
ABSTRACT
Numerous lines of evidence support the premise that the misfolding and subsequent accumulation of SNCA/α-synuclein (synuclein alpha) is responsible for the underlying neuronal pathology observed in Parkinson disease (PD) and other synucleinopathies. Moreover, the cell-to-cell transfer of these misfolded SNCA species is thought to be responsible for disease progression and the spread of cellular pathology throughout the brain. Previous work has shown that when exogenous, misfolded SNCA fibrils enter cells through endocytosis, they can damage and rupture the membranes of their endocytotic vesicles in which they are trafficked. Rupture of these vesicular membranes exposes intralumenal glycans leading to galectin protein binding, subsequent autophagic protein recruitment, and, ultimately, their introduction into the autophagic-lysosomal pathway. Increasing evidence indicates that both pathological and non-pathological SNCA species undergo autophagy-dependent unconventional secretion. While other proteins have also been shown to be secreted from cells by autophagy, what triggers this release process and how these specific proteins are recruited to a secretory autophagic pathway is largely unknown. Here, we use a human midbrain dopamine (mDA) neuronal culture model to provide evidence in support of a cellular mechanism that explains the cell-to-cell transfer of pathological forms of SNCA that are observed in PD. We demonstrate that LGALS3 (galectin 3) mediates the release of SNCA following vesicular damage. SNCA release is also dependent on TRIM16 (tripartite motif containing 16) and ATG16L1 (autophagy related 16 like 1), providing evidence that secretion of SNCA is mediated by an autophagic secretory pathway.
Introduction
Synucleinopathies are a category of neurodegenerative disorders associated with the accumulation of SNCA/α-synuclein (synuclein alpha) aggregates and progressive cell death throughout the central nervous system. Like many other neurodegenerative diseases, an integral aspect of Parkinson disease (PD) and other synucleinopathies is a self-propagating pathology which ultimately leads to physiological dysfunction, inflammation, and cell death in the affected tissues. In PD and Lewy body dementia (LBD), the accumulation of misfolded, insoluble SNCA in Lewy bodies (LBs) and neurites, collectively called Lewy pathology, are the histopathological hallmark of these diseases [Citation1], and numerous converging lines of experimental and genetic evidence support the premise that SNCA plays a central role in disease pathogenesis [Citation2,Citation3].
The cell-to-cell spread of misfolded SNCA species is thought to drive synucleinopathy disease progression. Multiple levels of evidence, including tissue culture [Citation4–6] and animal models [Citation7–11] of disease, as well as samples from human PD patients receiving therapeutic neuronal grafts to ameliorate disease [Citation12–14], indicate that transfer of misfolded SNCA between cells is associated with disease pathology. In addition to cell-to-cell transfer, another key aspect of disease progression is the ability of misfolded, pathological forms SNCA to permissively template or “seed” the conversion of endogenously expressed SNCA into misfolded forms. Indeed, the addition of exogenous, recombinant SNCA fibrils substantially increases detergent insoluble forms of SNCA that are akin to those observed in pathology as well as cell-to-cell transmission in vivo [Citation8–10Citation15,Citation16]. However, an understanding of the cellular mechanisms responsible for this cell-to-cell spread and propagation of further SNCA misfolding remains to be elucidated.
The autophagic-lysosomal pathway (ALP) is one of the primary degradative pathways in cells, responsible for the turnover of cytosolic content, such as protein aggregates, by lysosomal degradation [Citation17,Citation18]. In macroautophagy, the formation of a phagophore allows the engulfment of organelles and cytosolic proteins, and subsequently closes to form a vesicular compartment known as an autophagosome [Citation17,Citation18]. Degradation then occurs upon autophagosome fusion with the lysosome, exposing the engulfed contents to the hydrolytic enzymes within the lysosome [Citation17,Citation18]. Previous studies have revealed that SNCA is degraded by autophagic mechanisms, including macroautophagy (hereafter autophagy) and chaperone-mediated autophagy [Citation19–21].
The galectins are a family of proteins that share a carbohydrate recognition domain motif that interacts with β-galactoside glycans [Citation22]. Galectin proteins play a role in a broad range of cellular functions including immune responses, signaling, inflammation, and autophagy [Citation22,Citation23]. Several galectins, including LGALS1 (galectin 1), LGALS3, LGALS8 (galectin 8), and LGALS9 (galectin 9), have carbohydrate-rich domains that interact and adhere to the glycans present on the intraluminal membrane proteins of endosomes and lysosomes, such that damage to these vesicles leads to glycan exposure and galectin recruitment [Citation22,Citation24,Citation25]. Galectin recruitment to damaged vesicles leads to their recognition by autophagic adapter proteins and, subsequently, to their degradation via autophagy [Citation25–27]. We and others have demonstrated that fibrillar forms of SNCA, MAPT/tau and other amyloids can induce vesicle damage following endocytosis, leading to the recruitment of LGALS3, LGALS8, autophagic adaptors, and effector proteins [Citation6,Citation28–30]. When postmortem brain tissue from five PD patients was stained for LGALS3 and SNCA phosphorylated at serine 129 (p-S129) to identify LBs, a majority of the examined LBs displayed LGALS3 coronas [Citation28]. The presence of LGALS3 in LBs suggests a history of membrane damage.
Accumulating evidence reveals that the biological functions of galectin proteins are central to the ALP impairment that occurs in PD and other neurodegenerative diseases [Citation31–35]. The Deretic group demonstrated that the re-localization of LGALS3, LGALS8, and LGALS9 to damaged lysosomal compartments coordinates the cellular autophagic response [Citation36–40]. Specifically, in combination with ULK1, TRIM16 (tripartite motif containing 16), and ATG16L1, LGALS3 facilitates the recruitment of autophagic adaptors, receptors, and effectors to damaged lysosomal membranes [Citation38,Citation40]. A recent genome-wide association study reported that single nucleotide polymorphisms in LGALS3 (gene transcript) are associated with an increased risk of PD [Citation35]. Additionally, increased LGALS3 in the cerebrospinal fluid of PD patients has been reported [Citation31,Citation34,Citation41].
Recent studies have also demonstrated that galectins and proteins normally associated with ALP degradation also act to promote an unconventional secretory mechanism referred to as secretory autophagy [Citation39,Citation42,Citation43]. SNCA is known to be released via an unconventional secretory mechanism that is modulated by ALP perturbations [Citation5,Citation44–48]. Specifically, it is known that pharmacological inhibition of ALP degradation increases the secretion of both extracellular vesicle (EV) and non-EV associated SNCA [Citation45,Citation48–54]. In the context of the cell-to-cell spread of SNCA, the role of galectins in mediating secretory autophagy and their association with vesicles damaged by fibrillar SNCA and other amyloid proteins raises the possibility that galectins may promote the release of SNCA from cells, and thereby contribute to the pathological spread of SNCA between cells. Thus, we sought to determine whether LGALS3 contributes to the release of SNCA from cells. We observe that both endogenously expressed SNCA and exogenous SNCA fibrils colocalize in galectin-positive (+) intracellular vesicles and, ultimately, in EVs released from these cells. We also observe that the depletion of LGALS3 or the associated autophagic adaptor protein, TRIM16, as well as ATG16L1, reduce the release of SNCA from cells, including induced pluripotent stem cell (iPSC) derived midbrain dopamine (mDA) neurons.
Results
Exogenous SNCA fibrils are re-secreted with endogenous SNCA and LGALS3 by the ALP
To determine if changes in autophagy affect the secretion of SNCA, we utilized a complementation system similar to what others have used to detect the release of oligomerized SNCA species [Citation5,Citation7,Citation55–61]. This system uses complementing halves of Dual Split Protein (DSP; DSP1-7 and DSP8-11 [Citation62]), composed of two separate complementing fragments of Renilla luciferase (RLuc) and green fluorescent protein (GFP), which require complementation with the other corresponding fragment to function (hereafter referred to as DSP-A and DSP-B, )). We generated constructs that linked SNCA on to the N-terminal region of DSP-A and DSP-B and dually transduced HeLa and SH-SY5Y cells with the SNCA DSP-A and -B constructs to generate stable cell lines expressing both fusions. The advantages of this model include: (1) the ability to measure SNCA in a non-monomeric state, as the DSPs require complementation to gain activity; (2) the ability to visualize SNCA via its GFP signal and sensitively measure secretion by measuring RLuc activity in the culture supernatant; and (3) the ability to measure changes in cell expressed SNCA in response to exogenously added fibrillar SNCA, which can otherwise be difficult to discriminate from input fibrils. Both constructs were observed at their predicted molecular weights when the same lysates were run in parallel and evaluated with antibodies to SNCA, RLuc, and GFP, revealing that these constructs were expressed as intact fusions in these cells (Fig. S1(A-C)).
Figure 1. DSP-SNCA is secreted in response to lysosomal acidification inhibitors and exogenously added SNCA fibrils. (A) A cartoon depiction of SNCA linked to complementing halves A and B of the DSP construct. (B) Relative light units (RLUs) were measured from the RLuc signal in the cultured media from the stated cell lines 24 h after treatment with vehicle (0.1% DMSO) or Baf-A1 indicating the levels of complemented DSP-SNCA. (C) RLU signal detected in the cultured media from SH-SY5Y DSP-SNCA cells 24 h after control or SNCA fibril treatment. Statistical significance determined by a paired t-test. Data shows the mean value from at least three independent experiments where each data point indicates a biological replicate. Data are expressed as M ± SE. For all statistical tests *, **, ***, ****, p < 0.05, 0.01, 0.001, and 0.0001, respectively. (D) Z-stacks of untreated or fluorescently labeled SNCA fibril-treated HeLa DSP-SNCA and mCherry galectin-expressing cells. (E) The white arrow point to colocalized DSP-SNCA+ and mCherry LGALS3+ events in untreated cells and colocalized DSP-SNCA+, mCherry LGALS3+, and SNCA fibril+ events in the SNCA fibril-treated cells. (F) A representative z-stack of dually transduced DSP-SNCA SH-SY5Y cells from untreated and SNCA fibril-treated cells probed for LGALS3. Cell boundaries are shown as a white border. White arrows point to DSP-SNCA+, LGALS3+ double-positive events in untreated and DSP-SNCA+, LGALS3+, SNCA fibril+ triple-positive events in the SNCA fibril-treated conditions. (F) Extracellular colocalization of DSP-SNCA+ and mCherry LGALS3+ events observed in untreated cells.
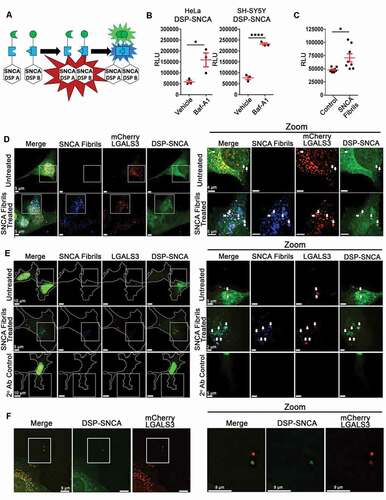
We next evaluated how SNCA fibrils treatment influenced the formation of higher molecular weight SNCA in the SNCA DSP-A + B SH-SY5Y cells (hereafter referred to as DSP-SNCA SH-SY5Y cells). Previous work suggests that fibrillar SNCA can recruit monomeric SNCA in cell culture, resulting in increased high molecular weight SNCA species [Citation63], such that following SNCA fibril treatment, endogenous SNCA represents the primary component of phosphorylated serine 129 (p-S129) SNCA species found within inclusions [Citation15]. To better understand the species of SNCA present in these cells before and after treatment with exogenous fibrils, we used antibodies specific for pS129 SNCA, aggregated conformation SNCA (MJFR-14-6-4-2) and monoclonal GFP (B-2). The same DSP-SNCA SH-SY5Y cells lysates were assessed by non-denaturing SDS-PAGE. In all cases, a band at ~48 kDa was observed for both the control and the SNCA fibril treated cells lysates, when p-S129 SNCA or aggregated conformation SNCA antibodies were used (Fig. S1(D)) as well as an intense band at ~33 kDa for GFP antibodies. However, higher molecular weight bands were also observed in all blots. When probed for p-S129 SNCA, SNCA fibril treatment resulted in more intense high molecular weight (> 150 kDa) bands (Fig. S1(D)). An increased band intensity at ~48 kDa was also observed in the SNCA fibril treated cell lysates when probed for GFP (Fig. S1(D)). Finally, increased band intensity was also observed in the SNCA fibril treated cell lysates relative to the control lysates when probed with the SNCA aggregate specific antibody (Fig. S1(D)). These data demonstrate that addition of exogenous SNCA fibrils induces phosphorylation of DSP-SNCA fusion proteins and leads to their accumulation on higher molecular weight species, consistent with studies examining the influence exogenous SNCA fibrils are known to have on native SNCA [Citation15].
To determine how ALP impairment alters the secretion of DSP-SNCA, DSP-SNCA HeLa and SH-SY5Y were treated with bafilomycin A1 (Baf-A1), a lysosomal acidification inhibitor that also prevents lysosome-autophagosome fusion [Citation64–66]. Baf-A1 treatment significantly increased complemented DSP-SNCA secretion ()), consistent with other studies examining native SNCA release and studies using similar complementation systems [Citation45–48,Citation52,Citation54,Citation67]. These data demonstrate that the increase in SNCA secretion observed in other studies is recapitulated by our DSP-SNCA cells, revealing it as a useful model to monitor SNCA release and characterize the pathway promoting secretion.
We next wanted to determine if the ALP dysfunction induced by exogenous SNCA fibrils can drive the release of endogenously expressed SNCA, which would provide a putative mechanism driving the cell-to-cell transfer of SNCA. To this end, we applied fluorophore labeled recombinant SNCA fibrils [Citation16,Citation28] to DSP-SNCA SH-SY5Y cells. Measuring the luciferase activity specific to the DSP-SNCA complementation, we observed an increase in the release of DSP-SNCA secretion after the SH-SY5Y cells were treated with SNCA fibrils ()). These data demonstrate that the cellular response to exogenous SNCA fibrils leads to an increase in the release of endogenously expressed SNCA.
In previous work, we found that SNCA fibrils treatment induced lysosomal rupture resulting in LGALS3 re-localization in cells over-expressing mCherry-LGALS3 [Citation6,Citation28]. We therefore examined the localization of DSP-SNCA in cells expressing mCherry-LGALS3 following treatment with SNCA fibrils. We observed numerous instances of colocalization between DSP-SNCA, SNCA fibrils, and mCherry-LGALS3 following fibril treatment ()). Some degree of colocalization was also observed between DSP-SNCA and mCherry-LGALS3 in the absence of SNCA fibrils ()). Similar results were obtained when the localization of endogenous LGALS3 expressed in these cells was assessed by immunofluorescence ()). We also observed instances of what appeared to be examples of extracellular accumulations of double positive DSP-SNCA+ and mCherry LGALS3+ independent of SNCA fibrils treatment ()). Given these observations, we next sought to determine if LGALS3 and DSP-SNCA were released from these cells in the context of EVs.
Exogenous and endogenous SNCA are released in LGALS3-containing EVs
It is known that SNCA is released in EVs and that these EVs have the potential to mediate the spread of pathological SNCA from cell-to-cell [Citation5,Citation47,Citation68]. Furthermore, treatment with either the lysosome acidification inhibitors Baf-A1 or chloroquine is known to affect EV secretion, increasing SNCA levels and impacting their composition, which leads to a hybrid “autophagosome-EV signature”, with increased levels of autophagic proteins including LC3-II [Citation47]. In addition to SNCA, LGALS3 is also secreted in EVs and has been shown to be increased during ALP impairment [Citation69,Citation70]. We, therefore, investigated the degree to which DSP-SNCA and LGALS3 colocalized among EVs released from cells and the impact of SNCA fibrils treatment on these EV populations. Nanoparticle tracking analysis (NTA) confirmed that the concentration of EVs from cultured media had a 202.3 ± 4 nm mean diameter size distribution while the most common diameter was 144.5 nm (Fig. S2(A)), consistent with previous observations in similar studies [Citation47,Citation71].
Following NTA validation, the contents of EVs released from our DSP-SNCA SH-SY5Y cells were characterized in response to SNCA fibrils alone or when co-treated with the commonly employed EV secretion inhibitor GW4869 which acts to impair SMPD3/neutral sphingomyelinase 2, preventing the formation of intraluminal vesicles in multivesicular bodies (MVBs), and, ultimately, blocking the release of EVs [Citation72]. The EVs from each condition were concentrated from equal volumes of cultured media, and both the concentrate EVs and lysates were assessed by non-reducing SDS-PAGE. GW4869 treatment reduced the levels of the canonical EV markers CD63 and PDCD6IP/Alix, as expected, as well as LGALS3 (Fig. S2(B)). (Fig. S2(B)). A more intense p-S129 SNCA band in the vehicle plus SNCA fibril EVs was observed compared to the other treatment conditions at molecular weights consistent with SNCA DSP-A and -B (Fig. S2(B)). Previous works indicates that GW4869 also affects the formation of autophagosomes and the secretion of LC3 [Citation73,Citation74]. Thus, we also compared LC3-II levels across all conditions. In agreement with these previous works GW4869 also reduced LC3-II levels compared to control as well as compared to GW4869 plus SNCA fibrils compared to SNCA fibrils treatment alone (Fig. S2(B)). GW4869 treatment impaired the release of p-S129 SNCA species of SNCA from these cells (Fig. S2(B)). No difference in cell viability among treatments were observed when measured by a lactate dehydrogenase (LDH) assay (Fig. S2(C)). Collectively, these data suggest that released EVs from DSP-SNCA cells contain LGALS3, the pathologically associated p-S129 SNCA, the autophagic marker LC3-II, and that their secretion is not stemming from treatment induced cell death.
We next imaged EVs released from HeLa DSP-SNCA cells, including following treatment with exogenous, fluorescently labeled SNCA fibrils to determine if DSP-SNCA could be detected in EVs in association with LGALS3. To this end, low speed centrifugation of DSP-SNCA cell culture supernatant was performed to adhere EVs released from cells onto coverslips in preparation for fluorescent microscopy imaging and analysis. Such low-speed centrifugation (i.e., 1,200 × g for 2 h) is sufficient to bind viral particles and EVs to coverslips but is insufficient to promote the binding of soluble proteins [Citation75,Citation76]. Following centrifugation, the bound EVs were fixed then stained with an antibody to LGALS3 to allow LGALS3+ EVs to be identified. The EVs were then imaged by wide-field fluorescence microscopy, which revealed puncta corresponding to the input ATTO 647 SNCA fibrils, LGALS3, and DSP-SNCA. We observed varying degrees of individual signal as well as examples double and triple colocalization in individual puncta ()). To characterize these differences among the released EVs from these cells, the EV multiplex analysis of colocalization (EV-MAC) [Citation75] workflow was used. This approach allows for the characterization of EVs at the single EV level to assess the relative degree of colocalization of individual EV constituents within the total EV population based on their cargoes, using a gating process similar to that used in flow cytometry. We applied this approach to determine the relative degree of colocalization of DSP-SNCA, SNCA fibrils, and LGALS3 puncta. We first assessed the degree to which DSP-SNCA+ EVs colocalized with LGALS3 or recombinant, input SNCA fibrils. Surface masks created around the DSP-SNCA signal reliably detected DSP SNCA+ puncta observed in these experiments ()). Analysis of the maximum intensity of LGALS3 and SNCA fibril signal within these DSP-SNCA masks from a representative image revealed colocalization of both LGALS3 and input fibrils in these puncta ()). Corresponding signal associated with secondary antibody controls or samples lacking recombinant fibrils was used to establish background thresholds for LGALS3 and SNCA fibril colocalization, respectively. The degree of colocalization of the DSP-SNCA surface masks with LGALS3 and SNCA fibrils from each of 20 images was calculated and, subsequently, averaged for each replicate. The averages among replicates were then compiled to determine the relative degree of colocalization of LGALS3 and SNCA fibrils with DSP-SNCA ()). In untreated cells, ~29% of the DSP-SNCA puncta were positive for LGALS3 (), left). Following SNCA fibrils treatment, ~30% of DSP-SNCA puncta colocalized with signal from the input SNCA fibrils, with ~66% of these events also being positive for LGALS3. Fibril treatment also increased the number of DSP-SNCA puncta that were positive for LGALS3 to ~60% (), middle). Furthermore, SNCA fibrils treatment increased the intensity of DSP-SNCA puncta released following treatment (Fig. S2(D)), suggesting larger accumulations of DSP-SNCA are released in EVs following SNCA fibrils treatment.
Figure 2. Secreted LGALS3 and DSP-SNCA colocalize with exogenously added SNCA fibrils. (A) A representative z-stack from a constructed z-stack MIP obtained from the cultured media of untreated HeLa DSP-SNCA and those treated with SNCA fibrils. Representative MIPs of z-stack images for cultured media obtained from untreated HeLa DSP-SNCA processed for immunofluorescence without the primary antibody (2° Ab treatment) are also given. The white arrows point to DSP-SNCA+, LGALS3+ double-positive events in untreated and DSP-SNCA+, LGALS3+, SNCA Fibril+ triple-positive events in the SNCA fibril treated conditions. (B) Demonstration of the Spots masking algorithm built around the DSP-SNCA channel from a z-stack MIP showing specificity for the desired channel. (C) Representative colocalization plots of the maximum fluorescence intensities of the LGALS3 (y-axis) and SNCA fibril (x-axis) signal found within each DSP-SNCA masking algorithm Spot pooled from a total of 15 images from a single treatment conditions’ coverslip. Each data point represents a single recognized DSP-SNCA+ spot within an image. Quadrant numbers indicate the percent colocalization of DSP-SNCA+ where the relative background signal was determined based on the LGALS3 and SNCA fibril maximum intensity signal measured in the 2° antibody control condition. The percent single DSP-SNCA+ positive, DSP-SNCA+ SNCA fibril+ double positive, DSP-SNCA+ LGALS3+ double positive, and DSP-SNCA+ SNCA fibril+ LGALS3+ triple positive are given in the bottom left, bottom right, top left, and top right quadrants, respectively. (D) Summarized average colocalization of DSP-SNCA+ masking algorithm Spots from four independent experiments. DSP-SNCA+ Spots that are single positive (green), DSP-SNCA+ SNCA fibril+ double positive (cyan), DSP-SNCA+ LGALS3+ double positive (yellow), and DSP-SNCA+ SNCA fibril+ LGALS3+ triple positive (white). (E) Representative colocalization plots of the maximum fluorescence intensities of the DSP-SNCA (y-axis) and SNCA fibril (x-axis) signal found within each LGALS3 masking algorithm Spot from the same images set of images. Colocalization plots are from a single image. The percent single LGALS3+ positive, LGALS3+ SNCA fibril+ double positive, LGALS3+ DSP-SNCA+ double positive, and LGALS3+ SNCA fibril+ DSP-SNCA+ triple positive are given in the bottom left, bottom right, top left, and top right quadrants, respectively. (F) Summarized average colocalization of LGALS3+ masking algorithm Spots from four independent experiments. LGALS3+ Spots that are single positive (red), LGALS3+ SNCA fibril+ double positive (magenta), LGALS3+ DSP-SNCA+ double positive (yellow), and LGALS3+ DSP-SNCA+ SNCA fibril+ triple positive (white). Summarized average colocalization of (D) DSP-SNCA or (F) LGALS3 masking algorithm Spots from four independent experiments. The degree of colocalization of DSP-SNCA or LGALS3 with the respective other channels was determined based on the background signal measured in the 2° Ab control. Data are expressed as M ± SE (n = 4 coverslips, where M was determined from 15 images).
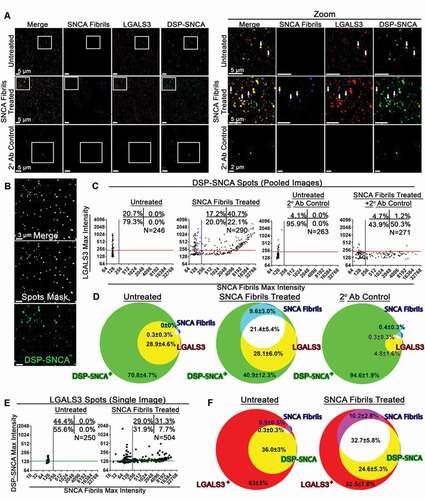
A reciprocal analysis was performed to determine the fraction of LGALS3+ EVs that also contained DSP-SNCA and fluorescent SNCA fibrils using the same images. This process was performed by creating surface masks around the LGALS3 signal and measuring the degree to which each LGALS3 mask was positive for DSP-SNCA or SNCA fibrils ()). This analysis revealed that ~36.0% of LGALS3+ puncta were positive for DSP-SNCA (), left). Fibril treatment increased the fraction of LGALS3+ EVs that were also positive for DSP-SNCA to ~57%, and a majority of these were also positive for the input fibrils (), right). Collectively, this indicates that: (1) EVs containing both LGALS3 and SNCA are released from cells; (2) Exogenous SNCA fibrils increased the relative abundance of DSP-SNCA in LGALS3+ EVs; and (3) Both endogenous and exogenous SNCA can be released from cells in the same EVs. Taken together with prior studies by us and others showing that SNCA fibrils can induce the rupture of lysosomes following endocytosis [Citation6,Citation28,Citation29,Citation77], this further suggests that lysosomal damage can promote the release of endogenously expressed SNCA as well as the original exogenous fibrillar species.
SNCA are also released in LGALS3-containing EVs from midbrain dopaminergic neurons
To better understand the impact of SNCA fibrils and lysosomal damage on SNCA release in cells expressing native, unmodified SNCA, we used midbrain dopaminergic (mDA) neurons derived from human induced pluripotent stem cells (iPSCs). We first validated that LGALS3 was also secreted from mDA neuron EVs. EVs from the cultured media of mDA neurons were concentrated by differential ultracentrifugation, stained with immunogold antibodies for the tetraspannin CD63 (10 nm), LGALS3 (15 nm), and SNCA (5 nm), and imaged using transmission electron microscopy analysis ()). Double- and triple-positive vesicles for the exosomal marker CD63, as well as LGALS3 and SNCA were observed ()). Further characterization by NTA determined the mean diameter of concentrated mDA EVs was 149.4 ± 7.4 nm, and the most common vesicle diameter was 106.5 nm ()). These results are consistent with previous reports for SNCA associated EVs [Citation47,Citation71,Citation78].
Figure 3. LGALS3 inhibition affects the composition of secreted extracellular vesicles from mDA neurons. (A) TEM from concentrated WT mDA culture media following immunogold labeling. Representative image shows SNCA+ (5 nm), CD63+ (10 nm), and LGALS3+ (15 nm) triple positive EVs. (B) NTA analysis of EVs from WT mDA neurons following differential ultracentrifugation. EV sizes binned in 1 nm increments with a bin center of 0.5 nm. NTA of resuspended mDA EVs were performed in technical quintuplicates, denoted as different shades of green, blue, or red for 3 independent samples. Each sample was from distinct mDA differentiation. (C) Representative z-stack from a constructed z-stack MIP obtained from concentrated EVs from mDA neuron cultured media. Concentrated EVs were stained for LGALS3, SNCA, and combined pool of mouse anti-TSPAN/tetraspanin antibodies against CD9, CD63, and CD81. Processed immunofluorescence with only the LGALS3 primary antibody and the secondary antibodies against all three channels (2° antibody control) was also performed to determine the background level of fluorescence. (D) Summarized average colocalization of LGALS3+ masking algorithm Spots from three independent experiments. LGALS3+ Spots that are single positive (green), LGALS3+ SNCA+ double positive (yellow), LGALS3+ TSPAN+ double positive (cyan), and LGALS3+ SNCA+ TSPAN+ triple positive (white). (E) Representative non-reducing SDS-PAGE of concentrated EVs and the corresponding lysates from mDA neurons treated with vehicle, TD139, SNCA fibrils, or TD139 + SNCA fibrils. (F) Relative SDS-PAGE band intensities of (E) in the EVs normalized to cell lysate GAPDH intensity. Data are expressed as M ± SE. Intensity data (C-D) (n = 3 independent experiments, 15 images per experiment, pooled colocalization data from each image among experiments). E, pooled cultured media from n = 3. F, n = 3 derived from the same samples as D.
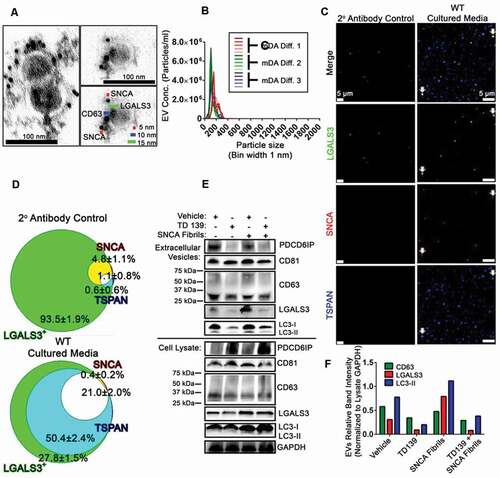
Using the EV-MAC workflow, concentrated EVs from mDA neurons were spinoculated and then probed with antibodies for either LGALS3 only or LGALS3, SNCA and the collective TSPAN EV markers CD9, CD63, and CD81 using pooled antibodies from the same species (hereafter referred to as a TSPAN). The same secondary antibodies were used in both conditions to determine the degree the LGALS3, SNCA, and TSPAN colocalized relative to the LGALS3 only (plus secondaries against SNCA and TSPAN) condition. Following imaging ()) and analysis, the degree to which LGALS3 colocalized with a TSPAN independent of SNCA was 50.4 ± 2.4%, and double colocalized with a TSPAN and SNCA was 21.0 ± 2.0% ()). Conversely, the percentage of LGALS3 colocalized with SNCA independent of the TSPANs was negligible and consistent with background levels measured in the absence of antibody and likely reflects background staining ()).
Given the results of our previous experiments using GW4869, EVs from mDA neurons were evaluated following an extended time course in which cells were subjected to vehicle or the LGALS3 inhibitor, TD139, treatment with or without an initial treatment of SNCA fibrils. The culture media was pooled from three independent cultures from the same, respective treatment condition, and concentrated to get a sufficient sample quantity. Non-reducing SDS-PAGE of the concentrated EVs and lysate was performed ()). The presence of the canonical EV markers PDCD6IP/ALIX (programmed cell death 6 interacting protein) and CD81 were observed in all treatment conditions. Similarly, CD63 was observed in all conditions. However, TD139 treatment lowered the observed CD63 levels in the EVs ()). TD139 treatment also reduced the LGALS3 and LC3-II band intensity relative to vehicle ()). This trend was also observed relative to the TD139 plus SNCA fibril treatment and vehicle plus SNCA fibril treatment conditions ()). Conversely LGALS3 band intensity was increased in the SNCA fibril treatment condition ()). No difference in cell viability between treatment conditions was observed (Fig. S2(E)) Collectively, these data support the notion that LGALS3 is released in mDA EVs and during LGALS3 inhibition, reduced autophagy associated EV release may occur.
SNCA fibrils treatment inhibits MTOR and upregulates autophagy in human mDA neurons
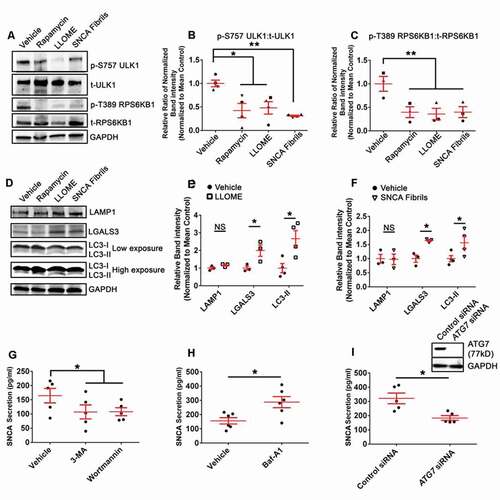
We have previously demonstrated that treating cells with SNCA fibrils results in uptake into endo-lysosomal compartments where they elicit membrane damage [Citation6,Citation28]. Recent studies have shown that lysosomal damage results in an upregulated cellular autophagic response in a variety of cell lines [Citation36–38,Citation40]. Thus, experiments were performed characterize the response of human mDA neurons to treatment with SNCA fibrils or L-leucyl-L-leucine methyl ester (LLOME; 2 µM; Santa Cruz Biotechology, sc-285992A), which pharmacologically induces lysosomal membrane damage. To assess the autophagic activation state in response to these treatments, phosphorylation of RPS6KB1/p70 S6K1 (ribosomal protein S6 kinase B 1) at threonine 389 (T389) and the phosphorylation of ULK1 at serine 757 (S757) was assessed, as MTOR suppresses autophagy by phosphorylating RPS6KB1 at T389 and ULK1 at S757. The ratio of each phosphorylated protein was compared to its respective total levels to control for possible differences in protein degradation or expression. Western blot analysis revealed that treatment with either SNCA fibrils, LLOME or the MTOR inhibitor rapamycin, reduced the relative band intensity of both phosphorylated ULK1 at S757 (p-S757 ULK1) and phosphorylated RPS6KB1 at T389 (p-T389 RPS6KB1) (-C)). No differences in cell viability were observed among treatment conditions (Fig. S3(A)). In agreement with these observations, we observed that SNCA fibril treatment or LLOME increased the levels of LC3-II in cells ()), while the levels of LAMP1 (lysosome associated membrane protein 1) were unaffected. We also observed an increase in LGALS3 expression levels following treatment with SNCA fibrils or LLOME, consistent with another study seeing an increase in LGALS3 levels following LLOME treatment in other cell types [Citation79]. ()). Collectively, these data suggest that SNCA fibrils induce similar changes in human mDA neurons as pharmacological lysosomal injury, leading to an increased cellular autophagic response in this cell type.
Figure 4. Lysosomal damage as a consequence of SNCA fibril or LLOME treatment elicits an autophagic response in mDA neurons. (A, D) Representative western blot of LAMP1, p-S757 ULK1, t-ULK1, p-T389 RPS6KB1, t-RPS6KB1, LGALS3, LC3-I, LC3-II, and GAPDH for mDA neurons treated with vehicle (0.1% DMSO), rapamycin, LLOME, or SNCA fibrils. (B, C) The quantification of p-T389 RPS6KB1, t-RPS6KB1, p-S757 ULK1, and t-ULK1. (E, F) Quantification of LAMP1, LGALS3, and LC3-II for LLOME (E) and SNCA fibrils treatment (F). The different symbols correspond to paired replicates (B, C). Cells were treated with SNCA fibrils, rapamycin, or vehicle for 24 h; cells were treated with LLOME for 4 h. (G, H) Secreted SNCA concentrations present in unlysed culture media following treatment with the early autophagy inhibitors Wor or 3-MA (G), the late-autophagy inhibitor Baf-A1 (H) or vehicle (0.1% DMSO) for 24 h as measured by SNCA sandwich ELISA. (I) A representative western blot demonstrating KD of ATG7 from mDA neurons and SNCA from unlysed, cultured media by ELISA. Cultured media was collected from 48 to 72 h post-transfection. Data are expressed as M ± SE (B, n = 4; E-F, n = 4; G, n = 5; H, n = 6; I, n = 5). Statistical significance was determined following natural log transformation and one-way ANOVA with Tukey’s post hoc tests. (G-I) Statistical significance was determined by paired two-tailed t-tests. For all statistical tests *, **, ***, ****, p < 0.05, 0.01, 0.001, and 0.0001, respectively.
Early inhibition of autophagy reduces autophagosome-associated SNCA secretion
Previous studies have found that inhibition autophagy of alters the release of SNCA in a number of cellular models, with some disparities that may be due to cell types used in these studies, and none of these studies examined this response in human mDA neurons [Citation45,Citation48,Citation50,Citation54]. To determine how impaired autophagosome formation influences SNCA secretion from mDA neurons, cells were treated with the early autophagy inhibitors, wortmannin (Wor) or 3-methyladenine (3-MA) and secreted SNCA was measured from culture media by enzyme-linked immunosorbent assay (ELISA). Wor and 3-MA prevent autophagy by inhibiting certain lipid kinases, including the class III phosphatidylinositol 3-kinase (PtdIns3K), required for initial phagophore formation. Treatment with Wor and 3-MA significantly reduced SNCA secretion ()). In contrast, late-stage inhibition by Baf-A1 increased SNCA secretion ()). Similar to the pharmacologically induced early autophagy inhibitors, siRNA-mediated depletion of ATG7 significant decreased SNCA secretion ()). Treatment with Wor, 3-MA, or Baf-A1 had no effect on cell viability (Fig. S3(B, C)) nor did ATG7 depletion (Fig. S3(D)).
To determine if LGALS3 secretion was similarly dependent on autophagic processes, SH-SY5Y cells were transduced to express HA-Firefly Luciferase-LGALS3 (FLuc LGALS3). After validating FLuc LGALS3 was not undergoing cleavage (Fig. S3(E)), cells were treated with Wor, 3-MA, and Baf-A1. Wor or 3-MA treatment significantly reduced the release of FLuc LGALS3 (Fig. S3(F)). Conversely, Baf-A1 treatment significantly increased FLuc LGALS3 secretion (Fig. S3(F)). These corresponding changes are in-line with what was observed for SNCA secretion (, H)) and consistent with other studies observing LGALS3 release via secretory autophagy [Citation70,Citation79]. Collectively, these data suggest that the secretion of SNCA is affected by early-stage or late-stage autophagic impairment in mDA neurons. More specifically, an impairment of autophagy at the early-stage of autophagosome formation reduced secretion while impairment at the late-stage of autophagosome-lysosome fusion increased secretion, consistent with previous studies in other cell types [Citation45,Citation48–54].
Secretion of SNCA is regulated by LGALS3 and autophagic adaptor and effector proteins
To determine if LGALS3 expression plays a functional role in SNCA secretion, mDA neurons were treated with the LGALS3 inhibitor, TD139. TD139 treatment significantly reduced SNCA secretion compared to vehicle ()). We also depleted LGALS3 using siRNA in mDA neurons, including both neurons differentiated from iPSCs in our lab as well as the commercially available human mDA iCell Dopaneurons. LGALS3 knockdown (KD) significantly decreased SNCA secretion in both mDA models, although the effect was modest in some experiments, as was LGALS3 depletion measured by western blot (), Fig. S4(A)). However, when the variability in the degree of LGALS3 depletion by siRNA was plotted against the relative SNCA release for each experiment, the degree of LGALS3 depletion positively correlated to the degree of reduction in SNCA release observed ()). Similarly, CRISPR-Cas9-mediated LGALS3 knockout (KO) in SH-SY5Y DSP-SNCA cells significantly decreased DSP-SNCA secretion ()). Corresponding measurements of the lysates revealed that DSP-SNCA levels were significantly greater in the SH-SY5Y DSP-SNCA LGALS3 KO cells compared to that observed in the lysates of the control KO cells ()), suggesting the change in SNCA secretion was not driven by differences in DSP-SNCA expression levels. No differences in cell viability were observed among the LGALS3 KO and the control KO cells (Fig. S4(B)).
Figure 5. Neuronal LGALS3 influences SNCA secretion and is reciprocally secreted during SNCA fibrils treatment. (A) Relative fold difference in SNCA from mDA cultured media treated with TD139 (5 μM) or vehicle (0.1% DMSO) treated control after 24 h measured by ELISA. (B) Mean fold difference in SNCA in human mDA neuron culture media 48–72 h post siRNA transfection, relative depletion of LGALS3, and representative KD. (B) Values were determined by ELISA and normalized to the mean of the control group (i.e. mean control). (C) A plot of the normalized to mean control data in (A) combined with (Fig. S4(A)) to illustrate the linear relationship between relative LGALS3 depletion (x-axis) and mean fold SNCA secretion (y-axis). (D) RLUs of RLuc activity of cultured media from CRISPR-Cas9-mediated KO of LGALS3 or control in SH-SY5Y DSP-SNCA cell-lines over a 24-h period, one week after selection, and demonstration of KO. (E) RLUs of RLuc activity from control or LGALS3 CRISPR-Cas9 mediated KO SH-SY5Y DSP-SNCA cell lysates after 24 h (n = 3). (F) Level of SNCA in the cultured media of mDA neuronal cultures at 48 h post-transduction measured by ELISA. (G) SNCA concentration present in empty vector control or FLuc LGALS3 transduced mDA neurons cell lysates after 96 h measured by ELISA. (H) RLU levels detected from cultured mDA neurons transduced with FLuc LGALS3 and either untreated or treated with SNCA fibrils at 48 h post-transduction. (I) Fold difference in LGALS3 present in the cultured media from the untreated control or SNCA fibrils treated mDA neurons after 24 h. (J, K) Lysosomal dysfunction assay shows the normalized RFUs from mDA neuronal cultures loaded with Magic Red CTSB dye. Data from J and K are from the same experiments; the data is displayed in 2 graphs to increase clarity. RFU normalization was calculated from the endpoint of the control siRNA plus vehicle (0.1% DMSO) condition. (L) Quantification of the area under the curve for each stated condition. The vehicle minus Baf-A1 was used to determine lysosomal activity. All data are expressed as M ± SE (A, n = 6; B, n = 3; C, n = 11; D and E, n = 6; F, n = 11; G, n = 4; H, n = 5; I, n = 6; J-L; pooled data from n = 4 independent experiments with 1–3 conditions per experiment). Statistical significance was determined by evaluating Pearson’s r (C), paired t-test (A, D, E, F, G, and H), paired one-way ANOVA with Dunnett’s post hoc tests (L), or ratio paired t-test (B and I) to better account for the distribution of values < 1. For all statistical tests *, **, ***, ****, p < 0.05, 0.01, 0.001, and 0.0001, respectively.
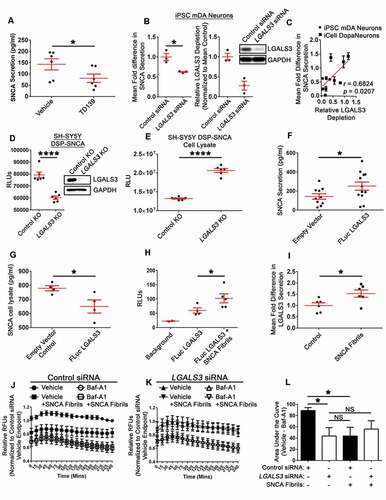
In a reciprocal fashion, we also asked if LGALS3 over-expression influenced endogenous SNCA secretion. mDA neurons were dually transduced with FLuc LGALS3 and RLuc (to control for transduction efficiency) lentiviral constructs, and then endogenous SNCA release was evaluated. FLuc LGALS3 expression increased SNCA secretion ()). Conversely, FLuc LGALS3 transduced mDA lysates had significantly less SNCA when measured by SNCA ELISA ()). Moreover, when the transduced human mDA neurons were treated with SNCA fibrils, increased FLuc LGALS3 was detected in the cultured media ()). The RLuc signal was comparable among the FLuc LGALS3 cells regardless of treatment, suggesting comparable transduction efficiency (Fig. S4(C)). An analysis of the lysate revealed the FLuc LGALS3 construct was not undergoing cleavage (Fig. S4(D)), and that cell viability was comparable among the empty vector control and the FLuc LGALS3 transduced cells (Fig. S4(E)). Finally, when wild type (WT) mDA neurons were treated with SNCA fibrils, increased LGALS3 secretion was also observed in the cultured media when measured by ELISA ()). These results demonstrate that LGALS3 expression levels are positively correlated to SNCA release, with decreases in LGALS3 expression leading to reduced SNCA release and increased LGALS3 expression leading to an increase in SNCA release.
LGALS3 depletion impairs lysosomal degradative function and reduces autophagic flux during SNCA fibril treatment
It has recently been shown that LGALS3 plays a role in maintaining lysosome membrane integrity and homeostasis, in addition to recruiting autophagic proteins to organelles that undergo extensive membrane damage [Citation38,Citation80]. As we observed that LGALS3 regulates the release of SNCA, even in the absence of acute vesicular damage, we reasoned that LGALS3 may be acting in a similar fashion to influence SNCA release from mDA neurons. To test this hypothesis, we measured CTSB (cathepsin B) activity in these cells following LGALS3 depletion and SNCA fibril treatment, and the impact of SNCA fibril treatment following LGALS3 depletion. CTSB activity was measured to evaluate lysosomal acidification based on the fluorescent Magic Red signal. To determine the relative impact of LGALS3 depletion or SNCA fibril treatment, mDA neurons were treated with either vehicle, to assess total cellular levels of CTSB activity, or with Baf-A1, to inhibit lysosome acidification, to determine the fraction of CTSB fluorescence dependent on lysosomal degradation. Fluorescence intensity was then measured every 15 min for 4 h (, K)), the area under the curve was integrated for each of the treatments, and the lysosomal contribution was calculated by subtracting the AUC for the vehicle treated condition from the AUC for the treatment condition ()). LGALS3 depletion alone decreased lysosomal degradative capacity in these cells ()), consistent with the observation that it acts to maintain lysosomal homeostasis in the absence of acute insult in other cell types [Citation38,Citation80]. Similarly, SH-SY5Y DSP-SNCA cells treated with the LGALS3 inhibitor, TD139, had significantly reduced lysosomal degradative activity relative to those treated with vehicle (Fig. S5(A, B)). SNCA fibrils treatment of cells transfected with control siRNA revealed that fibril treatment also reduced lysosomal degradative capacity, consistent with prior studies showing fibrils can induce damage to lysosomes following endocytosis [Citation6,Citation67,Citation81]. However, there was no significant difference in the degree of lysosomal degradative activity for mDA neurons that received a LGALS3 KD and were treated with SNCA fibrils compared to the mDA neurons that were LGALS3 depleted or were treated with SNCA fibrils ()). No differences in cell viability were observed following these treatments (Fig. S5(C)).
Afterward, the mDA neurons were lysed to evaluate autophagic flux by western blot (Fig. S5(D)) based on previously define criteria [Citation82]. This was performed by comparing the relative levels of SQSTM1/p62 and LC3-II among the lysosome uninhibited cells (vehicle) and lysosome inhibited cells (Baf-A1) cells with respect to control and LGALS3 siRNA treatment and in the presence or absence of SNCA fibrils. Based on the differences in LC3-II and SQSTM1 levels, one can determine if increased autophagosomes are the result of upregulated autophagy or reduced autophagic clearance. Baf-A1 treatment significantly increased SQSTM1 levels in control siRNA treated cells but had no effect during SNCA fibrils treatment (Fig. S5(E)). No difference in SQSTM1 levels were observed in the LGALS3 depleted cells following Baf-A1 or SNCA fibrils treatment. of the other conditions (Fig. S5(E)). No differences in SQSTM1 levels were observed in either the Control siRNA treated cells or LGALS3 siRNA treated cells when the SNCA fibrils treatment and the SNCA fibrils and Baf-A1 co-treatment conditions were compared (Fig. S5(E)). Notably, following SNCA fibril treatment in both LGALS3 depleted and non-depleted cells, SQSTM1 levels were more variable, with varying degrees of higher molecular weight bands observed. Both Baf-A1 and SNCA fibrils increased LC3-II, while LGALS3 KD decreased LC3-II (Fig. S5(F)). While it trended toward an increase, no significant difference in LC3-II was observed in Baf-A1 or SNCA fibrils among LGALS3-depleted mDA neurons. LC3-II levels were significantly greater for control siRNA treated than LGALS3 siRNA treated mDA neurons after Baf-A1 treatment (Fig. S5(F)). LC3-II levels were significantly greater for mDA neurons co-treated with Baf-A1 and SNCA fibrils compared to mDA neurons treated with SNCA fibrils only (Fig. S5(F)). In mDA neurons in which LGALS3 was depleted, there was no difference in LC3-II levels for those treated with SNCA fibrils and those co-treated with Baf-A1 and SNCA fibrils. No differences in LC3-II levels were observed between LGALS3 depleted mDA neurons and nondepleted neurons treated with SNCA fibrils and LGALS3 depleted mDA neurons and nondepleted neurons co-treated with Baf-A1 and SNCA fibrils. (Fig. S5(F)). Although treatment of mDA neurons with SNCA fibrils increased LGALS3 levels, no other between groups differences were observed (Fig. S5(G)). Finally, LGALS3 levels were significantly increased in the control siRNA SNCA fibrils treated cells compared to vehicle (Fig. S5(G)). Collectively, these results suggest that LGALS3 plays a role in governing lysosome function under basal conditions, that SNCA fibril treatment disrupts lysosome function and increases the cellular autophagy response in mDA neurons. Additionally, it suggests that LGALS3 depletion impairs autophagic flux, most consistent with early autophagic impairment as fewer autophagosomes appear to be forming, based on the changes in LC3-II, and no changes in SQSTM1 clearance were observed among the lysosome uninhibited and lysosome inhibited cells [Citation82].
Secretion of SNCA and LGALS3 is mediated by its specific autophagic adaptor and effector proteins TRIM16 and ATG16L1
Extrapolating from the LGALS3 depleted mDA cells impaired autophagic function, we theorized that SNCA secretion may be reduced during LGALS3 depletion through an impairment in the LGALS3 mediated autophagic response. Previous work suggests that LGALS3 recruits autophagic machinery through TRIM16 via ULK1 when bound to damaged endosomes and/or lysosomes [Citation36,Citation38,Citation83]. This process was shown to be mediated by connections with the autophagic protein ATG16L1 to clear damaged endosomes and/or lysosomes [Citation36,Citation38,Citation83]. To better understand the mechanism by which SNCA release is regulated by LGALS3, and in turn how these factors influence LGALS3 release, we depleted human mDA neuron cultures of TRIM16 and ATG16L1. TRIM16 depletion significantly decreased the level of both SNCA and LGALS3 in the cultured media ()). When the relative levels of TRIM16 depletion were compared following TRIM16 siRNA treatment, a positive correlation between the degree of TRIM16 depletion and the degree of inhibition of SNCA and LGALS3 release was observed ()). Similarly, following successful knockdown of ATG16L1, reduced secretion of both SNCA and LGALS3 occurred in mDA neurons ()). The relative levels of ATG16L1 depletion also correlated with the relative levels of SNCA and LGALS3 ()). No differences in cell viability were observed in response to TRIM16, ATG16L1, or LGALS3 depletion in mDA neurons (Fig. S5(H)), suggesting that changes in secretion were not the result of cell death. Collectively, these results indicate that the secretion of both SNCA and LGALS3 are regulated by TRIM16 and ATG16L1.
Figure 6. Neuronal Depletion of TRIM16 or ATG16L1 LGALS3 decreases SNCA and LGALS3 secretion. (A) Relative fold difference in SNCA (left) and LGALS3 (middle) in the cultured media of mDA neuronal cultures at 24 to 48 h post-transfection, relative depletion of TRIM16 (right), and representative KD (inset). Values were determined by ELISA and normalized to the mean of the control group (i.e. mean control). The different colors represent matched replicates. (B) A plot of the data shown in (A) illustrating the linear relationship between relative TRIM16 depletion (x-axis) and mean fold SNCA secretion (B, left, y-axis) or LGALS3 secretion (B, right, y-axis). (C) Relative fold difference in SNCA (left) and LGALS3 (middle) in the cultured media of mDA neuronal cultures at 48 to 72 h post-transfection, relative depletion of ATG16L1 (right), and representative KD (inset). (D) A plot of the data shown in (C) illustrating the linear relationship between relative ATG16L1 depletion (x-axis) and mean fold SNCA secretion (D, left, y-axis) or LGALS3 secretion (D, right, y-axis). (E) A representative z-stack from multiple z-stack MIPs of imaged mDA neurons’ cultured media. Cells were first transfected with siRNAs followed by either vehicle (0.1% DMSO) or Baf-A1 treatment 48 h post-transfection for 24 h. (F) A representative western blot that demonstrates successful depletion of LGALS3, TRIM16, or ATG7. (G, H) the Relative number of SNCA puncta (G) or LGALS3 puncta (H) from each image based on the same Imaris masking algorithm as a measure of relative differences between the conditions. Each data point is the mean of 20 images for each independent experiments. There were 4 independent experiments. (I-K) Relative fold difference in SNCA (I, left) and LGALS3 (I, right) in the cultured media of mDA cultures 4 h after vehicle (0.1% DMSO) or LLOME treatment measured by ELISA and normalized to the mean vehicle concentration. (J, K) Relative fold difference in SNCA from cultured media from either control and LGALS3 (J) or control and TRIM16 (K) siRNA transfected mDA neurons followed by 4 h of vehicle or LLOME treatment. Vehicle or LLOME treatment was performed 72 h post control or LGALS3 siRNA transfection, or 48 h post control or TRIM16 siRNA transfection. Representative mDA neuronal lysate western blots demonstrating successful knockdown of LGALS3 (J) or TRIM16 (K). A and C data are expressed as M ± SE, B and D data are expressed as M ± SD (A and B, n = 6 (left), n = 5 (middle), n = 6 (right); C and D, n = 8 (left), n = 7 (middle), control siRNA n = 6, ATG16L1 siRNA n = 8 (right), I, n = 4 (left), n = 6 (right), J and K, n = 5–6. Statistical significance was determined by evaluating Pearson’s r (B and D), ratio paired t-test (A, C, and I), a two-way ANOVA, with Sidak’s post hoc tests (G-H), or a one-way ANOVA with Dunnett’s post hoc tests (J-K) following natural log transformation to better account for the distribution of values < 1. (G-H) * indicates significance relative to control plus vehicle, # indicates significance relative to control plus Baf-A1. For all statistical tests *, **, ***, ****, or #, ##, ###, ####, p < 0.05, 0.01, 0.001, and 0.0001, respectively.
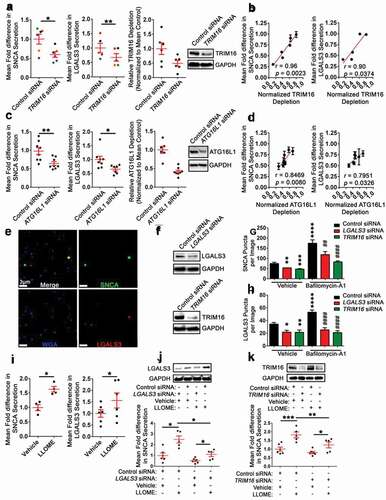
To further validate these changes in secretion, the EV-MAC workflow was used to simultaneously determine the relative levels of LGALS3 and SNCA EVs released from LGALS3 or TRIM16 depleted mDA neurons in the presence or absence of Baf-A1. The collected EVs from each condition were spinoculated onto coverslips, permeabilized, and SNCA, LGALS3, and wheat germ agglutinin (WGA), a lectin that has previously been used to recognize or isolate EVs from primary fluids [Citation84], were identified by immunofluorescence ()). Western blot analysis of cell lysates revealed reductions in LGALS3 and TRIM16 protein levels, although knockdown was incomplete ()). Using image analysis software (Imaris, Bitplane), a masking algorithm built around SNCA fluorescence was first used to quantify the relative number of SNCA EVs in each image for each condition. LGALS3 or TRIM16 KD significantly decreased the number of SNCA puncta per image in the context of vehicle treatment ()). mDA neurons that received control siRNA and were treated with Baf-A1, had increased SNCA puncta number per image compared to vehicle ()). For mDA neurons that received either a LGALS3 or a TRIM16 KD, Baf-A1 significantly increased the number of SNCA puncta per image relative to Baf-A1 control siRNA treated ()). Afterward, the same set of images were assessed for the relative number of LGALS3 EVs by building a Spots masking algorithm around the LGALS3 fluorescence signal. The changes in LGALS3 EVs among conditions mirrored those observed for SNCA ()). LGALS3 or TRIM16 siRNA treatment significantly decreased the number of LGALS3 puncta per image ()). Control siRNA treated cells had increased LGALS3 puncta per image when treated with Baf-A1 ()). LGALS3 or TRIM16 KD cells treated with Baf-A1 had significantly reduced LGALS3 puncta per image relative to the control siRNA Baf-A1 treated ()).
Lysosomal damage increases SNCA and LGALS3 secretion in association with increased autophagic activity
Given that lysosomal damage upregulated autophagy in mDA neurons, depletion of LGALS3 reduced SNCA secretion, and depletion of TRIM16 and ATG16L1 reduced both SNCA and LGALS3 secretion, we evaluated the effect lysosomal damage on the secretion of SNCA and/or LGALS3. When mDA neurons were treated with LLOME, significantly increased extracellular levels of both SNCA and LGALS3 were measured by ELISA ()). Similar results were observed following LLOME treatment of SH-SY5Y DSP-SNCA cells (Fig. S5(I)). To evaluate if LGALS3 or TRIM16 mediated this process, SNCA secretion was measured in response to LLOME treatment in mDA neuronal cultures first depleted of either LGALS3 or TRIM16. Western blot analysis of LGALS3 or TRIM16 levels demonstrated successful LGALS3 or TRIM16 knockdown ()). Treatment of mDA neurons with LLOME increased SNCA secretion in control siRNA, LGALS3 siRNA, and TRIM16 siRNA treated cells ()). However, SNCA secretion was reduced following siRNA depletion of LGALS3 and TRIM16 following LLOME treatment, relative to control siRNA transfection treated with ()). No difference in cell viability was observed among the vehicle and LLOME-treated control, LGALS3-, or TRIM16-depleted cells (Fig. S5(J)).
The autophagic activation state in response to LLOME in LGALS3 and TRIM16 depleted mDA neurons was also evaluated. In agreement with studies in other cell types [Citation38,Citation85,Citation86], LGALS3 and TRIM16 depletion did not affect the ratio of p-S757 ULK1 to total ULK1 or p-T389 RPS6KB1 to total RPS6KB1 (Fig. S6(A-D)), suggesting that LGALS3 or TRIM16 was not modulating MTOR signaling. TRIM16 depleted mDA neuronal lysates were also evaluated for LGALS3 to determine if the previously observed differences were due to the downregulation of LGALS3. However, no differences in LGALS3 were observed in either the vehicle or LLOME TRIM16 depleted cells with respect to the corresponding treatments in the control siRNA mDA neurons (Fig. S6(D)), consistent with a previous report in TRIM16-depleted human bone marrow-derived mesenchymal stem cells [Citation86].
LGALS3 or TRIM16 depleted cells were also evaluated for altered levels of lysosomes or autophagosomes, measured as differences in the lysosome associated LAMP1 and the autophagosome associated LC3-II proteins. No significant differences in LAMP1 were observed in the LGALS3 or TRIM16 depleted cells at both baseline and during LLOME treatment (Fig. S6(E-H)). Evaluation of autophagosome levels revealed that LGALS3 KD significantly decreased LC3-II levels in the vehicle-treated cells (Fig. S6(F)). TRIM16 KD had no effect on LC3-II levels (Fig. S6(H)). Whereas LLOME treatment increased LC3-II levels, LGALS3 KD slightly but significantly decreased LC3-II levels while TRIM16 KD did not affect the LLOME-induced increase in LC3-II levels (Fig. S6(F, H)). These changes in LC3-II levels among the LGALS3 and TRIM16 depleted mDA neurons in response to either vehicle or LLOME suggest the occurrence of abnormalities in autophagosome formation. These abnormalities led us to hypothesize that the observed changes in SNCA secretion in LGALS3, TRIM16, and ATG16L1 depleted mDA neurons may be due to specific, altered introduction of damaged endosomal and/or lysosomes into the autophagic pathway, a process mediated by LGALS3.
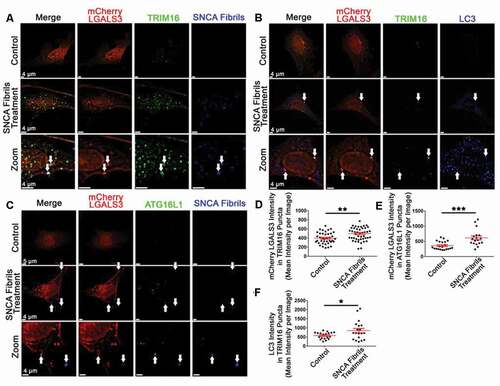
SNCA fibrils increase the association of TRIM16 and ATG16L1 with mCherry-LGALS3
To determine if TRIM16 and ATG16L1 were being recruited to damaged vesicles recognized by LGALS3 following SNCA fibril induced endosomal and/or lysosomal damage, we measured the change in colocalization of TRIM16 with mCherry-LGALS3, LC3, and SNCA fibrils in response to SNCA fibril treatment. Similarly, the colocalization of ATG16L1 with SNCA fibrils and mCherry-LGALS3 was measured following SNCA fibril treatment. Following validation that our mCherry-LGALS3 construct was not undergoing cleavage (Fig. S7(A)), SH-SY5Y expressing mCherry-LGALS3 were treated with SNCA fibrils. Following treatment with fluorescently labeled SNCA fibrils, immunofluorescent analysis of TRIM16 and ATG16L1 was performed to determine the extent to which TRIM16 or ATG16L1 puncta colocalized with the various markers of interest (-C)). Following SNCA fibrils treatment, SNCA fibrils signal was present in surface masks created around TRIM16 and ATG16L1 puncta (Fig. S7(B, C)). SNCA fibril treatment also significantly increased mCherry-LGALS3 signal within TRIM16 ()) and ATG16L1 puncta ()), respectively. Furthermore, when mCherry-LGALS3 cells were treated with unlabeled SNCA fibrils and stained for TRIM16 and the autophagosome marker LC3B, an increased in the LC3B fluorescence signal was observed within TRIM16 puncta following fibril treatment (, F)). Collectively, these data support the premise that following SNCA fibril induced endosomal/lysosomal membrane damage, LGALS3, TRIM16, and ATG16L1 are recruited to these vesicles to facilitate the formation of autophagosomes.
Figure 7. SNCA fibril treatment increases the colocalization of TRIM16 and ATG16L1 with mCherry LGALS3 and SNCA fibrils. (A-C) Representative images from mCherry LGALS3 SH-SY5Y cells treated with SNCA fibrils and stained for TRIM16 and LC3B (B), or ATTO 647 labeled SNCA Fibrils and stained for TRIM16 (A) or ATG16L1 (C). The white arrows point to instances of triple colocalization. (D-F) an Imaris masking algorithm built around TRIM16 (A-B, D, and F) or ATG16L1 puncta (C, E). Each data point represents the averaged maximum intensity from the total masked puncta in an individual image. The same algorithm was applied to all experiments and treatment conditions. Intensity data are expressed as M ± SE (n = 3 independent experiments, 7–10 images per experiment). Statistical significance was determined by unpaired t-tests. For all statistical tests *, **, ***, ****, p < 0.05, 0.01, 0.001, and 0.0001, respectively.
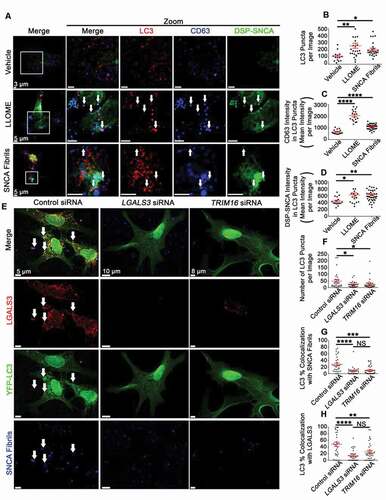
LGALS3 affects the formation of amphisomes containing SNCA fibrils in response to endo-lysosomal membrane damage
Previous work suggests that autophagy may modulate the secretion of EVs and SNCA through the formation of amphisomes, a hybrid organelle formed when autophagosomes and MVBs fuse, which subsequently releases its cargo upon further fusion with the plasma membrane [Citation87]. The formation of amphisomes can be measured based on the inclusion of the MVB marker, CD63, with the autophagosome marker, LC3B [Citation88–90]. To this end, we assessed whether LLOME or SNCA fibrils affected LC3B with CD63 colocalization by immunofluorescence staining and microscopy ()). Treatment with LLOME or SNCA fibrils significantly increased levels of LC3B puncta relative to vehicle ()). Additionally, both LLOME and SNCA fibrils significantly increased both DSP-SNCA and CD63 intensity in LC3B puncta (, D)). These data demonstrate that following endosomal and/or lysosomal membrane damage, autophagosome formation is increased and these autophagosomes are trafficked to compartments which are positive for amphisome markers.
Figure 8. Lysosomal rupture increases the formation of amphisomes DSP-SNCA SH-SY5Y cells and the depletion of LGALS3 or TRIM16 reduces the recruitment of SNCA fibrils to autophagosomes in mDA neurons. (A) Representative images from DSP-SNCA SH-SY5Y cells treated vehicle or LLOME for 4 h or SNCA fibrils for 24 h and subsequently stained for LC3B and CD63. The same end point was used for all conditions. The white arrows point to instances of triple labeled events. (B) Quantification of the number of LC3B puncta recognized by a masking algorithm among images. (C, D) Quantification of DSP-SNCA (C) or CD63 (D) intensity in masked LC3B puncta. Each data points represents the averaged maximum intensity from the total masked puncta in an individual image. The same algorithm was applied to all experiments and treatment conditions. (E) Representative images of induced YFP-LC3 (green) transduced mDA neuronal cultures treated with SNCA fibrils (blue) and stained for endogenous LGALS3 (red) shown at the same intensities for each of the channels for each condition. YFP-LC3 induction was initiated 24 h post-transfection. SNCA fibrils treatment was done 48 h post-transfection. Staining was conducted 72 h post-transfection. The white arrows point to triple colocalization of YFP-LC3, LGALS3, and SNCA fibrils. (F) Quantification of the number of YFP-LC3 puncta recognized by a masking algorithm among images for the indicated siRNA pretreated condition. (G, H) Quantification of the degree of masked YFP-LC3 puncta colocalized with SNCA fibrils puncta (G) and LGALS3 puncta (H). Each data point is the mean of 3 randomly selected coverslips (n = 15–20 images per coverslip). (A-D) Intensity data are expressed as M ± SE (n = 3 independent experiments, 7–10 images per experiment). Statistical significance was determined by one-way ANOVA with Dunnett’s post hoc tests (A-D) or with Tukey’s post hoc tests (E-H). For all statistical tests *, **, ***, ****, p < 0.05, 0.01, 0.001, and 0.0001, respectively.
We also measured the formation of amphisomes in CRISPR-Cas9 control and LGALS3 KO SH-SY5Y cells (Fig. S7(D, E)) in response to vehicle, LLOME, and fluorescently labeled SNCA fibril treatment. The number of LC3B puncta was unchanged in the LGALS3 KO cells among conditions yet were significantly lower compared to the control KO cells in response to either LLOME or SNCA fibril treatment (Fig. S7(F)). Increased colocalization of LGALS3 occurred in the control KO cells following LLOME treatment (Fig. S7(G)). While increased CD63 intensity in LC3B puncta occurred following LLOME or SNCA fibril treatment in the control KO cells, no change occurred in the LGALS3 KO cells, and these differences were at significantly lower levels when compared to the control KO for each treatment condition (Fig. S7(H)). Furthermore, significantly less SNCA fibril signal was observed in the LC3B puncta in the LGALS3 KO cells compared to the control KO cells (Fig. S7(I)). No difference in SNCA fibril uptake was observed among the LGALS3 KO and the control KO cells as measured by the SNCA fibrils fluorescence intensity using a plate reader immediately after treatment and following a media change 24 h later (Fig. S7(J)). As a whole, these data demonstrate that LGALS3 depletion reduces the formation of autophagosomes following SNCA fibril treatment and the recruitment of these autophagosomes to amphisomes.
Discussion
Previous studies, including work from our lab, have demonstrated that treating cells with SNCA fibrils results in their endocytosis into endo-lysosomal compartments where they induced rupture followed by their recruitment to the ALP [Citation6,Citation28,Citation29,Citation91,Citation92]. In this previous work from our lab, we observed LGALS3 immunoreactivity both within and surrounding LBs in brain sections obtained postmortem from PD patients. These findings suggest a link between SNCA aggregates, lysosomal rupture, and cellular pathology. In this study, we examined the secretory pathway of SNCA to understand how cellular perturbations of the ALP system may impact the release of SNCA. To do this, we began by examining how the release of DSP-SNCA fusion proteins was impacted by ALP stress, including ALP stress induced by exogenous SNCA fibrils. Using the differential labeling of the DSP-SNCA and exogenous SNCA fibrils, we were able to observe colocalization of these forms of SNCA in cells () and in EVs secreted from these cells ()). The DSP-SNCA model is valuable because it allows for the impact of exogenously added SNCA fibrils on the subsequent release of SNCA expressed in these cells to be evaluated. By leveraging this model, we observed that exogenous SNCA fibril treatment increases the release of DSP-SNCA from cells ()). We also observed that pharmacological induction of lysosomal damage with LLOME increases the release of SNCA from mDA neurons ()). Taken together, these observations provide a potential mechanism to explain how the cell-to-cell spread of SNCA occurs, with vesicular damage induced by endocytosed fibrillar forms of SNCA promoting the subsequent release of SNCA from that same cell following ALP perturbation induced by fibrillar SNCA. Consistent with this premise, we observed the colocalization of the added SNCA fibrils with DSP-SNCA in cells () and in the EVs they released (). The colocalization of these species of SNCA in ALP compartments is consistent with (1) studies showing that cellular SNCA is degraded in an ALP dependent mechanism [Citation93–95]; (2) prior studies showing vesicular damage and recruitment of the autophagic machinery to vesicles damaged by extracellular SNCA fibrils [Citation6,Citation28,Citation29,Citation91,Citation92]; and (3) that autophagy mediates both EV and non-EV associated SNCA secretion [Citation45,Citation48–54]. The colocalization of extracellular and endogenously expressed SNCA in these damaged degradative vesicles make it tempting to speculate that interactions between these two species of SNCA may promote the conversion of endogenous SNCA to misfolded, pathological species. In this regard, we have previously observed that vesicles damaged by SNCA fibrils can re-seal following damage, as measured by the accumulation of the acidophilic fluorophore lysotracker in LGALS3+ compartments containing fibrillar SNCA applied to cells [Citation96]. As it is known that low pH enhances the generation of fibrillar species of SNCA [Citation97,Citation98], the association of fibrillar and non-fibrillar species of SNCA in this low pH environment would be very favorable to promoting the misfolding of non-fibrillar SNCA.
We also extensively characterized the response of mDA neurons to SNCA fibrils. SNCA fibrils treatment induced an autophagic response similar to pharmacologically induced lysosomal damage by LLOME, including reduced MTOR-associated phosphorylation of ULK1 and RPS6KB1 comparable to rapamycin ()). Although the inability to distinguish between exogenous fibrils and endogenously expressed SNCA in mDA neuronal cells precluded us from determining how SNCA fibrils treatment impacts the endogenous SNCA release, we did observe that lysosomal damage induced by LLOME increased the endogenously expressed SNCA release (), Fig. S6(B)) and SNCA fibrils treatment increased the release of LGALS3 from mDA neurons ()), consistent with the data obtained from the DSP-SNCA SH-SY5Y cells. We also observed that inhibiting autophagy at early stages, using 3-MA, wortmannin or by ATG7 depletion, reduced the release of endogenously expressed SNCA from mDA neurons ()). However, inhibition of lysosomal degradation and autophagosome/lysosomal fusion by Baf-A1 increased SNCA release in mDA neurons ()), consistent with previous studies examining the effects of ALP impairment on SNCA degradation and/or secretion using other cellular models, with or without SNCA overexpression [Citation45,Citation48–54].
The fact that LGALS3 was released in a pattern that mirrored the release of SNCA in our experiments and was found to localize to EVs containing SNCA () prompted us to examine the role of LGALS3 in mediating SNCA release. These experiments show that depletion of LGALS3 reduced SNCA release from mDA neurons (). Subsequent interrogation of TRIM16 and ATG16L1, which have been implicated in LGALS3 dependent responses to vesicular damage in other studies [Citation36,Citation86,Citation99], showed that depletion of these proteins also reduced SNCA release from mDA neurons ()). Notably, this dependence on LGALS3, TRIM16 and ATG16L1 was observed in the absence of experimental stress to the ALP system. However, we also noted that depletion of LGALS3 from mDA neurons reduced the lysosomal degradative capacity of these cells (), Fig. S5). This observation suggests that there is some degree of incidental or chronic vesicular damage in mDA neurons that is recognized by LGALS3 to ensure lysosomal homeostasis. It is possible that some level of vesicular damage is common in neurons, reactive oxygen species have been shown to induce lysosomal membrane damage and the degradation of dopamine is inherently associated with reactive oxygen species production [Citation100–102]. It is also possible that cytoplasmic SNCA oligomers or protofibrils targeted for autophagic degradation damage membranes of the ALP pathway and require LGALS3 to maintain ALP homeostasis. In agreement with these possibilities, a recent paper from the Deretic group found LGALS3 coordinates a cellular system to facilitate lysosomal repair to acute insults as well as clearance during full-out rupture by way of the ALP system, to ensure lysosomal homeostasis in response to a wide range of lysosomal perturbations [Citation38].
Consistent with the functional data from mDA neurons showing that SNCA release is dependent on LGALS3, TRIM16, and ATG16L1, we also observe that these same proteins are recruited to vesicles damaged by SNCA fibrils in SH-SY5Y cells () and required for the recruitment of autophagosomal membranes containing LC3 to these damaged vesicles (). We also observe that following recruitment of the autophagic machinery to these vesicles, these complexes are ultimately trafficked to amphisomes containing CD63 (). This trafficking to a multivesicular body such as amphisomes is consistent with our previous study which used Structured Illumination Microscopy (SIM) to observe that LGALS3+ vesicles containing SNCA are often present in multivesicular structures. Although our data do not demonstrate that the release of SNCA containing EVs occurs from amphisomes, the release of damaged vesicles to amphisomes following recruitment of autophagic proteins would be consistent with our examination of these EVs () as well as another study finding the release of EVs with a “hybrid autophagosome-exosome-like phenotype” [Citation47]. A theoretical framework of these summarized events is depicted in .
Figure 9. A summary diagram of the hypothesized cellular mechanism. This diagram illustrates a hypothetical mechanism where endocytosed pathological SNCA enters the cell and induces lysosomal rupture. Lysosomal rupture results in MTOR inactivation subsequently increasing ULK1 and RPS6KB1 activity. Concurrently, lysosomal rupture exposures its inner-luminal glycans, which are recognized by LGALS3. Bound LGALS3 recruits TRIM16 and ULK1, and subsequently ATG16L1 to mediate further autophagic machinery assembly, increase cellular autophagy, and promote autophagosome development. This response, ultimately, increases SNCA secretion. (Bottom) LGALS3 recruitment to lysosomes during early membrane stress facilitate membrane repair.
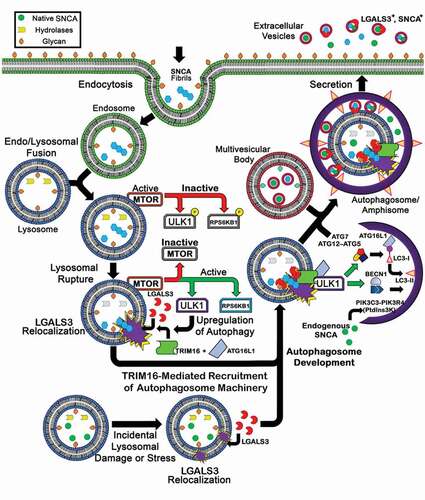
In conclusion, our study provides insight into the mechanisms by which the cell-to-cell propagation of SNCA may occur, finding that 1) following endocytosis, SNCA fibrils can induce lysosomal stress and induce autophagic responses that lead to the release of SNCA from the cell and 2) LGALS3, TRIM16 and ATG16L1 are novel host factors regulating the release of SNCA from cells. Continued delineation of the host factors responsible for promoting the cell-to-cell propagation of SNCA may provide opportunities to arrest the cell-to-cell spread of SNCA in animal models and in therapeutic approaches to treat PD. Moreover, given the fact that the ability to induce vesicle rupture following endocytosis is a conserved feature of amyloid proteins associated with other neurodegenerative disease [Citation81], the pathways and targets driving the cell-to-cell propagation of SNCA may also be relevant to other amyloid driven neurodegenerative diseases.
Methods
Culture of SH-SY5Y and HeLa cell lines
The SH-SY5Y human neuroblastoma cell line (CRL-2266) and the HeLa immortalized cell line (CRM-CCL-2) were acquired from the American Type Culture Collection. Cells were cultured in an incubator at 37°C with 5% CO2 in Dulbecco’s modified Eagle’s Medium (DMEM; Corning; Fischer Scientific, MT10017CV), supplemented with 10% fetal bovine serum (FBS; Hyclone, SH3007103HI), 10 µg/ml ciprofloxacin hydrochloride (Thermo Fischer, MP219902005), and 100 IU/ml penicillin and streptomycin (Thermo Fisher Scientific, 15-140-122).
Culture of iCell DopaNeurons
The iCell DopaNeurons were purchased from Fujifilm Cellular Dynamics (R1032). iCell DopaNeurons are a pre-differentiated culture of iPSC-derived human mDA neurons generated using protocols licensed and adapted from the Lorenz Studer lab. Cells were thawed, plated, and cultured using the protocol listed in the iCell DopaNeuron user guide. The cells were plated in 24-well plates onto coverslips (Thermo Fisher Scientific, 12-545-81) coated with 0.01% poly-L-ornithine (EMD Millipore, A-004-C) and laminin (Millipore Sigma, 23017015) at 2 × 105 cells per well. The plated cells were kept in culture for at least 7 days before any experiments were conducted to facilitate arborized cellular morphology. Cells were cultured in an incubator at 37°C with 5% CO2 and incubated in iCell Neural Base Medium containing iCell Nervous System Supplement 1 (Fujifilm Cellular Dynamics, M1031), iCell Neural Supplement B (Fujifilm Cellular Dynamics, M1029), and 100 IU/ml penicillin and streptomycin.
Culture of human iPSCs
The iPSC cell line used in the experiments described herein was obtained from Joseph R. Mazzulli laboratory (Northwestern University, Feinberg School of Medicine, Chicago, Il). This iPSC cell line was produced from primary human fibroblasts by retroviral expression of the reprogramming factors POU5F1, SOX2, MYC/cMYC, and KLF4 (described in [Citation103]). This cell line has been characterized through the expression of pluripotency markers (i.e. POU5F1, PODXL, SOX2, NANOG) [Citation104], genomic integrity through G-banding karyotype analysis (described in [Citation105]), teratoma analysis (described in [Citation106]), and RT-PCR analysis of pluripotency markers (described in [Citation106]). iPSCs (passage 50–60) were cultured on 6-well plates coated with either Matrigel (VMR, 47743–715) or Vitronectin XF (Stem Cell Technologies, 07180). Medium consisted of mTeSR1 basal medium containing 10% mTeSR1 5× supplement (Stem Cell Technologies, 05825) and was changed every other day. iPSCs were groomed by the manual removal of differentiated cells, passaged en bloc weekly by the manual dissection of iPSC colonies into ~2 mm2 chunks of cells and placement into a Matrigel- or Vitronectin XF-coated 6-well plate. Prior to floor plate induction, iPSCs were grown to 80–90% confluence. iPSC cultures were monitored daily with an in-hood EVOS Core XL cell imaging system (Thermo Fisher Scientific).
Induction of midbrain floor plate progenitors
iPSCs were incubated in knockout serum replacement media (KSRM; Thermo Fisher Scientific, 10828028) containing the Small Mothers Against Decapetaplegic inhibitors, LDN193189 (LDN, 100 nM; ReproCell, 04–0074) and SB431542 (SB, 10 µM; R & D System, 1614/1) for 24 h, KSRM containing LDN, SB, recombinant human SHH (sonic hedgehog signaling molecule; 100 pg/µl; R&D Systems, 1845-SH-025), purmorphamine (2 µM; ReproCell, 04–0009) and recombinant human FGF8 (fibroblast growth factor 8; 100 pg/µl; R&D Systems, 4745-F8-050) for 48 h, and KSRM containing LDN, SB, SHH, purmorphamine (Reprocell, 04–0009), FGF8, and CHIR99021 (CHIR, 3 µM; ReproCell, 04–0004) for 48 h. KSRM media was comprised of 82.20% KnockOut Dulbecco’s Modified Eagle’s Medium (Thermo Fisher Scientific, 10–829-018), 14.68% Knockout Serum Replacement (Thermo Fisher Scientific, 10–828-028), 0.98% 200 mM L-glutamine (Thermo Fisher Scientific, 35050061), 0.98% Minimum Essential Media Non-Essential Amino Acids solution (Thermo Fisher Scientific, 11140050), 0.18% 55 mM 2-mercopatoethanol, and 0.98% 10,000 U/ml penicillin-streptomycin solution (Thermo Fisher Scientific, 15–140-122). Next, iPSCs were incubated with a 3:1 ratio of KSRM to SM1 media containing LDN, SB, SHH, purmorphamine, FGF8, and CHIR for 48 h, a 1:1 ratio of KSRM to SM1 media containing LDN and CHIR for 48 h, and 1:3 ratio of KSRM to SM1 media containing LDN and CHIR for 48 h. SM1 media was comprised of 96.16% Neurobasal Medium (Thermo Fisher Scientific, 21103049), 1.92% NeuroCult SM1 Neuronal Supplement (Stem Cell Technologies, 05711), 0.96% 200 mM L-glutamine solution (Thermo Fisher Scientific, 35,050,061), and 0.96% 10,000 U/ml penicillin-streptomycin solution (Thermo Fisher Scientific, 15140122). The cultures were monitored daily with an in-hood EVOS Core XL cell imaging system (Thermo Fisher Scientific).
Differentiation of mDA neurons
Floor plate induced iPSCs were differentiated to mature mDA neurons by incubating them in SM1 media containing CHIR, 24-diamino-5-phenylthiazole (DAPT, 10 µM; ReproCell, 04–0041), recombinant human BDNF (brain derived neurotrophic factor protein, 20 pg/µl; R&D Systems, 248-BD8-050), ascorbic acid (AA, 200 µM; Millipore Sigma), recombinant human GDNF (glial derived neurotrophic factor, 5 pg/µl; R&D Systems, 212-GD-010), recombinant human TGFB3 (transforming growth factor beta 3, 1 pg/µl; PeproTech, 100–21-50ug), and dibutyryl cyclic adenosine monophosphate (cAMP, 500 µM; Enzo Life Sciences, BML-CN125-0100) for 48 h and SM1 media containing DAPT, BDNF, AA, GDNF, TGFB3, and cAMP for 24–48 h. The neuronal culture was allowed to expand by passaging them en bloc by mechanical dissociation of the thickened cell layer into ~2 mm2 squares, which were plated onto poly-d-lysine (66 µg/ml; Millipore Sigma, A-003-M)- and mouse laminin (1.25 µg/ml; Millipore Sigma, 23-017-015)-coated 10-cm culture dishes at 2 × 105 cells/cm2. The neurons were incubated in SM1 media containing the neuralization factors DAPT, BDNF, AA, GDNF, TGFB3, and cAMP for 10 days. The neurons were passaged by incubation with Accutase (Fisher Scientific, Corning, MT25058CI) then plated into poly-d-lysine (66 µg/ml)- and mouse laminin (1.25 µg/ml)-coated 24-, 48-, or 96-well dishes. The neurons were incubated in SM1 media containing DAPT, BDNF, AA, GDNF, TGFB3, and cAMP for 15 days; this media was changed every 3 days. The neurons were incubated in SM1 media without the aforementioned neuralization factors until analyses; this media was changed every 3 days. The neurons were considered terminally differentiated to mDA neurons 50 days after floor plate induction had begun. Neurons were monitored daily with an in-hood EVOS Core XL cell imaging system (Thermo Fisher Scientific).
mDA differentiation and characterization
This iPSC cell line was produced from primary human fibroblasts by retroviral expression of the reprogramming factors POU5F1, SOX2, MYC, and KLF4 (described in [Citation103]) and was characterized by measuring the expression of pluripotency markers (i.e., POU5F1, PODXL, SOX2, NANOG) [Citation104], genomic integrity through G-banding karyotype analysis (described in [Citation105]), teratoma analysis (described in Cooper et al. 2012) [Citation106], and reverse transcriptase-polymerase chain reaction (RT-PCR) analysis of pluripotency markers [Citation106]. Flow cytometry and immunofluorescence experiments show that, prior to the start of differentiation, greater than 85% of the human iPSCs express the pluripotency marker POU5F1, PODXL, and SOX2 (Fig. S8(A)). At day 14 post initial differentiation, we observed increased expression of LMX1A (LIM homeobox transcription factor 1 alpha) and the neural progenitor marker, NES (nestin; Fig. S8(A)), consistent with what has previously been described [Citation104,Citation107,Citation108]. Flow cytometry showed that 87 ± 4% and 88 ± 5% (M ± SE, n = 3 independent experiments) of the terminally differentiated cells were positive for the midbrain markers, FOXA2 (forkhead box A2) and LMX1A, respectively. Flow cytometry showed that 63 ± 17% and 70 ± 10% (M ± SE, n = 3 independent experiments) of the terminally differentiated cells were positive for the mDA neuron markers, NR4A2 (nuclear receptor subfamily 4 group A member 2) and TH (tyrosine hydroxylase), respectively. Immunofluorescence experiments showed that terminally differentiated cells expressed TH and the neuronal marker, TUBB3 (tubulin beta 3 class III; Fig. S8(A)). Consistent with a previous report in which synapses were observed in human mDA neurons derived from iPSCs [Citation109], TEM analysis of terminally differentiated cells revealed evidence of the formation of synapses (Fig. S8(B, C)). As previously described [Citation110,Citation111] and in accord with well recognized criteria [Citation112], a synapse was identified by the presence of presynaptic neurotransmitter vesicles and a synaptic cleft with parallel, electron dense, thickened pre- and postsynaptic membranes and the presence of electron dense content in the synaptic cleft. Consistent with the report that greater than 90% of the dopaminergic synapses in the human striatum are symmetric [Citation113] and are functionally inhibitory or modulatory, all of the synapses observed were symmetric synapses (Fig. S8(B, C)).
Flow cytometry
The procedure to prepare mature mDA neurons for analysis by flow cytometry was based on published protocol [Citation114] with minor modifications. A suspension of single cells was created by incubating the cells with Accutase (Fisher Scientific, Corning, MT25058CI). The single cell suspension was centrifuged at 220 × g for 5 min at room temperature then resuspended in 2% FBS in phosphate buffered saline (PBS; Fisher Scientific, Corning, MT21040CV). The cells were counted, distributed in aliquots, then fixed in 4% paraformaldehyde (Sigma-Aldrich, 158127) in PBS for 30 min at room temperature on a rotary mixer (Ted Pella, 1050). When the antigen was intracellular, the cells were permeabilized by incubation with a 0.5% saponin (Sigma Aldrich, 47,036) solution for 15 min at room temperature on a rotary mixer. This permeabilization step was omitted for surface antigens. After a PBS wash, the cells were incubating with a primary antibody solution containing primary antibody, 1% bovine serum albumin (BSA; Sigma Aldrich, A7906), 10% normal donkey serum (NDS; Millipore Sigma, S30-100ML) in PBS for 45 min at room temperature on a rotary mixer. When the antigen was intracellular, the primary antibody solution contained 0.5% saponin. When the antigen was expressed on the cell surface, saponin was omitted from the primary antibody solution. The primary antibodies used were TH (EMD Millipore, AB152), NR4A2/Nurr1 (Santa Cruz Biotechnology, sc-376984), LMX1A (Abcam, AB139726), and FOXA2/HNF-3B (Santa Cruz Biotechnology, SC-101060). After a PBS wash, the cells were incubated in a secondary antibody solution containing secondary antibody, 1% bovine serum albumin, 10% NDS in PBS for 45 min at room temperature on a rotary mixer. The secondary antibody was either Alexa Fluor 488 or Alexa Fluor 647 (Vector Laboratories, FI-1000-1, CY-1500-1, DI-1649-1, DI-1488-1.5). Cells were washed in PBS then resuspended in 2% FBS in PBS. Flow cytometry was performed on a FACS Canto II (BD Biosciences) and analyzed using FACS DIVA software (BD Biosciences). Cells were identified by light scatter for 10,000 gated events.
Transmission electron microscopy
Terminally differentiated mDA neurons, grown in 24-well culture dishes with each well containing one poly-d-lysine (66 µg/ml)- and mouse laminin (1.25 µg/ml)-coated cover glass (Carolina Biological Supply Company, 633009; circles, 12-mm diameter), were fixed by incubation in PBS containing 2% paraformaldehyde, 2% glutaraldehyde (Electron Microscopy Sciences, 16350), and 2% acrolein (Polysciences, Inc, 01679) for 30 min at room temperature. Cells were fixed in phosphate buffer containing 1% osmium tetroxide (Electron Microscopy Sciences, 19100) and 1.5% potassium ferricyanide (Electron Microscopy Sciences, 25154) for 1 h at room temperature in the dark. Next, the cells were dehydrated by incubation in an ascending series of hexylene glycol (25, 50, 75, 95, 100%; Electron Microscopy Sciences, 16740) followed by incubation in a 1:1 ratio of hexylene glycol to epoxy resin (comprised of a mixture of EMbed 812 (Electron Microscopy Sciences, 14,120), nadic methyl anhydride (Electron Microscopy Sciences, 19,000), dodecenyl succinic anhydride (Electron Microscopy Sciences, 13700), and 2,4,6-Tri(dimethylaminomethyl)phenol (Electron Microscopy Sciences, 13600) for 12 h at room temperature on a rotary mixer (Ted Pella, Inc). Next, the cells were incubated with 100% epoxy resin for 12 h at room temperature on a rotary mixer (Ted Pella, Inc). The epoxy resin was changed, and the cells were incubated for 2 h at room temperature on a rotary mixer (Ted Pella, Inc). The cover glass was removed from the culture dish, inverted, placed onto an embedding capsule filled with epoxy resin, then baked at 70°C for 12 h. The cells, cover glass, and embedding capsule filled with epoxy resin were immersed in liquid N2 to separate the cells from the cover glass. Ultrathin sections (70 nm) were cut with an ultramicrotome (EM UC7, Leica Microsystems), mounted on formvar- and carbon-coated 200 mesh copper grids then stained with filtered 2% uranyl acetate and Reynold’s lead citrate prior to imaging. Samples were imaged with a Philips CM 120 transmission electron microscope (TSS Microscopy) equipped with a BioSprint 16-megapixel digital camera (Advanced Microscopy Techniques).
Concentrated mDA neuron EV samples were dual (i.e., SNCA and CD63, SNCA and LGALS3, or CD63 and LGALS3) or triple (i.e., SNCA, CD63, and LGALS3) labeled then prepared for imaging by TEM according to published methods [Citation74,Citation103,Citation104] with minor modifications. For dual and triple labeling, mDA neuron EV samples were incubated with species specific primary antibodies (rabbit anti-SNCA, mouse anti-CD63, or rat anti-LGALS3) then incubated with secondary antibodies conjugated to different sized gold particles (i.e. goat anti-rabbit IgG conjugated to a 5-nm gold particle, goat anti-mouse IgG conjugated to a 10 nm gold particle, or goat anti-rat IgG conjugated to a 15-nm gold particle). The mDA neuron EV samples were fixed in 4% paraformaldehyde for 30 min at room temperature. Formvar- and carbon-coated 200 mesh nickel grids (Electron Microscopy Sciences, EMS200-Ni) were pretreated with 0.002% Alcian blue in 0.03% acetic acid, to increase the hydrophilicity of the grids, then floated on top of 50 µl drops of fixed mDA neuron EV samples for 20 min at room temperature. The grids were washed thrice in PBS then incubated with 50 mM glycine to quench free aldehyde groups. Next, the grids were washed thrice in PBS then incubated in PBS containing 5% bovine serum albumin, 0.1% cold water fish skin gelatin (Sigma-Aldrich, G7765-1 L), and 5% normal goat serum (Electron Microscopy Sciences, NS02L) for 30 min at room temperature. Grids were thoroughly washed with PBS containing 0.2% acetylated bovine serum albumin (Electron Microscopy Sciences, B2518) then incubated with PBS containing 0.2% acetylated bovine serum albumin, 0.1% saponin, and either rabbit anti-SNCA (MJFR1, 1:50; Abcam, ab138501) or mouse anti-CD63 (1:50; BD Bioscience, 556019) for 2 h in a humidity chamber at room temperature. The grids were thoroughly washed in PBS containing 0.2% acetylated bovine serum albumin then incubated with either goat anti-rabbit IgG conjugated to a 5 nm gold particle (1:50; Cytodiagnostics, AC-5-01) or goat anti-mouse IgG conjugated to a 10-nm gold particle (Electron Microscopy Sciences, 25128) for 2 h in a humidity chamber at room temperature. The grids were incubated with 1% glutaraldehyde for 15 min at room temperature to fix the gold particles to the surface of the grid then thoroughly washed with deionized water. The EVs were again permeabilized by incubating them with PBS containing 0.1% saponin for 30 min at room temperature, washed thrice in PBS, then incubated in PBS containing 5% bovine serum albumin, 0.1% cold water fish skin gelatin, and 5% normal goat serum for 30 min at room temperature. Grids were thoroughly washed with PBS containing 0.2% acetylated bovine serum albumin then incubated with PBS containing 0.2% acetylated bovine serum albumin, 0.1% saponin, and either mouse anti-CD63 (1:50, BD Bioscience, 556019) or rat anti- LGALS3 (1:50; M3/38; Millipore Sigma, 125402) for 2 h in a humidity chamber at room temperature. The grids were thoroughly washed in PBS containing 0.2% acetylated bovine serum albumin then incubated with either goat anti-mouse IgG conjugated to a 10-nm gold particle (1:50, Electron Microscopy Sciences, 25149) or goat anti-rat IgG conjugated to a 15-nm gold particle (Electron Microscopy Sciences, 25193) for 2 h in a humidity chamber at room temperature. The grids were incubated with 1% glutaraldehyde for 15 min at room temperature to fix the gold particles to the surface of the grid then thoroughly washed with deionized water. For the EVs that were triple labeled, the EVs were again permeabilized by incubating them with PBS containing 0.1% saponin for 30 min at room temperature. The grids were washed thrice in PBS then incubated in PBS containing 5% bovine serum albumin, 0.1% cold water fish skin gelatin, and 5% normal goat serum (Electron Microscopy Sciences) for 30 min at room temperature. Grids were thoroughly washed with PBS containing 0.2% acetylated bovine serum albumin (Electron Microscopy Sciences) then incubated with PBS containing 0.2% acetylated bovine serum albumin, 0.1% saponin, and rat anti-LGALS3 (1:50) for 2 h in a humidity chamber at room temperature. The grids were thoroughly washed in PBS containing 0.2% acetylated bovine serum albumin then incubated with goat anti-rat IgG conjugated to a 15 nm gold particle (Electron Microscopy Sciences, 25193) for 2 h in a humidity chamber at room temperature. The grids were incubated with 1% glutaraldehyde for 15 min at room temperature to fix the gold particles to the surface of the grid then thoroughly washed with deionized water. Next, the mDA neuron EV samples were negatively stained by floating each grid on a 50 µl drop of uranyl-oxalate (pH 7) for 5 min at room temperature followed by floating the grid on a 50 µl drop of a methyl cellulose-uranyl acetate-phosphotungstic acid solution (700 µl 2% methyl cellulose, 100 µl 3% uranyl acetate (pH 3.5), 25 µl phosphotungstic acid (pH 7.2), and 75 µl deionized water) for 10 min on wet ice. The grids were removed from the methyl cellulose-uranyl acetate-phosphotungstic acid solution with nichrome loops (3.5 mm internal diameter, Ted Pella, Inc) then blotted against a sheet of Whatman filter paper so that a thin layer of film was left on the EV side of the grid. The sample was placed into a grid storage box and allowed to dry for 12 h prior to imaging with a Philips CM120 transmission electron microscope (TSS Microscopy) equipped with a BioSprint 16-megapixel digital camera (Advanced Microscopy Techniques).
Characterization of SNCA fibrils
Human wild-type SNCA was expressed in E. coli BL21 DE3 CodonPlus cells (Stratagene, 230245) and purified as described previously [Citation115]. Monomeric human wild-type SNCA (100 µM) was assembled into fibrils by incubation in 50 mM Tris HCl, pH 7.5, 150 mM KCl at 37°C under continuous shaking in an Eppendorf Thermomixer set at 600 rpm for 5 days [Citation63]. The assembly reaction was monitored by withdrawing aliquots (20 µl) from the assembly reaction at different time intervals, mixing them with Thioflavin T (10 µM, final concentration) and recording the fluorescence increase on a Cary Eclipse Fluorescence Spectrophotometer (Varian Medical Systems Inc.) using an excitation wavelength = 440 nm, an emission wavelength = 480 nm and excitation and emission slits set at 5 and 10 nm, respectively. To label SNCA fibrils with extrinsic fluorophores, the fibrils were centrifuged twice at 15,000 × g for 10 min and re-suspended twice in PBS at 1,446 g/L and two molar equivalents of ATTO-488 NHS-ester (Atto-Tec GmbH) fluorophore in DMSO were added. The mix was incubated for 1 h at room temperature. The labeling reactions were arrested by addition of 1 mM Tris, pH 7.5. The unreacted fluorophore was removed by a final cycle of two centrifugations at 15,000 × g for 10 min and resuspensions of the pellets in PBS. The fibrillar nature of SNCA was assessed by TEM after adsorption of the fibrils onto carbon-coated 200 mesh grids and negative staining with 1% uranyl acetate using a JEOL 1400 transmission electron microscope. The images were recorded with a Gatan Orius CCD camera. The resulting SNCA fibrils were fragmented by sonication for 20 min in 2-ml microcentrifuge tubes in a Vial Tweeter powered by an ultrasonic processor UIS250v (250 W, 2.4 kHz; Hielscher Ultrasonic) to generate fibrillar particles with an average size 42–52 nm as assessed by TEM analysis. The fingerprint of the resulting fibrils was obtained by subjecting them to limited proteolysis using proteinase K (Fisher Scientific, 71-049-4) [Citation116]. The fibrillar polymorphs were diluted to 100 µM in PBS and incubated at 37°C with proteinase K (3.8 µg/mL). At different time intervals, samples were withdrawn from the degradation reaction and supplemented with protease inhibitor phenylmethanesulfonyl fluoride (Sigma-Aldrich, P7626-1 G) final concentration of 3.3 µM). Samples were then frozen in liquid nitrogen and dehydrated. Fibrils were then disassembled by treatment with 100% hexafluoro-2-propanol for 1 h. Samples were then air-dried, dissolved in 15 µl of Laemmli sample buffer and denatured for 5 min at 80°C. Samples were then analyzed on Tris-Glycine-SDS-polyacrylamide (15%) gel (SDS-PAGE), stained by Coomassie Brilliant Blue and imaged using a ChemiDocTM MP (BioRad).
Concentration of extracellular vesicles
Cultured media from DSP-SNCA SH-SY5Y or iPSC mDA neurons was spun in a 15 ml or 50 ml conical tube in a tabletop centrifuge at 300 × g for 10 min at 4°C. The supernatant was collected and added to either Beckman Coulter polycarbonate centrifuge tubes and spun at 10,000 × g with either SW41 TI or SW28 Beckman rotors, respectively, in an Optima L-90 K ultracentrifuge at 4°C for 30 min. Subsequently, the supernatant was collected and ultracentrifuged at 100,000 × g for 150 min at 4°C using new centrifuge tubes. Afterward, the supernatant was discarded, and the pellet was resuspended in PBS. The resuspended pellet was subjected to another round of 100,000 × g centrifugation with same rotor and machine for 150 min at 4°C. The supernatant was discarded, and the pellet was resuspended in 50 µl of PBS when used for TEM or EV-MAC. When used for non-reducing SDS-PAGE, pellet was immediately lysed in lysis buffer plus protease inhibitor cocktail (Roche). The resuspended pellets were stored at 4°C and used within two weeks. Concentrated EVs from was collected from DSP-SNCA SH-SY5Y cells plated in 10-cm plates at an initial confluency of 60–70%. Culture media was collected from iPSC mDA neurons plated in a 24-well plate. Collected samples from multiple wells (n ≥ 3) undergoing the same conditions were used for vehicle, TD139, SNCA fibril, and SNCA plus TD139 conditions to acquire sufficient sample size to assess EVs by non-reducing SDS-PAGE.
Cell viability assay
Cell viability was measured in accord with a published LDH assay [Citation117]. In brief, assay buffer components A, composed of 4 mM iodonitrotetrazolium chloride in 0.2 M Tris HCl, pH 8.2, and assay buffer B, composed of 6.4 mM nicotinamide adenine dinucleotide (Sigma-Aldrich, N0632-5 G), 320 mM lithium lactate (Sigma-Aldrich, L2250-10 G), in 0.2 M Tris-HCl buffer, pH 8.2 were made, frozen at −20°C, and thawed at the time of the experiment. 1-Methoxyphenazine methosulfate (150 mM) in Tris buffer, pH 8.2, was also made and frozen at −20°C. Activate assay buffer was made by adding 5 ml of assay buffer A, 5 ml of assay buffer B, and 0.5 μl of 150 mM 1-methoxyphenazine methosulfate in Tris buffer. To measure released LDH, 50 μl of a conditioned sample cultured media was plated in duplicate in a clear, flat-bottom 96-well plate. 50 μl of active assay buffer was added by multichannel pipette, incubated in the dark for one hour at room temperature, and subsequently quenched with the addition of 50 μl 1 M acetic acid. Signal was measured by reading absorbance at λ = 490 nm on a Synergy HTX Multi-Mode plate reader (BioTek Instruments) in conjunction with Gen5 software. To determine relative cell viability, secreted culture media LDH was compared to untreated lysed cells in lysis buffer composed of 9% Triton X-100 in water. The same volume of sample cell cultured media to be used in the experiment was used to lyse cells to evaluate differences at 1:1 ratio. Viability was calculated by taking the background subtracted condition sample absorbance and dividing it by the background subtracted lysed cell absorbance. This value was subtracted by 1 and multiplied by 100 to determine the percent viability. All reagents used were purchased from Sigma.
Generation of DSP-SNCA constructs, FLuc LGALS3, mCherry LGALS3 and an inducible YFP-LC3B plasmid
Expression plasmids were all generated by PCR-based cloning and restriction enzyme strategies. The initial generation SNCA DSP constructs A and B were made by first inserting the generating PCR primers against DSP1-7 and DSP8-11, which have previously been characterized in detail [Citation62], and inserting them into the lentiviral pLVX plasmid after restriction digest. Primers against SNCA were generated and the amplified sequence was then inserted on the C-terminal of the DSP1-7 and DSP8-11 sequence. The pLVX DSP1-7 SNCA is the SNCA DSP-A construct and the pLVX DSP8-11 SNCA is the SNCA DSP-B construct. Similarly, primers against FLuc were used to first amplify by PCR and then insert into the pLVX lentiviral construct by restriction digest. Primers against LGALS3 were also generated to amplify the LGALS3 sequence, which was then subcloned by adding it to the C-terminal of the now pLVX FLuc construct by restriction digest. pLVX mCherry LGALS3 construct was made using primers to amplify LGALS3 and inserting it at the C-terminal of the pLVX-mCherry-C1 lentiviral construct. The generation of the tetracycline inducible YFP-LC3B lentiviral plasmid was conducted by generating primers against YFP-LC3B, amplifying the sequence by PCR, and inserting it into the EZ-TET-pLKO-puromycin plasmid by restriction digest.
Generation of stable SNCA DSP A + B, FLuc LGALS3, and mCherry LGALS3 cell lines
To generate stable SH-SY5Y cell lines, lentiviral particles were generated by overnight polyethylenimine (PEI) transfection of HEK 293 T cells using equal concentrations of VSV-g, ΔNRF or psPax2, and FLuc LGALS3 or mCherry LGALS3. The next morning, the cell medium was changed. The cultured medium was collected 48 h later, and lentiviral particles were collected and purified using a 0.45 µm (Millipore Sigma, CLS431231) syringe filter. The purified FLuc LGALS3 or mCherry LGALS3 lentiviral particles were added to SH-SY5Y cells and spinoculated at 13°C for 2 h at 1200 × g. At 72 h post-transduction, cells were treated with supplementing DMEM with puromycin (5 µg/ml; Hyclone, Thermo Fisher Scientific, A1113803) for selection. The generation of SNCA DSP A + B SH-SY5Y and HeLa cell lines occurred by the simultaneous transduction of the SNCA DSP A and SNCA DSP B constructs with purified lentiviral particles. The lentiviral particles were generated by transfection of HEK 293 T cells using equal concentrations of VSV-g, ΔNRF or psPax2, and pLVX SNCA DSP A or pLVX SNCA DSP B and purified as stated previously. The purified SNCA DSP A and SNCA DSP B lentiviral particles were added in equal concentration to either SH-SY5Y or HeLa cells and spinoculated. At 72 h post-transduction, cells were treated with puromycin supplementing DMEM. Approximately 96 h later, the puromycin-selected cells were further selected by flow cytometry based on the GFP signal.
Generation of LGALS3 CRISPR plasmids and KO cell lines
The initial generation of CRISPR plasmids was conducted by identifying guide sequences, which was accomplished through the use of the MIT CRISPR design tool and the GeCKO library tool (http://guides.sanjanalab.org/#/). The oligonucleotide guide sequences targeting LGALS3, and the control sequence are 5ʹ-CACCGGGGAAGGGAAGAAAGACAGT-3ʹ and 5ʹ-CACCGGCACTACCAGAGCTAACTCA-3ʹ, respectively. The guide sequence oligonucleotides were annealed and inserted into the Lenti-CRISPRV2 plasmid (Addgene, 52961; deposited by Feng Zhang) by BsmBI digestion and PCR reaction using the published protocol [Citation118]. PEI transfection of HEK 293 T to generate, collect, and purify lentiviral particles was then performed as described in the previous section. Lentiviral particles were then added to generate lgals3 knockouts in the SNCA DSP A + B SH-SY5Y or WT SH-SY5Y cells. The transduced SH-SY5Y were then selected by supplementing DMEM with hygromycin (5 µg/ml, Hyclone, MT30240CR) 72 h post-transduction. Following a 1 week-long selection period in hygromycin, knockout was validated with respect to the control transduced cells by western blots. The heterogeneously selected cells were then used for experiments. Western blots demonstrating successful knockouts of the respective cell lines show the cells following the end of all experiments.
SNCA fibril and drug concentrations and treatment paradigm
The experiments involving SNCA fibril treatment were performed at a final concentration of 200 nM. For the experiments involving drug treatments, the respective culture media was supplemented with the stated drug at the listed final concentration: vehicle (0.1% dimethyl sulfoxide [DMSO]), rapamycin (100 µM; Apex Bio, A8167-25), Baf-A1 (100 nM; Cayman Chemical Company, 11038), 3-MA (5 mM; Sigma-Aldrich, M9281-100 MG), Wor (100 nM; InvivoGen, tlrl-wtm), TD139 (1 µM; MedChem Express, HY-19940), GW4869 (10 µM; Cayman Chemical Company, 13127), and LLOME (2 µM; Santa Cruz Biotechology, sc-285992A). All experiments involving drug treatments, if not otherwise stated, were performed for 24 h except for those in which LLOME was used. LLOME treatments were conducted for 4 h. In the case of the MTOR activation experiments, vehicle, rapamycin, and SNCA fibril treatment were conducted for 24 h, and LLOME treatment was conducted for 4 h so that all treatments finished at the same time. All drugs were purchased from Sigma Aldrich unless otherwise stated.
siRNA transfection
SMARTpool Accell siRNA for control, LGALS3, TRIM16, ATG16L1, and ATG7 were purchased from Horizon Discovery to generate knockdowns in mDA neuronal cultures and iCell DopaNeurons. siRNAs were initially resuspended in 1× siRNA Buffer at a stock concentration of 100 µM as described by Horizon Discovery’s siRNA Resuspension Protocol (https://horizondiscovery.com/-/media/Files/Horizon/resources/Protocols/basic-sirna-resuspension-protocol.pdf). Stock siRNAs were then aliquoted and frozen at −20°C to minimize the effects of multiple freeze-thaw cycles. Before use, aliquots were thawed at room temperature and 5 µl was added to 500 µl of room temperature Accell siRNA Delivery Media (B-005000-500) and gently mixed and added to the neuronal culture as stated in the Dharmacon Accell siRNA delivery protocol (https://horizondiscovery.com/-/media/Files/Horizon/resources/Protocols/accell-delivery-protocol.pdf). The transfection media was changed to SM1 Neurobasal media or iCell Neural Base Medium 24 h later. Experiments including transfections using iCell DopaNeurons were conducted at initial cell concentrations of 2 × 105 cells per well at 72 h post-transfection for all targets. Experiments involving mDA neuronal cultures were performed at 72 h post-transfection for all targets, except for the evaluation of SNCA and LGALS3 levels in the cultured medium from TRIM16 KD cells, which was performed at 48 h post-transfection. To evaluate KD of each target, cells were removed from their adhered well with either the addition of TrypLE express Enzyme 1×, no phenol red (Gibco, Thermo Scientific, 12605010) or trypsin and incubated at 37°C for 7 min and pipetted into 1-ml microcentrifuge tubes. The wells were washed twice with an equal volume of 10% FBS supplemented DMEM and added to the microcentrifuge tube to quench trypsin. The cells were then pelleted by sequential centrifugation at 4°C at 300 × g for 5 min followed by 10,000 × g for 10 min, and the supernatant was carefully aspirated in order to not disrupt the pellet.
Non-denaturing and denaturing SDS-PAGE, western blots, and western blot antibodies
Proteins from pelleted cells or concentrated media were isolated by the addition of lysis buffer composed of 100 mM Tris, pH 8.0, 1% NP-40 (Thermo Fischer Scientific, 85124) 150 mM sodium chloride and protease inhibitor cocktail (Roche, 11836145001) on ice for 30 min. The lysates were centrifuged at 4°C for 10 min at 10,000 × g and afterward the supernatant was collected. The collected supernatants’ protein concentrations were determined by a Pierce BCA protein assay kit (Thermo Fisher Scientific, 23227) or at equal cell number for non-dividing cells. For denaturing SDS-PAGE, an equal fraction of SDS solution was added to the proteins and the contents were boiled on a dry block for 10 min. For non-denaturing SDS-PAGE, an equal fraction of cell lysates to SDS solution with no 2-mercaptoethanol was put on a dry block at 90°C for 7 min. Subsequently, the protein contents were equally loaded and ran on a 10% polyacrylamide gel or a precast 4–20% gradient polyacrylamide gel (Mini-PROTEAN TGX Precast Polyacrylamide Gels, Bio-Rad, 4561096) for SDS-PAGE. After separation, the proteins were transferred to a nitrocellulose membrane (Bio-Rad, 162–0115) and probed overnight at 4°C with the respective primary antibodies diluted in powdered milk block solution at 2.5 g/50 mL of Tris-buffered saline, 0.1% Tween 20 (Sigma-Aldrich, P7949-500ML). The primary antibodies were rabbit anti-ATG7 (1:350; Invitrogen, PIPA535203), rabbit anti-LAMP1 (1:2,000; Abcam, ab24170), mouse anti-GAPDH (1:3,000; Santa Cruz Biotechnology, sc-365062), rabbit anti-p-S757 ULK1 (1:500; Cell Signaling Technology, 6888S), rabbit anti-ULK1 (1:1,000; Cell Signaling Technology, 8054S), anti-p-T389 RPS6KB1/p70 S6K1 (1:1,000; Cell Signaling Technology, 9234S), anti-RPS6KB1/p70 S6K1 (1:1,000; Cell Signaling Technology, 2708S), mouse anti-SNCA/alpha-synuclein (211, 1:2,000; Santa Cruz Biotechnology, sc-12767), rabbit anti-SNCA/alpha-synuclein (MJFR1, 1:2,000; Abcam, ab138501), rabbit polyclonal anti-GFP (1:1000; Thermo Scientific, A6455), mouse anti-GFP (B-2, 1:2000; Santa Cruz Biotechology, sc-9996), rabbit anti-SNCA/alpha-synuclein (p-S129) ([MJF-R13 (8–8)], 1:2,000; Abcam, ab168381), rabbit anti-SNCA/alpha-synuclein aggregate antibody (MJFR-14-6-4-2, 1:2,000; Abcam, ab209538), rabbit anti-TRIM16/EBBP (1:2,000; Bethyl Laboratory, A301-159A), mouse anti-TRIM16 (1:500; Santa Cruz Biotechology, sc-398851), mouse anti-ATG16L1 (1:500; Santa Cruz Biotechology, sc-393274), mouse anti-LGALS3/galectin 3 (1:2,000; BD Pharmingen, 556904), rat anti-HA HRP conjugated (1:2000; Roche, 12013819001), mouse anti-PDCD6IP/Alix (1:500; BioLegend, 634502), mouse anti-CD63 (1:1000; BD Bioscience, 556019), mouse anti-CD81 (1:1000; BD Bioscience, 555675), mouse and rabbit anti-LC3B (1:2,000; Sigma-Aldrich, L7543-100UL).
The nitrocellulose was washed in Tris-buffered saline, 0.1% Tween 20 and probed with the respective horseradish peroxidase (HRP)-conjugated Goat anti-mouse (Thermo Fisher Scientific, 12–349) or anti-rabbit secondary antibody (Thermo Fisher Scientific, 12–348) diluted in milk block solution at 1:10,000 for 30 min. HRP was detected with the addition of SuperSignal West Femto Chemiluminescent Substrate (Thermo Fisher Scientific, PI34096) Chemiluminescence levels were measured using the FlourchemE Imaging System (Protein Simple) containing a CCD camera with 5 logs of bitrate.
Quantification of band intensities were determined using the Gel Analyzer function of ImageJ. The band intensities were plotted and normalized to the respective sample’s GAPDH intensity. The normalized control values were then averaged, and each value was divided by the mean control value.
Renilla luciferase assay
RLuc experiments were performed with HeLa or SH-SY5Y SNCA DSP A + B cells that were equally plated in a 24-well plate at a density of 0.65–2.1 × 105 cells per well depending on the experiment. The cells were then treated with the stated drug at the previously stated concentration. Vehicle or Baf-A1 were treated simultaneously with 200 nM SNCA fibrils for 24 h. Afterward, the cultured media was collected and centrifuged at 4°C for 5–10 min and at 1,200 × g. Next, the supernatant was collected and plated in triplicate at 50 µl per well, in a 96-well, for a luciferase assay. Depending on the experiment, 50–100 µl of coelenterazine (NanoLight, 303–1) solution was added via machine administration Veritas microplate luminometer (Turner BioSystems, Inc) in combination with GloMax software (version 1.9.3). Each experiment was performed in triplicate unless otherwise specified.
mDA neuronal culture transduction and firefly luciferase assay
mDA neuronal culture experiments were performed by co-transducing mDA neuronal cultures with lentiviral pLVX RLuc and either pLVX FLuc LGALS3 or an empty pLVX plasmid. PEI transfection of HEK 293 T cells to generate, collect, and purify lentiviral particles was performed as described previously, using equal DNA concentrations of VSV-g and psPax2, as well as either pLVX RLuc, empty pLVX, or pLVX FLuc LGALS3 plasmid. After purifying lentiviral particles with a 0.45-µm syringe filter, 5-ml aliquots of the particle containing media were concentrated by overnight centrifugation at 5,000 × g. The media was then aspirated and the pellet was immediately resuspended in 1 ml of SM1 Neurobasal media by pipetting. The resuspended pellet was then gently rocked on ice in a 4°C cold room for 1 h to facilitate redistribution of particles. The media was then allowed to come to room temperature and added to 3 wells of a 24-well plate of mDA neurons. The media was changed 72 h after transduction and subsequently collected to evaluate SNCA levels by an SNCA ELISA. Afterward, firefly LGALS3 cells were treated with 200 nM SNCA fibrils or left untreated for 48 h and then culture media was collected in preparation for a FLuc assay. To evaluate the firefly signal, cultured media was centrifuged at 4°C for 10 min and 10,000 × g to remove any cells. Afterward, the supernatant was collected and 150 µl was plated in triplicate in a 96-well plate. The 96-well plate was then added to a Veritas microplate Luminometer and 150 µl of firefly luciferin substrate was added by machine injection and immediately read in combination with GloMax software (version 1.9.3). Background levels were determined by measuring the cultured media from the empty vector cells.
Cell treatment and EV-MAC immunofluorescence staining
The experiments involving SNCA DSP A + B HeLa cells were performed by treating cells with 200 nM SNCA fibrils labeled with ATTO 647 NHS-ester (Atto-Tec GmbH) or leaving them untreated for 24 h. Similarly, in the experiments using the mCherry LGALS3 SH-SY5Y cells, cells were treated with 200 nM SNCA fibrils labeled with ATTO 647 for 24 h, or vehicle (0.1% DMSO) or LLOME (2 µM) for 4 h. For amphisome experiments, control or LGALS3 KO SH-SY5Y cells were treated with 200 nM SNCA fibrils labeled with ATTO 550 for 24 h, vehicle (0.1% DMSO) or LLOME (2 µM) for 4 h. For TFEB experiments, the procedure was identical to that described above with the exception of the use of unlabeled SNCA fibrils. The same endpoint was used for all treatment conditions. For the experiments involving the iCell DopaNeurons, the cells were transfected with siRNA as previously described and cultured media was obtained at 24–72 h post-transfection. Collected cultured media was initially centrifuged at 1,200 × g for 5 min at 4°C to remove cellular debris. Next, 500 µl of the supernatant cultured media was added to a well of a 24-well plate containing a glass coverslip (Thermo Fisher Sciences). Coverslips were stored long-term in a 50 ml conical containing 70% ethanol and were added to individual wells of a 24-well tissue culture plate and subsequently washed thrice with PBS. The final round of PBS was left in the well and aspirated immediately before continuing. In order for the contents of the cultured media to adhere to the coverslips, the cultured media was spinoculated by centrifugation at 13°C for 2 h at 1,200 × g and subsequently fixed in a solution of 0.1 M piperazine-N-N′bis[2-ethanesulfonic acid] buffer containing 3.7% formaldehyde (Polysciences, 04018–1) for 15 min and washed thrice with PBS. The coverslips were permeabilized with a 0.1% solution of saponin in block solution, composed of 500 ml of PBS supplemented with 10% NDS and 0.01% sodium azide, for 5 min. After washing 3 times, the coverslips were incubated with either mouse anti-LGALS3 (1:1,000; BD Pharmingen, 556904), mouse anti-CD9 (1:1,000; BD Bioscience, 555370), mouse anti-CD63 (1:1,000), mouse anti-CD81 (1:1,000; BD Bioscience, 555675), or rabbit anti-SNCA (MJFR1, 1:1,500) for 1 h at room temperature and then washed three times with PBS. Where applicable, biotin conjugated WGA (Vector Laboratories, B-1025) was used at a working concentration of 5 μg/ml for 1 h at room temperature after incubation with the solution containing primary antibody. Afterward, primary antibodies were labeled using fluorophore conjugated donkey anti-mouse or donkey anti-rabbit antibodies (1:400; Jackson ImmunoResearch Laboratories, Alexa Fluor 488: 715-545-150, 711-545-152; Alexa Fluor 594: 715-585-150, 711-585-152; Alexa Fluor 647: 715-605-151, 711-605-152) for 30 min at room temperature. Depending on the set of experiments, the conjugated fluorophore color varied. Alexa Fluor 594 donkey anti-mouse (Jackson ImmunoResearch Laboratories, 715-585-150) was used to label the LGALS3 antibody from SNCA DSP A + B culture media. Alexa Fluor 647 donkey anti-mouse (Jackson ImmunoResearch Laboratories, Alexa Fluor 647: 715-605-151) and Alexa Fluor 594 donkey anti-rabbit (Jackson ImmunoResearch Laboratories, 711-585-152) were used to label the LGALS3 and SNCA antibodies, respectively. Afterward, the coverslips were washed with PBS. If the coverslips underwent lectin staining, biotin-conjugated WGA was labeled using PBS-diluted fluorescein isothiocyanate conjugated streptavidin (SAV, 1:1,000; BD Pharmingen, 554060) for 1 h at room temperature. Afterward, coverslips were fixed and mounted with Fluoro-Gel (Electron Microscopy Sciences, 17985–11) onto slides (Fisher Scientific, 12-550-15).
Fixation and immunofluorescence staining of cells
In preparation for immunofluorescence antibody staining, cells were fixed in a solution of 4% formaldehyde in Dulbecco’s phosphate buffered saline (DPBS) for 15 min and gently washed twice in DPBS. Cells were permeabilized, blocked, and probed with primary antibody in a block solution composed of DPBS supplemented with 2% FBS, 2% NDS, and 0.2% Triton X-100 sterile filtered through a 0.22 µm filter. The following immunofluorescence antibodies were used at the specified concentrations: mouse anti-galectin 3 (1:1,000, BD Pharmingen, 556904), mouse anti-OCT3/4 (1:500; Santa Cruz Biotechology, sc-5279), rabbit anti-LMX1A (1:1,000; EMD Millipore, AB10533), mouse anti-NES/nestin (1:500; Santa Cruz Biotechology, sc-23927), rabbit anti-TH/tyrosine hydroxylase (1:1,000; EMD Millipore, AB152), rabbit anti-TRIM16/EBBP (1:1,000; Bethyl Laboratory, A301-159A), mouse anti-TRIM16 (1:500; Santa Cruz Biotechology, sc-398851), rabbit anti-LC3B (1:400; Sigma-Aldrich, L7543-100UL), mouse anti-ATG16L1 (1:500; Santa Cruz Biotechology, sc-393274), mouse anti-CD63 (1:1,000; BD Bioscience, 556019), and mouse anti-beta-III tubulin (1:500; Santa Cruz Biotechology, sc-80005). Cells were incubated with primary antibody solution at room temperature for 1 h and washed thrice in DPBS. Non-overlapping conjugated donkey anti-mouse and/or anti-rabbit were used at either 488, 594, or 647 (1:400) in blocking buffer. When used, 4′,6-diamino-2-phenylindole (DAPI; Sigma-Aldrich, D9542) and/or phalloidin (Sigma-Aldrich, P1951) was added to the secondary antibody solution. Cells were incubated with secondary antibody solution for 30 min at room temperature and subsequently washed thrice with DPBS. Afterward, coverslips were fixed and mounted onto slides (Globe Scientific Inc.).
Wide-field fluorescence deconvolution microscopy
Imaging of cultured media or cells was performed on a DeltaVision wide-field fluorescence microscope (Applied Precision, Inc.) outfitted with a digital camera (CoolSNAP HQ2; Photometrics), while using an oil immersion Olympus Plan Apo 60× objective lens (1.42 numerical aperture) with 1.5150 refraction index ResolveTM immersion oil (Richard Allen Scientific). Direct excitation with a 250-watt Xenon Arc lamp from the back of the microscope was focused from below onto the coverslip held on an Olympus IX-71 stage. The exposure times were varied and depended on the staining conditions.
Fluorescence imaging paradigm
The number of images taken per experiment is stated in the figure legend. Data was collected by z-stack imaging and was analyzed as maximum intensity projections (MIPs). Cells were imaged using z-stacks with 0.5 µm between each stack and a total of 30 z-stacks. The imaging of YFP-LC3B was conducted by visually identifying YFP-LC3B positive cells. A subset of non-transduced YFP-LC3B cells were also imaged to help evaluate background fluorescence levels in the siRNA-treated control cells. Similarly, DSP-SNCA, control KO, and LGALS3 KO experiments were conducted with secondary only antibody controls to determine the level of background fluorescence. The same exposure conditions were used for all coverslips and experiments.
Calibration of the microscope and a more extensive description of the EV-MAC workflow was previously described [Citation75]. In brief, imaged cultured media was performed from different locations on the coverslip using the “panels” function of the SoftWoRx software (Applied Precision, Inc.) to obtain a non-biased, representative sample of images from the total population. The same set of panels was applied to each coverslip and the z-stack distance was manually recalibrated for each coverslip to ensure proper focus of the images. Each image was taken with 30 z-stacks at 0.2 µm between each stack. Coverslips were imaged so that z-stacks started and ended on either side of the focal plane. Z-stack imaging was used to facilitate the constrained iterative deconvolution performed by SoftWoRx (Applied Precision, Inc.) using the prerecorded and empirically generated optical transfer function for the 60 × 1.42 numerical aperture Olympus Plan Apo objective (Applied Precision, Inc).
Imaging analysis
The collected z-stack images were used as reconstructed 3-dimensional MIPs for analysis with Imaris software (version 7.6.4, Bitplane) in order to use the 3-dimensional masking algorithm function. The same masking algorithm was applied to all images of a single experiment regardless of condition to permit valid between groups comparisons using the Batch Coordinator tool (Imaris, Bitplane).
In cultured media experiments, non-representative images where the focal plane was not correctly imaged were excluded. No more than 2 images per condition, per replicate were excluded. The relative background levels of maximum intensity for each respective channel were determined based on the secondary antibody controls for mouse, rabbit, and SAV (where applicable) by determining the 95th percentile’s maximum fluorescence intensity from each image and, subsequently, averaging them.
Lysosome dysfunction assay and measurement of autophagic flux
An equal concentration of mDA neuronal culture cells were plated in a clear bottom, black 96-well plate (Costar) during the penultimate steps of the differentiation process to mature until day 50 post-initial differentiation at ~100,000 cells per well. Cells were treated with control or LGALS3 siRNA as previously described. The media was changed 24 h post-treatment. At 48 h post-transfection, the media was changed and either supplemented with SNCA fibrils or left untreated. At 72 h post-transfection, the wells were washed thrice then treated with vehicle (0.1% DMSO) or 200 nM Baf-A1 diluted in SM1 Neurobasal (Gibco, 21103049 media for 2 h then subsequently loaded with reconstituted Magic Red cathepsin B (ImmunoChemistry Technologies, 937) for 30 min. Magic Red cathepsin B was reconstituted and loaded based on the manufacturers’ protocol. Cells were then washed thrice with clear Neurobasal media (Gibco, 12348017) at room temperature, and clear Neurobasal media supplemented with vehicle or Baf-A1 was added to cells, based on previous treatments. The cells were then placed in a plate reader warmed to 37°C and supplemented with 5% CO2. The Magic Red fluorescence signal was measured using a 528 nm excitation wavelength and 628 nm emission wavelength at baseline and every 15 min over a 4-h period. At the end of the 4 h, the cells were lysed and saved for western blot analysis. A total of 6–8 wells were used for each of the vehicle treatments (control/LGALS3 siRNA and ± SNCA fibril) and a total of 2–4 wells for the Baf-A1 treatments (control/LGALS3 siRNA and ± SNCA fibril in each experiment). The graphed data was normalized to the endpoint relative fluorescence units (RFU) of the control siRNA plus vehicle minus that of the SNCA fibril and subsequently extrapolated to all time points and conditions within each row of the 96-well plate, which contains one of every possible variable combination. Each row was considered a biological replicate and was pooled with all other replicates from a total of 4 independent experiments (n = 17–25 among conditions). Lysate from 2 wells from each treatment condition was pooled to obtain enough protein, ran on an SDS-PAGE gel, then transferred for western blot analysis. Wells in which insufficient KD was observed (< 40% LGALS3 depletion) relative to that of the comparable treatments’ control siRNA condition were excluded. Western blot quantifications were used to determine autophagic flux: n = 8, control siRNA plus vehicle conditions; n = 8, LGALS3 siRNA plus vehicle; n = 6, control siRNA plus Baf-A1; n = 6, LGALS3 siRNA plus Baf-A1; n = 6, control siRNA plus SNCA fibrils; n = 6, LGALS3 siRNA plus SNCA fibrils; n = 6, control siRNA plus SNCA fibrils + Baf-A1; and n = 6, LGALS3 siRNA plus SNCA fibrils + Baf-A1.
mDA neuronal culture transduction and YFP-LC3B colocalization assay
The PEI transfection of HEK 293 T cells to generate, collect, and purify lentiviral particles was performed as described previously, using equal DNA concentrations of either pLVX inducible YFP-LC3B plasmid, VSV-g and psPax2. After purifying lentiviral particles with a 0.45 µm syringe filter, the particles were concentrated by overnight centrifugation of 5 ml of lentiviral particle-containing media at 5,000 × g. The media was then aspirated and the pellet was resuspended in 1 ml of SM1 Neurobasal media (Gibco) by pipetting. The resuspended pellet was then gently rocked on ice in a 4°C cold room for 1 h to facilitate redistribution of particles. The media was then allowed to come to room temperature and added to 3 wells of a 24-well plate of mDA neuronal cultures. Experiments were conducted at least 72 h post-transduction.
To perform experiments, cells were transfected with siRNAs as previously described. At 24 h after transfection, the media was changed to SM1 Neurobasal media supplemented with doxycycline monohydrate (Sigma-Aldrich, 324385–1 GM) at a working concentration of 1:10,000 for 24 h to induce the expression of YFP-LC3B. Afterward, the media was changed, and the cells were treated with 200 nM ATTO 647 NHS-ester SNCA fibrils for 24 h (48 h post-transfection). Cells were then fixed in 4% formaldehyde solution diluted in DPBS at 72 h post-siRNA transfection. They were subjected to immunofluorescence staining as previously described.
SNCA and LGALS3 sandwich ELISA
Evaluation of SNCA or LGALS3 in the culture media was determined by an in-house sandwich ELISA using commercially available antibodies. When serial dilutions were added to uncultured media, a standard curve was generated for SNCA (Fig. S9(A)) and LGALS3 (Fig. S9(B)). The characterization of antibodies used in an SNCA sandwich ELISA has been previous reviewed in-depth including their specific binding epitopes [Citation119–123]. Similarly, epitope mapping to specific regions of LGALS3 with the following antibodies has been previously described and/or used in direct ELISA: anti-LGALS3/galectin 3 antibodies B2C10 (Santa Cruz Biotechology, sc-32790), A3A12 (Santa Cruz Biotechology, sc-53127), and M3/38 (Millipore Sigma, 125402) [Citation31,Citation124–127].
For the SNCA sandwich ELISA, mouse anti-SNCA (BD Biosciences, 610786) and LGALS3 sandwich ELISA mouse anti-LGALS3 B2C10 or A3A12 (Santa Cruz Biotechology, SC-32790, SC-53127) antibodies were diluted in pH 9.6 carbonate buffer to a final concentration of 1 µg/ml and added to wells of a 96-well Maxisorp ELISA plate (Nunc, 44-2404-22) at 4°C for 16 h on an orbital shaker to coat the wells. The wells were then washed with PBS containing Tween-20 (PBS-T) three times. Afterward, the wells were blocked in equal portions of 10% bovine serum albumin supplemented DMEM and PBS for 2 h at room temperature and subsequently washed thrice with PBS-T. For the SNCA and LGALS3 sandwich ELISA, 50 µl or 100 µl of cultured media, respectively, from each sample as well as serial dilutions of recombinant human SNCA monomer (Proteos, rp-001) or LGALS3 (Abcam, ab89487) were added in triplicate for 24 h at 4°C. Wells were then washed thrice with PBS-T. HRP conjugated mouse anti-SNCA (211; Santa Cruz Biotechology, SC-12767 HRP) was diluted to a final concentration of 1 µg/ml or biotin conjugated rat anti-LGALS3 (M3/38; Millipore Sigma, 125402) was diluted to a final concentration of 500 ng/ml in 1× ELISA Diluent solution (Thermo Fisher Scientific, 00-4202-56) and then added to wells for 24 h at 4°C on an orbital shaker. Afterward, the wells were washed thrice with PBS-T. For the LGALS3 ELISA, HRP conjugated SAV was diluted to 1 µg/ml in 1× ELISA Diluent solution and added for 30 min at room temperature on a rocker. The plate was then washed 5 times and with PBS-T and the HRP signal was detected with the addition of 1 × 3,3′, 5, 5-tetramethylbenzidine (Invitrogen, 00-4201-56) and the reaction was quenched with 2 N sulfuric acid. The absorbance was read at 450 nm on a PowerWave XS plate reader (BioTek Instruments) in conjunction with Gen5 software.
Statistical analysis
All statistical analyses were conducted with, and graphs were made with Prism (version 6.0, GraphPad Software, Inc.). Data from a single cultured media preparation or coverslip are given as M ± SD. Data from experiments in which multiple cultured media preparations and independent coverslips were imaged are given as M ± SE. The statistical test and post hoc analysis for each experiment are stated in the figure legend. Each graph shows the mean value, and each data point indicates a biological replicate and is the mean value when a sample was tested multiple times. For all statistical tests *, **, ***, ****, p < 0.05, 0.01, 0.001, and 0.0001, respectively.
Supplemental Material
Download MS Word (50.8 MB)Acknowledgments
This work was support by grants from the Michael J Fox Foundation and the Emerald Foundation. NanoSight300 data was acquired with the assistance of the Analytical bioNanoTechnology Core Facility of the Simpson Querrey Institute at Northwestern University.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
References
- Galloway DA, Phillips AEM, Owen DRJ, et al. Phagocytosis in the Brain: homeostasis and Disease. Front Immunol. 2019;10:790.
- Hernandez DG, Reed X, Singleton AB. Genetics in Parkinson disease: mendelian versus non-Mendelian inheritance. J Neurochem. 2016 Oct;139(Suppl 1):59–74.
- Visanji NP, Brotchie JM, Kalia LV, et al. alpha-Synuclein-Based animal models of Parkinson’s Disease: challenges and opportunities in a New Era. Trends Neurosci. 2016 Nov;39(11):750–762.
- Lee SJ, Desplats P, Lee HJ, et al. Cell-to-cell transmission of alpha-synuclein aggregates. Methods Mol Biol. 2012;849:347–359.
- Danzer KM, Kranich LR, Ruf WP, et al. Exosomal cell-to-cell transmission of alpha synuclein oligomers. Mol Neurodegener. 2012 Aug 24;7(1):42.
- Freeman D, Cedillos R, Choyke S, et al. Alpha-synuclein induces lysosomal rupture and cathepsin dependent reactive oxygen species following endocytosis. PLoS One. 2013;8(4):e62143.
- Hansen C, Angot E, Bergstrom AL, et al. alpha-Synuclein propagates from mouse brain to grafted dopaminergic neurons and seeds aggregation in cultured human cells. J Clin Invest. 2011 Feb 1;121(2):715–725.
- Luk KC, Kehm VM, Zhang B, et al. Intracerebral inoculation of pathological alpha-synuclein initiates a rapidly progressive neurodegenerative alpha-synucleinopathy in mice. J Exp Med. 2012 May 7;209(5):975–986.
- Peelaerts W, Bousset L, Van der Perren A, et al. alpha-Synuclein strains cause distinct synucleinopathies after local and systemic administration. Nature. 2015 Jun 18;522(7556):340–344.
- Rey NL, Steiner JA, Maroof N, et al. Widespread transneuronal propagation of alpha-synucleinopathy triggered in olfactory bulb mimics prodromal Parkinson’s disease. J Exp Med. 2016 Aug 22;213(9):1759–1778.
- Desplats P, Lee HJ, Bae EJ, et al. Inclusion formation and neuronal cell death through neuron-to-neuron transmission of alpha-synuclein. Proc Natl Acad Sci U S A. 2009 Aug 4;106(31):13010–13015.
- Kordower JH, Chu Y, Hauser RA, et al. Lewy body-like pathology in long-term embryonic nigral transplants in Parkinson’s disease. Nat Med. 2008 May;14(5):504–506.
- Kordower JH, Chu Y, Hauser RA, et al. Transplanted dopaminergic neurons develop PD pathologic changes: a second case report. Mov Disord. 2008 Dec 15;23(16):2303–2306.
- Li JY, Englund E, Widner H, et al. Characterization of Lewy body pathology in 12- and 16-year-old intrastriatal mesencephalic grafts surviving in a patient with Parkinson’s disease. Mov Disord. 2010 Jun 15;25(8):1091–1096.
- Luk KC, Song C, O’Brien P, et al. Exogenous alpha-synuclein fibrils seed the formation of Lewy body-like intracellular inclusions in cultured cells. Proc Natl Acad Sci U S A. 2009 Nov 24;106(47):20051–20056.
- Luk KC, Kehm V, Carroll J, et al. Pathological alpha-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science. 2012 Nov 16;338(6109):949–953.
- Frake RA, Ricketts T, Menzies FM, et al. Autophagy and neurodegeneration. J Clin Invest. 2015 Jan;125(1):65–74.
- Weidberg H, Shvets E, Elazar Z. Biogenesis and cargo selectivity of autophagosomes. Annu Rev Biochem. 2011;80:125–156.
- Cuervo AM, Stefanis L, Fredenburg R, et al. Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science. 2004 Aug 27;305(5688):1292–1295.
- Tofaris GK. Lysosome-dependent pathways as a unifying theme in Parkinson’s disease. Mov Disord. 2012 Sep 15;27(11):1364–1369.
- Tofaris GK, Kim HT, Hourez R, et al. Ubiquitin ligase Nedd4 promotes alpha-synuclein degradation by the endosomal-lysosomal pathway. Proc Natl Acad Sci U S A. 2011 Oct 11;108(41):17004–17009.
- Johannes L, Jacob R, Leffler H. Galectins at a glance. J Cell Sci. 2018 May 1;131(9). DOI:https://doi.org/10.1242/jcs.208884.
- Tan Y, Zheng Y, Xu D, et al. Galectin-3: a key player in microglia-mediated neuroinflammation and Alzheimer’s disease. Cell Biosci. 2021 Apr 27;11(1):78.
- Paz I, Sachse M, Dupont N, et al. Galectin-3, a marker for vacuole lysis by invasive pathogens. Cell Microbiol. 2010 Apr 1;12(4):530–544.
- Thurston TL, Wandel MP, von Muhlinen N, et al. Galectin 8 targets damaged vesicles for autophagy to defend cells against bacterial invasion. Nature. 2012 Feb 16;482(7385):414–418.
- Randow F, MacMicking JD, James LC. Cellular self-defense: how cell-autonomous immunity protects against pathogens. Science. 2013 May 10;340(6133):701–706.
- Randow F, Youle RJ. Self and nonself: how autophagy targets mitochondria and bacteria. Cell Host Microbe. 2014 Apr 09;15(4):403–411.
- Flavin WP, Bousset L, Green ZC, et al. Endocytic vesicle rupture is a conserved mechanism of cellular invasion by amyloid proteins. Acta Neuropathol. 2017 Oct;134(4):629–653.
- Bussi C, Peralta Ramos JM, Arroyo DS, et al. Alpha-synuclein fibrils recruit TBK1 and OPTN to lysosomal damage sites and induce autophagy in microglial cells. J Cell Sci. 2018 Nov 30;131(23).
- Falcon B, Noad J, McMahon H, et al. Galectin-8-mediated selective autophagy protects against seeded tau aggregation. J Biol Chem. 2018 Feb 16;293(7):2438–2451.
- Ashraf GM, Baeesa SS. Investigation of Gal-3 expression pattern in serum and cerebrospinal fluid of patients suffering from neurodegenerative disorders. Front Neurosci. 2018;12:430.
- Rousseaux MWC, Vazquez-Velez GE, Al-Ramahi I, et al. A druggable genome screen identifies modifiers of alpha-synuclein levels via a tiered cross-species validation approach. J Neurosci. 2018 Oct 24;38(43):9286–9301.
- Rusmini P, Cortese K, Crippa V, et al. Trehalose induces autophagy via lysosomal-mediated TFEB activation in models of motoneuron degeneration. Autophagy. 2019 Apr;15(4):631–651.
- Diogo D, Tian C, Franklin CS, et al. Phenome-wide association studies across large population cohorts support drug target validation. Nat Commun. 2018 Oct 16;9(1):4285.
- Billingsley KJ, Barbosa IA, Bandres-Ciga S, et al. Mitochondria function associated genes contribute to Parkinson’s Disease risk and later age at onset. NPJ Parkinsons Dis. 2019;5:8.
- Chauhan S, Kumar S, Jain A, et al. TRIMs and galectins globally cooperate and TRIM16 and galectin-3 Co-direct autophagy in endomembrane damage homeostasis. Dev Cell. 2016 Oct 10;39(1):13–27.
- Jia J, Abudu YP, Claude-Taupin A, et al. Galectins control mTOR in response to endomembrane damage. Mol Cell. 2018 Apr 5;70(1):120–135 e8.
- Jia J, Claude-Taupin A, Gu Y, et al. Galectin-3 coordinates a cellular system for lysosomal repair and removal. Dev Cell. 2020 Jan 6;52(1):69–87 e8.
- Kimura T, Jia J, Kumar S, et al. Dedicated SNAREs and specialized TRIM cargo receptors mediate secretory autophagy. EMBO J. 2017 Jan 04;36(1):42–60.
- Kumar S, Chauhan S, Jain A, et al. Galectins and TRIMs directly interact and orchestrate autophagic response to endomembrane damage. Autophagy. 2017 Jun 3;13(6):1086–1087.
- Wang X, Zhang S, Lin F, et al. Elevated Galectin-3 levels in the serum of patients with Alzheimer’s Disease. Am J Alzheimers Dis Other Demen. 2015 Dec;30(8):729–732.
- Dupont N, Jiang S, Pilli M, et al. Autophagy-based unconventional secretory pathway for extracellular delivery of IL-1beta. EMBO J. 2011 Nov 08;30(23):4701–4711.
- Ponpuak M, Mandell MA, Kimura T, et al. Secretory autophagy. Curr Opin Cell Biol. 2015 Aug;35:106–116.
- Christensen DP, Ejlerskov P, Rasmussen I, et al. Reciprocal signals between microglia and neurons regulate alpha-synuclein secretion by exophagy through a neuronal cJUN-N-terminal kinase-signaling axis. J Neuroinflammation. 2016 Mar 08;13(1):59.
- Poehler AM, Xiang W, Spitzer P, et al. Autophagy modulates SNCA/alpha-synuclein release, thereby generating a hostile microenvironment. Autophagy. 2014;10(12):2171–2192.
- Alvarez-Erviti L, Seow Y, Schapira AH, et al. Lysosomal dysfunction increases exosome-mediated alpha-synuclein release and transmission. Neurobiol Dis. 2011 Jun;42(3):360–367.
- Minakaki G, Menges S, Kittel A, et al. Autophagy inhibition promotes SNCA/alpha-synuclein release and transfer via extracellular vesicles with a hybrid autophagosome-exosome-like phenotype. Autophagy. 2018;14(1):98–119.
- Lee HJ, Cho ED, Lee KW, et al. Autophagic failure promotes the exocytosis and intercellular transfer of alpha-synuclein. Exp Mol Med. 2013;45:e22.
- Hasegawa T, Konno M, Baba T, et al. The AAA-ATPase VPS4 regulates extracellular secretion and lysosomal targeting of alpha-synuclein. PLoS One. 2011;6(12):e29460.
- Fussi N, Hollerhage M, Chakroun T, et al. Exosomal secretion of alpha-synuclein as protective mechanism after upstream blockage of macroautophagy. Cell Death Dis. 2018 Jul 9;9(7):757.
- Gudbergsson JM, Johnsen KB. Exosomes and autophagy: rekindling the vesicular waste hypothesis. J Cell Commun Signal. 2019 Dec;13(4):443–450.
- Jang A, Lee HJ, Suk JE, et al. Non-classical exocytosis of alpha-synuclein is sensitive to folding states and promoted under stress conditions. J Neurochem. 2010 Jun;113(5):1263–1274.
- Borland H, Vilhardt F. Prelysosomal compartments in the unconventional secretion of amyloidogenic seeds. Int J Mol Sci. 2017 Jan 23;18(1):227.
- Ejlerskov P, Rasmussen I, Nielsen TT, et al. Tubulin polymerization-promoting protein (TPPP/p25alpha) promotes unconventional secretion of alpha-synuclein through exophagy by impairing autophagosome-lysosome fusion. J Biol Chem. 2013 Jun 14;288(24):17313–17335.
- Cai W, Feng D, Schwarzschild MA, et al. Bimolecular fluorescence complementation of alpha-synuclein demonstrates its oligomerization with dopaminergic phenotype in mice. EBioMedicine. 2018 Mar;29:13–22.
- Outeiro TF, Putcha P, Tetzlaff JE, et al. Formation of toxic oligomeric alpha-synuclein species in living cells. PLoS One. 2008 Apr 2;3(4):e1867.
- Kiechle M, von Einem B, Hofs L, et al. In vivo protein complementation demonstrates presynaptic alpha-synuclein oligomerization and age-dependent accumulation of 8-16-mer oligomer species. Cell Rep. 2019 Nov 26;29(9):2862–2874 e9.
- Eckermann K, Kugler S, Bahr M. Dimerization propensities of Synucleins are not predictive for Synuclein aggregation. Biochim Biophys Acta. 2015 Aug;1852(8):1658–1664.
- Holmqvist S, Chutna O, Bousset L, et al. Direct evidence of Parkinson pathology spread from the gastrointestinal tract to the brain in rats. Acta Neuropathol. 2014 Dec;128(6):805–820.
- Aelvoet SA, Ibrahimi A, Macchi F, et al. Noninvasive bioluminescence imaging of alpha-synuclein oligomerization in mouse brain using split firefly luciferase reporters. J Neurosci. 2014 Dec 03;34(49):16518–16532.
- Danzer KM, Ruf WP, Putcha P, et al. Heat-shock protein 70 modulates toxic extracellular alpha-synuclein oligomers and rescues trans-synaptic toxicity. FASEB J. 2011 Jan;25(1):326–336.
- Kondo N, Miyauchi K, Meng F, et al. Conformational changes of the HIV-1 envelope protein during membrane fusion are inhibited by the replacement of its membrane-spanning domain. J Biol Chem. 2010 May 7;285(19):14681–14688.
- Bousset L, Pieri L, Ruiz-Arlandis G, et al. Structural and functional characterization of two alpha-synuclein strains. Nat Commun. 2013;4:2575.
- Mauvezin C, Nagy P, Juhasz G, et al. Autophagosome-lysosome fusion is independent of V-ATPase-mediated acidification. Nat Commun. 2015 May;11(6):7007.
- Mauvezin C, Neufeld TP. Bafilomycin A1 disrupts autophagic flux by inhibiting both V-ATPase-dependent acidification and Ca-P60A/SERCA-dependent autophagosome-lysosome fusion. Autophagy. 2015;11(8):1437–1438.
- Yamamoto A, Tagawa Y, Yoshimori T, et al. Bafilomycin A1 prevents maturation of autophagic vacuoles by inhibiting fusion between autophagosomes and lysosomes in rat hepatoma cell line, H-4-II-E cells. Cell Struct Funct. 1998 Feb;23(1):33–42.
- Fernandes HJ, Hartfield EM, Christian HC, et al. ER Stress and autophagic perturbations lead to elevated extracellular alpha-synuclein in GBA-N370S Parkinson’s iPSC-derived dopamine neurons. Stem Cell Reports. 2016 Mar 8;6(3):342–356.
- Kunadt M, Eckermann K, Stuendl A, et al. Extracellular vesicle sorting of alpha-Synuclein is regulated by sumoylation. Acta Neuropathol. 2015 May;129(5):695–713.
- Banfer S, Jacob R. Galectins in intra- and extracellular vesicles. Biomolecules. 2020 Aug 24;10(9):1232.
- Ohman T, Teirila L, Lahesmaa-Korpinen AM, et al. Dectin-1 pathway activates robust autophagy-dependent unconventional protein secretion in human macrophages. J Immunol. 2014 Jun 15;192(12):5952–5962.
- Grey M, Dunning CJ, Gaspar R, et al. Acceleration of alpha-synuclein aggregation by exosomes. J Biol Chem. 2015 Jan 30;290(5):2969–2982.
- Catalano M, O’Driscoll L. Inhibiting extracellular vesicles formation and release: a review of EV inhibitors. J Extracell Vesicles. 2020;9(1):1703244.
- Leidal AM, Huang HH, Marsh T, et al. The LC3-conjugation machinery specifies the loading of RNA-binding proteins into extracellular vesicles. Nat Cell Biol. 2020 Feb;22(2):187–199.
- Back MJ, Ha HC, Fu Z, et al. Activation of neutral sphingomyelinase 2 by starvation induces cell-protective autophagy via an increase in Golgi-localized ceramide. Cell Death Dis. 2018 Jun 4;9(6):670.
- Burbidge K, Zwikelmaier V, Cook B, et al. Cargo and cell-specific differences in extracellular vesicle populations identified by multiplexed immunofluorescent analysis. J Extracell Vesicles. 2020;9(1):1789326.
- O’Doherty U, Swiggard WJ, Malim MH. Human immunodeficiency virus type 1 spinoculation enhances infection through virus binding. J Virol. 2000 Nov;74(21):10074–10080.
- Sandhof CA, Hoppe SO, Druffel-Augustin S, et al. Reducing INS-IGF1 signaling protects against non-cell autonomous vesicle rupture caused by SNCA spreading. Autophagy. 2020 May;16(5):878–899.
- Doyle LM, Wang MZ. Overview of extracellular vesicles, their origin, composition, purpose, and methods for exosome isolation and analysis. Cells. 2019 Jul 15;8(7):727.
- Stewart SE, Menzies SA, Popa SJ, et al. A genome-wide CRISPR screen reconciles the role of N-linked glycosylation in galectin-3 transport to the cell surface. J Cell Sci. 2017 Oct 1;130(19):3234–3247.
- Skowyra ML, Schlesinger PH, Naismith TV, et al. Triggered recruitment of ESCRT machinery promotes endolysosomal repair. Science. 2018 Apr 6;360(6384):eaar5078.
- Frahm S, Melis V, Horsley D, et al. Alpha-Synuclein transgenic mice, h-α-SynL62, display α-Syn aggregation and a dopaminergic phenotype reminiscent of Parkinson’s disease. Behav Brain Res. 2018 Feb 26;339:153–168.
- Yoshii SR, Mizushima N. Monitoring and measuring autophagy. Int J Mol Sci. 2017 Aug 28;18(9):1865.
- Jena KK, Kolapalli SP, Mehto S, et al. TRIM16 controls assembly and degradation of protein aggregates by modulating the p62-NRF2 axis and autophagy. EMBO J. 2018 Sep 14;37(18). DOI:https://doi.org/10.15252/embj.201798358.
- Echevarria J, Royo F, Pazos R, et al. Microarray-based identification of lectins for the purification of human urinary extracellular vesicles directly from urine samples. Chembiochem. 2014 Jul 21;15(11):1621–1626.
- Jia J, Abudu YP, Claude-Taupin A, et al. Galectins control MTOR and AMPK in response to lysosomal damage to induce autophagy. Autophagy. 2019 Jan;15(1):169–171.
- Chen WT, Zhang F, Zhao XQ, et al. Galectin-3 and TRIM16 coregulate osteogenic differentiation of human bone marrow-derived mesenchymal stem cells at least partly via enhancing autophagy. Bone. 2020 Feb;131:115059.
- Cohen MJ, Chirico WJ, Lipke PN. Through the back door: unconventional protein secretion. The Cell Surface. 2020;6:100045.
- Salimi L, Akbari A, Jabbari N, et al. Synergies in exosomes and autophagy pathways for cellular homeostasis and metastasis of tumor cells. Cell Biosci. 2020 May 13;10(1):64.
- Chen Y-D, Fang Y-T, Cheng Y-L, et al. Exophagy of annexin A2 via RAB11, RAB8A and RAB27A in IFN-γ-stimulated lung epithelial cells. Sci Rep. 2017 Jul 18;7(1):5676.
- Hurwitz SN, Cheerathodi MR, Nkosi D, et al. Tetraspanin CD63 bridges autophagic and endosomal processes to regulate exosomal secretion and intracellular signaling of Epstein-Barr Virus LMP1. J Virol. 2018;92(5):e01969–17.
- Jiang P, Gan M, Yen SH, et al. Impaired endo-lysosomal membrane integrity accelerates the seeding progression of alpha-synuclein aggregates. Sci Rep. 2017 Aug 9;7(1):7690.
- Hoffmann AC, Minakaki G, Menges S, et al. Extracellular aggregated alpha synuclein primarily triggers lysosomal dysfunction in neural cells prevented by trehalose. Sci Rep. 2019 Jan 24;9(1):544.
- Hou X, Watzlawik JO, Fiesel FC, et al. Autophagy in Parkinson's Disease. J Mol Biol. 2020 Apr 3;432(8):2651–2672
- Stefanis L, Emmanouilidou E, Pantazopoulou M, et al. How is alpha-synuclein cleared from the cell? J Neurochem. 2019;150(5):577–590.
- Yu WH, Dorado B, Figueroa HY, et al. Metabolic activity determines efficacy of macroautophagic clearance of pathological oligomeric alpha-synuclein. Am J Pathol. 2009 Aug;175(2):736–747.
- Flavin WP, Bousset L, Green ZC, et al. Endocytic vesicle rupture is a conserved mechanism of cellular invasion by amyloid proteins. Acta Neuropathol. 2017 May 19;134(4):629–653.
- Uversky VN, Li J, Fink AL. Evidence for a partially folded intermediate in alpha-synuclein fibril formation. J Biol Chem. 2001 Apr 6;276(14):10737–10744.
- Hoyer W, Antony T, Cherny D, et al. Dependence of alpha-synuclein aggregate morphology on solution conditions. J Mol Biol. 2002 Sep 13;322(2):383–393.
- Fraiberg M, Elazar Z. A TRIM16-Galactin3 complex mediates autophagy of damaged endomembranes. Dev Cell. 2016 Oct 10;39(1):1–2.
- Nilsson E, Ghassemifar R, Brunk UT. Lysosomal heterogeneity between and within cells with respect to resistance against oxidative stress. Histochem J. 1997 Nov-Dec;29(11–12):857–865.
- Johansson AC, Appelqvist H, Nilsson C, et al. Regulation of apoptosis-associated lysosomal membrane permeabilization. Apoptosis. 2010 May;15(5):527–540.
- Meiser J, Weindl D, Hiller K. Complexity of dopamine metabolism. Cell Commun Signaling. 2013 May 17;11(1):34.
- Seibler P, Graziotto J, Jeong H, et al. Mitochondrial Parkin recruitment is impaired in neurons derived from mutant PINK1 induced pluripotent stem cells. J Neurosci. 2011 Apr 20;31(16):5970–5976.
- Mazzulli JR, Zunke F, Isacson O, et al. alpha-Synuclein-induced lysosomal dysfunction occurs through disruptions in protein trafficking in human midbrain synucleinopathy models. Proc Natl Acad Sci U S A. 2016 Feb 16;113(7):1931–1936.
- Mazzulli JR, Xu YH, Sun Y, et al. Gaucher disease glucocerebrosidase and alpha-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell. 2011 Jul 08;146(1):37–52.
- Cooper O, Seo H, Andrabi S, et al. Pharmacological rescue of mitochondrial deficits in iPSC-derived neural cells from patients with familial Parkinson’s disease. Sci Transl Med. 2012 Jul 4;4(141):141ra90.
- Pitcairn C, Wani WY, Mazzulli JR. Dysregulation of the autophagic-lysosomal pathway in Gaucher and Parkinson’s disease. Neurobiol Dis. 2019 Feb;122:72–82.
- Kriks S, Shim JW, Piao J, et al. Dopamine neurons derived from human ES cells efficiently engraft in animal models of Parkinson’s disease. Nature. 2011 Nov 06;480(7378):547–551.
- Perrier AL, Tabar V, Barberi T, et al. Derivation of midbrain dopamine neurons from human embryonic stem cells. Proc Natl Acad Sci U S A. 2004 Aug 24;101(34):12543–12548.
- Zhang Y, Meredith GE, Mendoza-Elias N, et al. Aberrant restoration of spines and their synapses in L-DOPA-induced dyskinesia: involvement of corticostriatal but not thalamostriatal synapses. J Neurosci. 2013 Jul 10;33(28):11655–11667.
- Rademacher DJ, Mendoza-Elias N, Meredith GE. Effects of context-drug learning on synaptic connectivity in the basolateral nucleus of the amygdala in rats. Eur J Neurosci. 2015 Jan;41(2):205–215.
- Peters A, Palay SL, Webster H. Neurons and their supporting cells. New York: Oxford University Press; 1991. (The Fine Structure of the Nervous System:).
- Kung L, Force M, Chute DJ, et al. Immunocytochemical localization of tyrosine hydroxylase in the human striatum: a postmortem ultrastructural study. J Comp Neurol. 1998;390(1):52–62.
- Menon V, Thomas R, Ghale AR, et al. Flow cytometry protocols for surface and intracellular antigen analyses of neural cell types. J Vis Exp. 2014 Dec;18(94):52241.
- Ghee M, Melki R, Michot N, et al. PA700, the regulatory complex of the 26S proteasome, interferes with alpha-synuclein assembly. FEBS J. 2005 Aug;272(16):4023–4033.
- Rey NL, Bousset L, George S, et al. alpha-Synuclein conformational strains spread, seed and target neuronal cells differentially after injection into the olfactory bulb. Acta Neuropathol Commun. 2019 Dec 30;7(1):221.
- Kaja S, Payne AJ, Naumchuk Y, et al. Quantification of lactate dehydrogenase for cell viability testing using cell lines and primary cultured astrocytes. Curr Protoc Toxicol. 2017 May 2;72:2 26 1–2 26 10.
- Sanjana NE, Shalem O, Zhang F. Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods. 2014 Aug;11(8):783–784.
- Dhillon JS, Riffe C, Moore BD, et al. A novel panel of alpha-synuclein antibodies reveal distinctive staining profiles in synucleinopathies. PLoS One. 2017;12(9):e0184731.
- Schmid AW, Fauvet B, Moniatte M, et al. Alpha-synuclein post-translational modifications as potential biomarkers for Parkinson disease and other synucleinopathies. Mol Cell Proteomics. 2013 Dec;12(12):3543–3558.
- Lassen LB, Gregersen E, Isager AK, et al. ELISA method to detect alpha-synuclein oligomers in cell and animal models. PLoS One. 2018;13(4):e0196056.
- Lee HJ, Bae EJ, Jang A, et al. Enzyme-linked immunosorbent assays for alpha-synuclein with species and multimeric state specificities. J Neurosci Methods. 2011 Aug 15;199(2):249–257.
- Vaikath NN, Majbour NK, Paleologou KE, et al. Generation and characterization of novel conformation-specific monoclonal antibodies for alpha-synuclein pathology. Neurobiol Dis. 2015 Jul;79:81–99.
- Giuseppe Pugliese FP, Leto G, Amadio L, et al. The diabetic milieu modulates the advanced glycation end product–receptor complex in the mesangium by inducing or upregulating Galectin-3 expression. DIABETES. 2000;49:1249–1257.
- Liu FT, Hsu DK, Zuberi RI, et al. Modulation of functional properties of galectin-3 by monoclonal antibodies binding to the non-lectin domains. Biochemistry. 1996 May 14;35(19):6073–6079.
- Delacour D, Greb C, Koch A, et al. Apical sorting by galectin-3-dependent glycoprotein clustering. Traffic. 2007 Apr;8(4):379–388.
- Pieter Muntendam SW. inventor GALECTIN-3 Immunoassay. (United States patent US 2017/0370944A1). 2017.