute of Technology) containing ATG5 or CCN2 shRNA were constructed by EcoRI and AgeI digestion and subsequent T4 ligation. After transformation, monoclonal colonies were selected for sequencing identification. The right plasmid was amplified and used for lentivirus production. Lentivirus production was conducted by cotransfection of HEK293 FT cells with three plasmids as follows: a packaging defective helper construct (pRΔ8.9, kindly provided by Dr. Lingtao Wu from University of California; 5 µg), a construct expressing a heterologous envelope protein (pMDG-VSVG, kindly provided by Dr. Lingtao Wu from University of California; 1 µg), and pLKO.1 vector harboring a specific shRNA sequence (pLKO.1-shRNA; 5 µg). After 72 h of infection, conditioned medium was collected and filtered through a 0.45-µm filter (Millipore, HAWP04700). CCC-HEH-2 cells were infected with lentiviral-conditioned medium overnight and then collected for further research.
The following shRNA oligonucleotides targeting the indicated genes were obtained from GenePharma:
CCN2 shRNA #1 (5`-3`):
GAGAACATTAAGAAGGGCAAACTCGAGTTTGCCCTTCTTAATGTTCTCTTTTTG
CCN2 shRNA #2 (5`-3`):
GCATGAAGACATACCGAGCTACTCGAGTAGCTCGGTATGTCTTCATGCTTTTTG
ATG5 shRNA #1 (5`-3`):
CATCTGAGCTACCCGGATATTCTCGAGAATATCCGGGTAGCTCAGATGTTTTTG
Immunofluorescence assay
Cells grown on poly-D-lysine-coated coverslips were washed with phosphate-buffered saline (PBS; Gibco, 10,010,023) and fixed with fresh 4% paraformaldehyde (Sigma-Aldrich, P6148) in PBS for 20 min at room temperature. The cells were then permeabilized with 0.1% Triton X-100 in PBS for 10 min at 4°C and blocked with 1% bovine serum albumin (BSA; Sigma-Aldrich, B2064) in PBS for 30 min.
Hearts were collected and fixed with fresh 4% paraformaldehyde overnight. Then the hearts were dehydrated with 30% sucrose (Sinopharm Chemical Reagent, 10,021,418) and embedded with Optimal Cutting Temperature (Sakura Finetek, 4583) to make frozen sections (8 µm). The sections were washed with 1x PBS 3 times and blocked with 1% BSA in PBS with 0.1% Tween-20 (Aladdin, T104863) for 30 min. Cells or sections were incubated with primary antibodies at 4°C overnight. After washing by PBS with 0.1% Tween-20, the cells or sections were incubated with Alexa Fluor 488- or Alexa Fluor 568-conjugated secondary antibodies (Thermo Fisher Scientific, A21202, A10042; 1:200) for 2 h, stained with 4ʹ,6-diamidino-2-phenylindole (DAPI; Dojindo, D212) for 5 min and mounted for fluorescence microscopy (Olympus IX81-FV1000, Japan). The following primary antibodies were used: anti-LC3 (Santa Cruz Biotechnology, sc-376,404; 1:200), anti-ATG7 (Cell Signaling Technology, 2631s; 1:200) anti-HMGB1 (Huabio, ET1601-2; 1:200), anti-ACTN2 (Sigma, A7811; 1:300), anti-CCN2 (Proteintch, 23,936-1-AP; 1:200), anti-TOLLIP (Santa Cruz Biotechnology, sc-292,758; 1:100) and anti-c-CASP3 (Cell Signaling Technology, 9664s; 1:200).
Autophagic flux assay
Cells expressing mCherry-GFP-LC3 were grown overnight on poly-D-lysine-coated coverslips before Hanks’ balanced salt solution (HBSS; Gibco, 14,025,092) starvation or treatment with sunitinib. Cells were fixed with 4% paraformaldehyde in PBS for 15 min at room temperature. The cells were then permeabilized with 0.2% Triton X-100 in PBS for 15 min. After washing with PBS, the cells were stained with DAPI for 5 min and mounted for analysis by confocal microscopy. Images were acquired randomly, and cells with intermediate total mCherry-GFP-LC3 fluorescence were chosen for further analysis. Cells were outlined manually, and each channel was independently autothresholded using the same settings across all images from each channel. mCherry and GFP puncta were quantified using the Analyze Particles plugin in ImageJ.
Immunoprecipitation assay
Cells were collected in lysis buffer (pH 7.5): 50 mM Tris-HCl, 150 mM NaCl, 1 mM EDTA, 1% NP-40; protease inhibitor cocktail (Cell Signaling Technology, 5871) was added before use. The lysate was centrifuged and immunoprecipitated with 20 µL protein A/G Plus-agarose (Santa Cruz Biotechnology, sc-2003) and a primary antibody at 4°C overnight. The following primary antibodies were used: anti-CCN2 (Santa Cruz Biotechnology, sc-14,939; 1 µg per 500 µg of total protein). Precipitated proteins as well as initial whole-cell lysates were analyzed using western blots, as described above.
Lysosome isolation and proteomics analysis
Lysosomes were separated using a lysosome isolation kit (Sigma-Aldrich, LYSISO1). Briefly, following the co-treatment of sunitinib, pepstatin A (Sigma-Aldrich, P5318) and E-64d (Sigma-Aldrich, E8640), cells were collected by centrifugation at 500 g for 10 min. Next, cells were resuspended in ice-cold isolation buffer and homogenized with a Dounce type homogenizer (Kimble-chase, 885,300–0002). The sample was centrifuged at 20,000 g for 20 min to remove the nuclear compartment. Next, the pellet containing lysosomes was further centrifuged at 20,000 g for 60 min. The location of the lysosomal fractions was verified using an acid phosphatase assay kit (Sigma-Aldrich, CS0740). The samples were labeled with iTRAQ labeling reagents (ABSCIEX, 4,381,663) and then subjected to Liquid Chromatography and tandem MS analysis. Protein analysis and relative iTRAQ quantification services were undertaken by Oebiotech (Shanghai, China).
Flow cytometry analysis
Sub-G1 analysis after propidium iodide (PI; Sigma-Aldrich, P4170) staining was employed to assess the apoptosis caused by sunitinib. After treatment or not with sunitinib at the indicated concentrations for 24 h, cells were harvested and fixed with 75% ethanol at -20°C. The yellow fluorescence of sunitinib was removed by ethanol. PI staining followed by flow cytometry was performed by methods described previously.
ANXA5-PI staining was employed to assess the apoptosis caused by CCN2 knockdown. Cells were treated with CCN2 shRNA lentivirus, harvested and washed with PBS. The cells were stained with Pharmingen™ FITC Annexin V Apoptosis Detection Kit I (BD Pharmingen, 556,547) according to the manufacturer´s instructions.
MMP was detected by JC-1 staining assays. After treatment with sunitinib for the indicated times, cells were collected by trypsinization and stained with JC-1 (5 µM; Sigma-Aldrich, T4069) for 30 min at 37°C in the dark. MMP was analyzed by FACS-Calibur cytometer (BD Biosciences, USA) using a 520-nm filter for the observation of green fluorescence and 590 nm for red fluorescence.
For each sample, 1 × 104 cells were analyzed using FACS-Calibur cytometer (BD Biosciences, USA), and the data were analyzed using Cell Quest software.
Statistical analysis
Statistical analysis was performed using GraphPad Prism 9 software. All data are expressed as the mean value ± standard deviation (SD). When comparing two groups, Student’s t-test (unpaired, two-tailed) was performed. Dunnett’s T3 test was used to assess the significance of differences among treatment groups with a single control mean, and a Sidak test was used to assess the significance of differences among various pairs of groups.
Supplemental Material
Download MS Word (25.8 MB)Acknowledgments
We would like to thank Dr. Lingtao Wu (University of California, USA) for providing pRΔ8.9 and pMDG-VSVG plasmid and Dr. Ronggui Hu (University of Chinese Academy of Sciences, China) for providing the pcDNA3.0-HA and pcDNA3.0-FLAG plasmids. We would like to thank Dr. Wei Liu (Zhejiang University, China) for providing thoughtful suggestions for this work. We would also like to thank experimenter Wei Yin from the Image Center, Core Facilities, School of Medicine, Zhejiang University for the confocal laser scanning microscope images.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
References
- Gao GY, Chen WW, Yan MJ, et al. Rapamycin regulates the balance between cardiomyocyte apoptosis and autophagy in chronic heart failure by inhibiting mTOR signaling. Int J Mol Med. 2020;45:195–209.
- Wang J, Liu SJ, Heallen T, et al. The Hippo pathway in the heart: pivotal roles in development, disease, and regeneration. Nat Rev Cardiol. 2018;15:672–684.
- Del Re DP, Amgalan D, Linkermann A, et al. Fundamental mechanisms of regulated cell death and implications for heart disease. Physiol Rev. 2019;99:1763–1817.
- Xu ZF, Jin Y, Yan H, et al. High-mobility group box 1 protein-mediated necroptosis contributes to dasatinib-induced cardiotoxicity. Toxicol Lett. 2018;296:39–47.
- Catanzaro MP, Weiner A, Kaminaris A, et al. Doxorubicin-induced cardiomyocyte death is mediated by unchecked mitochondrial fission and mitophagy. Faseb J. 2019;33:11096–11108.
- Jin Y, Xu ZF, Yan H, et al. A Comprehensive Review of Clinical Cardiotoxicity Incidence of FDA-Approved Small-Molecule Kinase Inhibitors. Front Pharmacol. 2020;11:891.
- Valentim L, Lawrence KM, Townsend PA, et al. Urocortin inhibits Beclin1-mediated autophagic cell death in cardiac myocytes exposed to ischaemia/reperfusion injury. J Mol Cell Cardiol. 2006;40:846–852.
- Marino G, Pietrocola F, Kong YL, et al. Dimethyl alpha-ketoglutarate inhibits maladaptive autophagy in pressure overload-induced cardiomyopathy. Autophagy. 2014;10:930–932.
- Luo PH, Zhu Y, Chen M, et al. HMGB1 contributes to adriamycin-induced cardiotoxicity via up-regulating autophagy. Toxicol Lett. 2018;292:115–122.
- Shirakabe A, Zhai PY, Ikeda Y, et al. Drp1-Dependent mitochondrial autophagy plays a protective role against pressure overload-induced mitochondrial dysfunction and heart failure. Circulation. 2016;133:1249–1263.
- Matsui Y, Takagi H, Qu XP, et al. Distinct roles of autophagy in the heart during ischemia and reperfusion - Roles of AMP-activated protein kinase and Beclin 1 in mediating autophagy. Circ Res. 2007;100:914–922.
- Nakai A, Yamaguchi O, Takeda T, et al. The role of autophagy in cardiomyocyte in the basal state and in response to hemodynamic stress. Nat Med. 2007;13:619–624.
- Lavandero S, Chiang M, Rothermel BA, et al. Autophagy in cardiovascular biology. J Clin Invest. 2015;125:55–64.
- Nezis IP, Shravage BV, Sagona AP, et al. Autophagic degradation of dBruce controls DNA fragmentation in nurse cells during late Drosophila melanogaster oogenesis. J Cell Biol. 2010;190:523–531.
- Yu L, Wan FY, Dutta S, et al. Autophagic programmed cell death by selective catalase degradation. Proc Natl Acad Sci U S A. 2006;103:4952–4957.
- Raymond E, Dahan L, Raoul JL, et al. Sunitinib malate for the treatment of pancreatic neuroendocrine tumors. New Engl J Med. 2011;364:501–513.
- Motzer RJ, Hutson TE, Tomczak P, et al. Overall survival and updated results for sunitinib compared with interferon alfa in patients with metastatic renal cell carcinoma. J Clin Oncol. 2009;27:3584–3590.
- Chu TF, Rupnick MA, Kerkela R, et al. Cardiotoxicity associated with tyrosine kinase inhibitor sunitinib. Lancet. 2007;370:2011–2019.
- Khakoo AY, Kassiotis CM, Tannir N, et al. Heart failure associated with sunitinib malate - A multitargeted receptor tyrosine kinase inhibitor. Cancer-Am Cancer Soc. 2008;112:2500–2508.
- Telli ML, Witteles RM, Fisher GA, et al. Cardiotoxicity associated with the cancer therapeutic agent sunitinib malate. Ann Oncol. 2008;19:1613–1618.
- Gan HK, Seruga B, Knox JJ. Sunitinib in solid tumors. Expert Opin Inv Drug. 2009;18:821–834.
- Zhao YQ, Xue T, Yang XC, et al. Autophagy plays an important role in Sunitinib-mediated cell death in H9c2 cardiac muscle cells. Toxicol Appl Pharm. 2010;248:20–27.
- Kimura T, Uesugi M, Takase K, et al. Hsp90 inhibitor geldanamycin attenuates the cytotoxicity of sunitinib in cardiomyocyte via inhibition of the autophagy pathway. Toxicol Appl Pharm. 2017;329:282–292.
- Tang DL, Kang R, Livesey KM, et al. Endogenous HMGB1 regulates autophagy. J Cell Biol. 2010;190:881–892.
- Sun XF, Tang DL. HMGB1-dependent and -independent autophagy. Autophagy. 2014;10:1873–1876.
- Andrassy M, Volz HC, Igwe JC, et al. High-mobility group box-1 in ischemia-reperfusion injury of the heart. Circulation. 2008;117:3216–3226.
- Volz HC, Seidel C, Laohachewin D, et al. HMGB1: the missing link between diabetes mellitus and heart failure. Basic Res Cardiol. 2010;105:805–820.
- Luo PH, Xu ZF, Li GQ, et al. HMGB1 represses the anti-cancer activity of sunitinib by governing TP53 autophagic degradation via its nucleus-to-cytoplasm transport. Autophagy. 2018;14(12):2155–2170. .
- Mizushima N. Methods for monitoring autophagy. Int J Biochem Cell B. 2004;36:2491–2502.
- Kliosnky D. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition) (vol 12, pg 1, 2015). Autophagy. 2016;12:443.
- Luo PH, Yan H, Du JX, et al. PLK1 (polo like kinase 1)-dependent autophagy facilitates gefitinib-induced hepatotoxicity by degrading COX6A1 (cytochrome c oxidase subunit 6A1). Autophagy. 2020;1–17. doi:https://doi.org/10.1080/15548627.2020.1851492.
- Komatsu M, Waguri S, Ueno T, et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol. 2005;169:425–434.
- Hou W, Xie YC, Song XX, et al. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy. 2016;12:1425–1428.
- Nicolas-Avila JA, Lechuga-Vieco AV, Esteban-Martinez L, et al. A Network of Macrophages Supports Mitochondrial Homeostasis in the Heart. Cell. 2020;183:94-+.
- Tong MM, Saito T, Zhai PY, et al. Mitophagy is essential for maintaining cardiac function during high fat diet-induced diabetic cardiomyopathy. Circ Res. 2019;124(9):1360-1371.
- Yu L, Strandberg L, Lenardo MJ. The selectivity of autophagy and its role in cell death and survival. Autophagy. 2008;4:567–573.
- Wang WJ, Wang Y, Chen HZ, et al. Orphan nuclear receptor TR3 acts in autophagic cell death via mitochondrial signaling pathway. Nat Chem Biol. 2014;10:133–140.
- Ramage L, Junne T, Hahne K, et al. Functional cooperation of mitochondrial protein import receptors in yeast. Embo J. 1993;12:4115–4123.
- Cheng MY, Hartl FU, Martin J, et al. Mitochondrial heat-shock protein HSP60 is essential for assembly of proteins imported into yeast mitochondria. Nature. 1989;337:620–625.
- Battula VL, Chen Y, Cabreira MD, et al. Connective tissue growth factor regulates adipocyte differentiation of mesenchymal stromal cells and facilitates leukemia bone marrow engraftment. Blood. 2013;122:357–366.
- Guo YZ, Lu XH, Wang H. Downregulation of miR-18a induces CTGF and promotes proliferation and migration of sodium hyaluronate treated human corneal epithelial cells. Gene. 2016;591:129–136.
- Liu XW, Cai TY, Zhu H, et al. Q6, a novel hypoxia-targeted drug, regulates hypoxia-inducible factor signaling via an autophagy-dependent mechanism in hepatocellular carcinoma. Autophagy. 2014;10:111–122.
- Rogov V, Dotsch V, Johansen T, et al. Interactions between autophagy receptors and ubiquitin-like proteins form the molecular basis for selective autophagy. Mol Cell. 2014;53:167–178.
- Zellner S, Schifferer M, Behrends C. Systematically defining selective autophagy receptor-specific cargo using autophagosome content profiling. Mol Cell. 2021;81(6):1337-1354.
- Zellner S, Behrends C. Autophagosome content profiling reveals receptor-specific cargo candidates. Autophagy. 2021;17:1281–1283.
- Lin CY, Nozawa T, Minowa-Nozawa A, et al. Autophagy receptor tollip facilitates bacterial autophagy by recruiting galectin-7 in response to group a streptococcus infection. Front Cell Infect Mi. 2020;10:583137.
- Limana F, Esposito G, D’Arcangelo D, et al. HMGB1 attenuates cardiac remodelling in the failing heart via enhanced cardiac regeneration and miR-206-mediated inhibition of TIMP-3. Plos One. 2011;6(6):e19845.
- Yamada S, Maruyama I. HMGB1, a novel inflammatory cytokine. Clin Chim Acta. 2007;375:36–42.
- Takahashi T, Shishido T, Kinoshita D, et al. Cardiac nuclear high-mobility group box 1 ameliorates pathological cardiac hypertrophy by inhibiting DNA damage response. Jacc-Basic Transl Sci. 2019;4:234–247.
- Tang DL, Kang R, Livesey KM, et al. High-mobility group box 1 is essential for mitochondrial quality control. Cell Metab. 2011;13:701–711.
- Kang R, Livesey KM, Zeh HJ, et al. Metabolic regulation by HMGB1-mediated autophagy and mitophagy. Autophagy. 2011;7:1256–1258.
- Kumada H. Long-term treatment of chronic hepatitis C with glycyrrhizin [stronger neo-minophagen C (SNMC)] for preventing liver cirrhosis and hepatocellular carcinoma. Oncology-Basel. 2002;62:94–100.
- Lau A, Wang S, Liu WH, et al. Glycyrrhizic acid ameliorates HMGB1-mediated cell death and inflammation after renal ischemia reperfusion injury. Am J Nephrol. 2014;40:84–95.
- Ho CY, Lopez B, Coelho-Filho OR, et al. Myocardial fibrosis as an early manifestation of hypertrophic cardiomyopathy. New Engl J Med. 2010;363:552–563.
- Bouitbir J, Alshaikhali A, Panajatovic MV, et al. Mitochondrial oxidative stress plays a critical role in the cardiotoxicity of sunitinib Running title: sunitinib and oxidative stress in hearts. Toxicology. 2019;426:152281.
- Doherty KR, Wappel RL, Talbert DR, et al. Multi-parameter in vitro toxicity testing of crizotinib, sunitinib, erlotinib, and nilotinib in human cardiomyocyte. Toxicol Appl Pharm. 2013;272:245–255.
- Yang Y, Li N, Chen TS, et al. Sirt3 promotes sensitivity to sunitinib-induced cardiotoxicity via inhibition of GTSP1/JNK/autophagy pathway in vivo and in vitro. Arch Toxicol. 2019;93:3249–3260.
- Yang Y, Li N, Chen TS, et al. Trimetazidine ameliorates sunitinib-induced cardiotoxicity in mice via the AMPK/mTOR/autophagy pathway. Pharm Biol. 2019;57:625–631.
- Galluzzi L, Bravo-San Pedro JM, Levine B, et al. Pharmacological modulation of autophagy: therapeutic potential and persisting obstacles. Nat Rev Drug Discov. 2017;16:487–511.
- Berg TO, Fengsrud M, Stromhaug PE, et al. Isolation and characterization of rat liver amphisomes - Evidence for fusion of autophagosomes with both early and late endosomes. J Biol Chem. 1998;273:21883–21892.
- Lamb CA, Dooley HC, Tooze SA. Endocytosis and autophagy: shared machinery for degradation. Bioessays. 2013;35:34–45.
- Itakura E, Kishi-Itakura C, Mizushima N. The hairpin-type tail-anchored SNARE syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell. 2012;151:1256–1269.
- Accornero F, van Berlo JH, Correll RN, et al. Genetic analysis of connective tissue growth factor as an effector of transforming growth factor beta signaling and cardiac remodeling. Mol Cell Biol. 2015;35:2154–2164.
- Rutkovskiy A, Sagave J, Czibik G, et al. Connective tissue growth factor and bone morphogenetic protein 2 are induced following myocardial ischemia in mice and humans. Scand J Clin Lab Inv. 2017;77:321–331. .
- Wu CK, Wang YC, Lee JK, et al. Connective tissue growth factor and cardiac diastolic dysfunction: human data from the Taiwan Diastolic Heart Failure Registry and molecular basis by cellular and animal models. Eur J Heart Fail. 2014;16:163–172.
- Ahmed MS, Gravning J, Martinov VN, et al. Mechanisms of novel cardioprotective functions of CCN2/CTGF in myocardial ischemia-reperfusion injury. Am J Physiol-Heart C. 2011;300:H1291–H1302. .
- Gravning J, Ahmed MS, von Lueder TG, et al. CCN2/CTGF attenuates myocardial hypertrophy and cardiac dysfunction upon chronic pressure-overload. Int J Cardiol. 2013;168:2049–2056.
- Yan L, Sadoshima J, Vatner DE, et al. Autophagy in ischemic preconditioning and hibernating myocardium. Autophagy. 2009;5:709–712.
- Dosenko VE, Nagibin VS, Tumanovskaya LV, et al. Postconditioning prevents apoptotic, necrotic and autophagic cardiomyocyte cell death in culture. Fiziolohichnyi Zhurnal (Kiev). 2005;51:12–17.
- Hariharan N, Zhai PY, Sadoshima J. Oxidative stress stimulates autophagic flux during ischemia/reperfusion. Antioxid Redox Sign. 2011;14:2179–2190.
- Zhai PY, Sciarretta S, Galeotti J, et al. Differential roles of GSK-3 beta during myocardial ischemia and ischemia/reperfusion. Circ Res. 2011;109:502–U123.
- Li Y, Ma Y, Song LQ, et al. SIRT3 deficiency exacerbates p53/Parkin-mediated mitophagy inhibition and promotes mitochondrial dysfunction: implication for aged hearts. Int J Mol Med. 2018;41:3517–3526.
- Huang CY, Pai PY, Kuo CH, et al. p53-mediated miR-18 repression activates HSF2 for IGF-IIR-dependent myocyte hypertrophy in hypertension-induced heart failure. Cell Death Dis. 2017;8(8):e2990.
- Yoshida Y, Shimizu I, Katsuumi G, et al. P53-induced inflammation exacerbates cardiac dysfunction during pressure overload. Eur Heart J. 2015;36:551.
- Stuhlmiller TJ, Zawistowski JS, Chen X, et al. Kinome and transcriptome profiling reveal broad and distinct activities of erlotinib, sunitinib, and sorafenib in the mouse heart and suggest cardiotoxicity from combined signal transducer and activator of transcription and epidermal growth factor receptor inhibition. J Am Heart Assoc. 2017;6(10):e006635.
- Livesey KM, Kang R, Vernon P, et al. p53/HMGB1 complexes regulate autophagy and apoptosis. Cancer Res. 2012;72:1996–2005.
- Liu RZ, Li T, Zhao GQ. Cytosolic HMGB1 mediates autophagy activation in an emulsified isoflurane anesthesia cell model. Neurochem Res. 2019;44:1090–1100.
- Petrovic A, Bogojevic D, Korac A, et al. Oxidative stress-dependent contribution of HMGB1 to the interplay between apoptosis and autophagy in diabetic rat liver. J Physiol Biochem. 2017;73:511–521.
- Kong Q, Xu LH, Xu W, et al. HMGB1 translocation is involved in the transformation of autophagy complexes and promotes chemoresistance in leukaemia. Int J Oncol. 2015;47:161–170.
- Zhao MY, Yang MH, Yang LC, et al. HMGB1 regulates autophagy through increasing transcriptional activities of JNK and ERK in human myeloid leukemia cells. Bmb Rep. 2011;44:601–606.
- Bangert A, Andrassy M, Muller AM, et al. Critical role of RAGE and HMGB1 in inflammatory heart disease. Proc Natl Acad Sci U S A. 2016;113:E155–E164.
- Ouyang F, Huang H, Zhang MY, et al. HMGB1 induces apoptosis and EMT in association with increased autophagy following H/R injury in cardiomyocyte. Int J Mol Med. 2016;37:679–689.
- Wu RN, Yu TY, Zhou JC, et al. Targeting HMGB1 ameliorates cardiac fibrosis through restoring TLR2-mediated autophagy suppression in myocardial fibroblasts. Int J Cardiol. 2018;267:156–162.
- Kim J, Kundu M, Viollet B, et al. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13:132–U171.
- Jang M, Park R, Kim H, et al. AMPK contributes to autophagosome maturation and lysosomal fusion. Sci Rep-Uk. 2018;8(1):12637.
- Liang B, Guo XL, Jin J, et al. Glycyrrhizic acid inhibits apoptosis and fibrosis in carbon-tetrachloride-induced rat liver injury. World J Gastroenterol. 2015;21:5271–5280.
- Qu LH, Chen C, He W, et al. Glycyrrhizic acid ameliorates LPS-induced acute lung injury by regulating autophagy through the PI3K/AKT/mTOR pathway. Am J Transl Res. 2019;11:2042–2055.
- Zhai CL, Zhang MQ, Zhang Y, et al. Glycyrrhizin protects rat heart against ischemia-reperfusion injury through blockade of HMGB1-dependent phospho-JNK/Bax pathway. Acta Pharmacol Sin. 2012;33:1477–1487.
- Yang J, Shi YF, Chen H, et al. Glycyrrhizic acid attenuates myocardial injury: involvement of RIP140/NF-kB Pathway. Biomed Pharmacother. 2017;95:62–67.
- Li MY, Wen ZS, Xue YR, et al. Cardioprotective effects of glycyrrhizic acid involve inhibition of calcium influx via L-type calcium channels and myocardial contraction in rats. N-S Arch Pharmacol. 2020;393:979–989.
- Prasad KMR, Xu YQ, Yang ZQ, et al. Topoisomerase inhibition accelerates gene expression after adeno-associated virus-mediated gene transfer to the mammalian heart. Mol Ther. 2007;15:764–771.
- Trouet A, Passioukov A, Van Derpoorten K, et al. Extracellularly tumor-activated prodrugs for the selective chemotherapy of cancer: application to doxorubicin and preliminary in vitro and in vivo studies. Cancer Res. 2001;61:2843–2846.
- Li DX, Wu J, Bai Y, et al. Isolation and culture of adult mouse cardiomyocyte for cell signaling and in vitro cardiac hypertrophy. Jove-J Vis Exp. 2014. DOI:https://doi.org/10.3791/51357.