
ABSTRACT
Mutations in the macroautophagy/autophagy gene EPG5 are responsible for Vici syndrome, a human genetic disease characterized by combined immunodeficiency. Previously, we found that epg5−/- mice exhibit hyperinflammation in the lungs mediated by IL1B/IL-1β and TNF/TNFα, resulting in resistance to influenza. Here, we find that disruption of Epg5 results in protection against multiple enteric viruses including norovirus and rotavirus. Gene expression analysis reveals IFNL/IFN-λ responsive genes as a key alteration. Further, mice lacking Epg5 exhibit substantial alterations of the intestinal microbiota. Surprisingly, germ-free mouse studies indicate Epg5-associated inflammation of both the intestine and lung is microbiota-independent. Genetic studies support IFNL signaling as the primary mediator of resistance to enteric viruses, but not of microbial dysbiosis, in epg5−/- mice. This study unveils an important role, unexpectedly independent of the microbiota, for autophagy gene Epg5 in host organism protection by modulating intestinal IFNL responses.
Abbreviations: CTNNB1: catenin (cadherin associated protein), beta 1; DAPI: 4′,6-diamidino-2-phenylindole; EPG5: ectopic P-granules autophagy protein 5 homolog (C. elegans); FT: fecal transplant; IFI44: interferon-induced protein 44; IFIT1: interferon-induced protein with tetratricopeptide repeats 1; IFNG/IFN-γ: interferon gamma; IFNL/IFN-λ: interferon lambda; IFNLR1: interferon lambda receptor 1; IL1B/IL-1β: interleukin 1 beta; ISG: interferon stimulated gene; GF: germ-free; LEfSe: linear discriminant analysis effect size; MAP1LC3/LC3: microtubule-associated protein 1 light chain 3; MNoV: murine norovirus; MX2: MX dynamin-like GTPase 2; OAS1A: 2’-5’ oligoadenylate synthetase 1A; RV: rotavirus; SPF: specific-pathogen free; SQSTM1/p62: sequestosome 1; STAT1: signal transducer and activator of transcription 1; STING1: stimulator of interferon response cGAMP interactor 1; TBK1: TANK-binding kinase 1; TNF/TNFα: tumor necrosis factor
Introduction
Vici syndrome is a rare multisystem disorder characterized by combined immunodeficiency, abnormalities in autophagy, corpus callosum agenesis, and other multisystemic features [Citation1]. Patients with Vici syndrome develop frequent and severe lung and gastrointestinal infections which, along with cardiomyopathy, are the main causes of death at a median age of 24 months [Citation2,Citation3]. Vici syndrome has been linked to causative mutations in human EPG5, which is essential for basal autophagy [Citation1].
Autophagy is an evolutionarily-conserved process that is essential for both the maintenance of homeostasis and environmental stress responses – it recycles cellular constituents and removes cytotoxic threats such as protein aggregates and damaged organelles [Citation4]. This process is executed through the formation of double-membrane autophagosomes and their delivery to lysosomes for degradation [Citation5]. EPG5 deficiency impairs autophagic flux by blocking the maturation of autophagosomes into degradative autolysosomes, leading to accumulation of SQSTM1/p62 aggregates and ubiquitin-positive inclusions in neurons and glial cells [Citation6–8]. These cellular defects, along with key features of Vici syndrome, are recapitulated in epg5−/- mice [Citation8]. Unlike disruptions in other autophagy-related genes, mutation of Epg5 may be non-lethal because EPG5 acts at such a late stage in autophagy or is redundant with other effectors [Citation7,Citation9].
Autophagy serves as a key cellular response to pathogen infection, and autophagy adaptors serve as their own category of pattern recognition receptors, helping to control intracellular microbes via direct capture, stimulation of inflammatory signaling, and delivery of antimicrobial peptides [Citation10–12]. Recently, appreciation for a role for autophagy in interactions with commensal microbial populations has also been growing [Citation13]. The disruption of autophagy genes has been linked to a range of different disorders related to immune system modulation in the gut, from Crohn disease [Citation14–16] to microbiota-related colorectal tumorigenesis [Citation17]. Multiple autophagy genes, including Atg5 and Atg7, have been shown to regulate the gut microbiota with effects on host immune function [Citation16,Citation17]. The effects of autophagy on the gut microbiota are thus imperative to explore since autophagy is potentially a key means by which the immune system and the microbiota interface.
We previously reported that mice deficient in Epg5 have increased baseline innate immune lung inflammation and are highly resistant to influenza infection, phenotypes also observed in mice lacking Atg5, Atg7, Atg14, and Rb1cc1/Fip200 in myeloid cells [Citation18–20]. Enhanced expression of IL1B and TNF in the lung, but not other tissues, is correlated with the influenza resistance of epg5−/- mice [Citation18]. The role of autophagy genes in suppressing innate inflammation and conferring susceptibility to various pathogens remains to be fully elucidated, but it has been linked to mediation of quiescence of tissue-resident macrophages by limiting the effects of systemic IFNG (interferon gamma) [Citation21,Citation22]. Spontaneous inflammation in Epg5-deficient mice was speculated to be due to the inadequate control of commensal or environmental microbes, because the administration of antibiotics ameliorates pulmonary inflammation in some autophagy mutants [Citation20]. While autophagy-deficient mice and cells have been reported to fail to appropriately sense and respond to immunomodulatory products from commensal bacteria, thereby leading to colitis [Citation23], the recent observation that autophagy proteins prevent spontaneous type I interferon responses driven by the microbiota in the intestine also supports a role for the commensal microbes in driving inflammation [Citation24]. Thus, much remains to be explored regarding the interactions between autophagy, innate immune responses, and the microbiota.
In this study, we used well-characterized enteric viruses murine norovirus and rotavirus to interrogate the effects of Epg5 in the intestine. We found that epg5−/- mice are resistant to enteric viruses, and we identified increased colonic ISG (interferon-stimulated gene) signatures associated with dysregulation of the epg5−/- intestinal microbiota and intestinal epithelial cell SQSTM1 accumulation. Intriguingly, we determined that the inflammatory intestinal phenotype, as well as the pulmonary inflammatory phenotype, in epg5−/- mice are fully independent of the commensal microbiota. Finally, the ISG signatures responsible for limiting viral infection in epg5−/- mice were dependent upon intact IFNL/IFN-λ signaling. Our study indicates that Epg5 limits the IFN-mediated antiviral state and maintains microbial homeostasis in the gut independent of detection of the microbiota itself.
Results
Epg5-deficient mice are resistant to multiple enteric viral infections
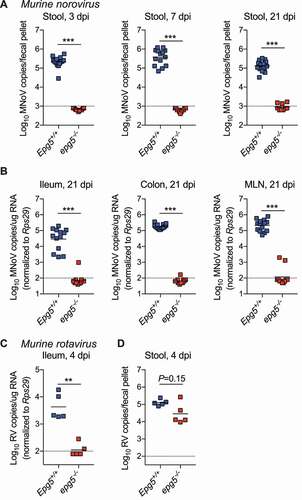
Based upon its previously-described role in regulating inflammation and viral infection in the lung [Citation18], we hypothesized that Epg5 deficiency may affect viral pathogenesis at another important mucosal site: the intestine. To explore this possibility, we infected epg5−/- mice or littermate controls with two enteric viruses, MNoV (murine norovirus) and murine RV (rotavirus). First, mice were infected with MNoVCR6, a persistent strain exhibiting intestinal tropism [Citation25,Citation26]. Strikingly, epg5−/- mice exhibited complete resistance to MNoVCR6 at acute (three and seven days post-infection (dpi)) and persistent (21 dpi) time points. No viral shedding from epg5−/- mice was detected at any time point (). Viral levels in the ileum, colon, and mesenteric lymph node were negligible in epg5−/- mice (). We also tested acute strain MNoVCW3, which can spread systemically in wild-type mice, and observed inhibited viral infection in epg5−/- mice in the spleen at three dpi (Figure S1A). In vitro, MNoVCW3 replicated equivalently in wild-type and epg5−/- bone marrow-derived macrophage cells, and was sensitive to IFNG in both, suggesting there is not a cell-intrinsic defect in MNoV replication in the absence of Epg5 (Figure S1B). Next, we challenged epg5−/- mice or littermate controls with the EC strain of murine rotavirus (RVEC), and examined viral replication and shedding at an acute time point (4 dpi). Similar to the MNoVCR6 results, epg5−/- mice exhibited a near-complete absence of viral replication in the ileum (), with an associated trend toward decreased viral shedding into the stool (). Thus, Epg5-deficiency provides resistance to multiple enteric viruses.
Figure 1. Epg5−/- mice are resistant to infection by persistent murine norovirus and murine rotavirus. (A,B) Epg5+/+ and epg5−/- littermates were orally inoculated with 106 PFU of MNoVCR6, and stool MNoV levels at 3, 7, and 21 dpi (A), and tissue viral levels at 21 dpi (B) were analyzed. N = 11–14 mice per group over two independent experiments. (C,D) Epg5+/+ and epg5−/- littermates were orally inoculated with 105 SD50 of RVEC, and ileum (C) and stool (D) viral levels were quantified at 4 dpi. Statistical analyses performed using Mann-Whitney test.
Inflammatory intestinal gene expression in Epg5-deficient mice
Because resistance to infection in the lung of epg5−/- mice is associated with autophagy defects, including accumulation of SQSTM1 and inflammation, we sought to explore these phenotypes in the intestine in the absence of infectious stimuli. No overt morphological or cellular changes were evident by histological analysis (), and flow cytometric analysis of intestinal intraepithelial lymphocytes, T regulatory cells, and colonic macrophages revealed only subtly decreased CD8+ intraepithelial lymphocytes and decreased ADGRE1/F4/80 staining in macrophages, potentially associated with increased activation [Citation27,Citation28] (Figure S1C-G). However, epg5−/- mice exhibited dramatic SQSTM1 accumulation in the epithelial layer of the colon (), suggesting impaired autophagy in the intestine as was previously observed in lung [Citation18]. We also detected LC3-II accumulation by western blot in ileum and colon samples from epg5−/- mice, consistent with defects in autophagosome turnover (Figure S2A). We confirmed there were no overt defects in tuft cell numbers, the cellular reservoir of persistent MNoV infection [Citation29] (Figure S2B,C), or robust SQSTM1 accumulation in tuft cells in epg5−/- mice (Figure S2C,D). Colonic epithelial cell proliferation was also similar between epg5−/- mice and control littermates (Figure S2E).
Figure 2. Epg5−/- mice exhibit SQSTM1 accumulation and inflammatory signatures in the colon. (A) Histological analysis of wild-type and epg5−/- colons, stained for epithelial cell marker CTNNB1 and autophagy substrate SQSTM1 with DAPI counterstaining. Representative photomicrographs are shown (N = 4 mice per group). (B-D) RNAseq analysis was performed using proximal colon tissue from Epg5+/+ and epg5−/- littermates at 9–12 weeks of age. (B) Heatmap of significantly upregulated or downregulated genes. Gene set enrichment analysis identified significantly enriched hallmark gene sets (C), with the IFNL pathway being the most enriched in epg5−/- colons (D). N = 6 mice per group.
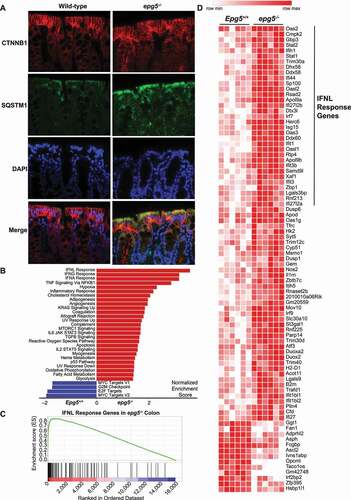
We next assessed the epg5−/- intestine for any gene expression changes by performing RNAseq on proximal colon sections from naïve epg5−/- mice and wild-type littermates (). While the overall number of significantly differentially-expressed genes in the epg5−/- colon was limited (85 genes upregulated; 13 downregulated) (), gene set enrichment analysis revealed the most robust expression signatures to be consistent with IFN and TNF inflammatory responses (). Of particular interest was the top signature, that of IFNL response, which is known to be potently antiviral in controlling both MNoV and RV [Citation30–34]. We validated this signature by analyzing ISGs by qPCR, and found canonical ISGs to be significantly upregulated in the epg5−/- intestine (Figure S3A). We also assessed expression of type I, II, and III IFNs themselves by qPCR, and detected elevated Ifna4 and Ifna11 expression as well as a trend toward elevated Ifnl2 and Ifnl3 expression in epg5−/- mice (Figure S3B). Ifnb and Ifng were equivalent between epg5−/- mice and wild-type littermates (Figure S3B). MNoV infection did not alter ISG expression in epg5−/- mice (Figure S3C). Associated with enhanced intestinal ISG expression, we detected increased phosphorylated STAT1 in the ileum and colon of epg5−/- mice, indicating enhanced activation of this transcription factor responsible for ISG expression in response to IFNs (Figure S3D). Levels of IFNL protein were similar in the serum and colon between epg5−/- mice and controls, but in ileum we observed a trend toward increased IFNL levels in epg5−/- mice (Figure S3E). Consistent with this observation, we found enhanced levels of phosphorylated protein kinase TBK1, a key upstream signaling molecule in the induction of type I and III IFNs [Citation35], in the ileum but not the colon of epg5−/- mice (Figure S3F). We tested whether the inflammatory ISG signature observed in the epg5−/- intestine was associated with differential sensitivity to colitis, but did not observe substantial differences in weight loss or survival after dextran sodium sulfate administration in epg5−/- mice (Figure S4A,B). In summary, Epg5-deficiency in mice leads to accumulation of autophagic flux indicator SQSTM1 in the villi and associated dysregulated IFN responses in the intestine.
The microbiota is altered in mice lacking Epg5
With the finding of an inflammatory transcriptional profile in the intestine of epg5−/- mice, we sought to determine if dysregulated cross-talk with the microbiota could be a factor. Canonical autophagy pathway genes Atg5 and Atg7 can regulate intestinal microbial populations [Citation16,Citation36], and we thus assessed whether late autophagy gene Epg5 was similarly involved in microbiota control. Cross-sectional analysis of the fecal bacterial microbiota of adult epg5−/- mice compared to their Epg5+/+ littermates revealed subtly increased absolute bacterial levels in epg5−/- mice, as well as significant overall community composition differences between these groups (), a finding consistent with prior studies in mice lacking autophagy genes in intestinal epithelial cells [Citation16,Citation36]. To determine whether development of microbiota differences required maintenance of distinct genotypes in a co-housed environment, we extended our initial analysis by performing a longitudinal comparison of Epg5+/+ and epg5−/- littermates who were initially co-housed en masse, then singly-housed for the duration of the experiment to prevent mouse-to-mouse microbial transfer. Over four weeks of being singly-housed, we again observed significant differences in overall community composition by adulthood (). We performed LEfSe analysis to identify putative biomarkers, and observed numerous taxonomic changes from the phylum to genus level in epg5−/- mice (). epg5−/- mice exhibited loss of protective microbe Akkermansia muciniphila (), a finding also reported in mice lacking Atg5 in intestinal epithelial cells [Citation16]. A significant decrease in Bacteroidetes and concomitant rise in Firmicutes was observed in epg5−/- mice by cross-sectional analysis (); a non-significant trend in the same direction was observed after singly-housing (). Thus, deficiency in Epg5 is associated with gradual development of distinct intestinal bacterial populations, independent of the specific method of housing. We also analyzed the bacterial microbiota of two other mouse lines with intestinal epithelial cell-specific defects in autophagy, atg7f/f-Vil-Cre and atg16l1f/f-Vil-Cre mice, and found that both exhibited significant differences in overall community composition from control littermates (Figure S4C,D). Of interest, these lines exhibited loss of Akkermansia muciniphila as well as Allobaculum stercoricanis, a microbe in the Erysipelotrichaceae family which is also decreased in epg5−/- mice (Figure S4E,F), suggesting that defects in autophagy are associated with consistent changes in microbiota composition.
Figure 3. fThe intestinal bacterial microbiota is altered in epg5−/- mice. (A,B) Epg5+/+ and epg5−/- littermates were separated by genotype at 4–5 weeks of age and maintained in cages of 2–5 mice/cage. Fecal pellets were collected at 8–10 weeks of age, and sequencing analysis of the 16S rRNA gene V4 region was performed. QPCR was performed on extracted DNA to determine 16S rRNA gene copies/fecal pellet, and richness and Faith’s phylogenetic diversity analyses were performed on V4 sequencing data (A). Principal components analysis was also performed using weighted UniFrac distances (B). N = 16–21 mice per group over two independent experiments. (C) Fecal samples were collected from Epg5+/+ and epg5−/- littermates around time of weaning (4 weeks of age), after mice were subsequently co-housed in hamster cages by sex for 4 weeks (8 weeks), and then after they were subsequently singly-housed for an additional 4 weeks (12 weeks). Principal components analysis was performed. N = 9 mice per group. (D) LEfSe analysis of 16S rRNA gene sequencing of Epg5+/+ and epg5−/- littermates (cohorts in B and C) identified statistically significant bacterial taxa biomarkers. (E, F) Relative abundances of Bacteroidetes and Firmicutes phyla from (B) and (C). Statistical analyses performed using Mann-Whitney test (A, E, F) or permutational MANOVA (ADONIS) (B, C). **, P < 0.01; *, P < 0.05; ns, not significant.
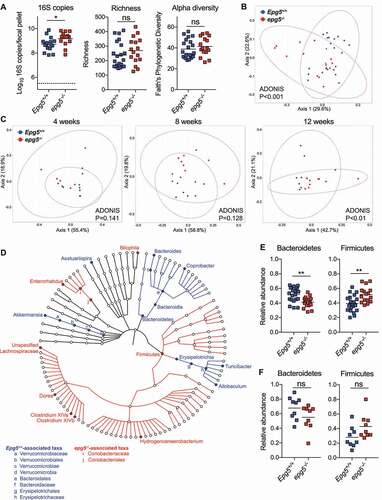
Interestingly, while performing our microbiota analysis, we observed that epg5−/- mice exhibited significantly smaller fecal pellets than wild-type littermates (Figure S4G). We also found significantly delayed intestinal transit time in epg5−/- mice via assessment of Evan’s blue dye transit (Figure S4H). Thus, in addition to enhanced intestinal inflammatory signatures, Epg5-deficiency is also associated with altered commensal bacterial populations and differential intestinal function.
The microbiota does not regulate the basal inflammatory state of the epg5−/- colon
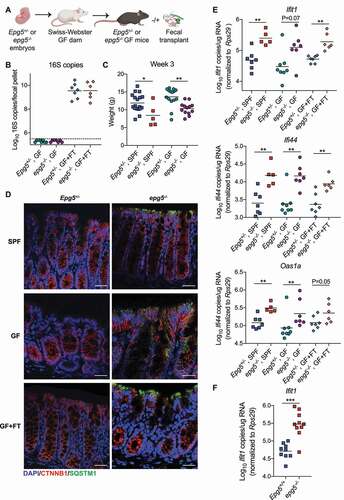
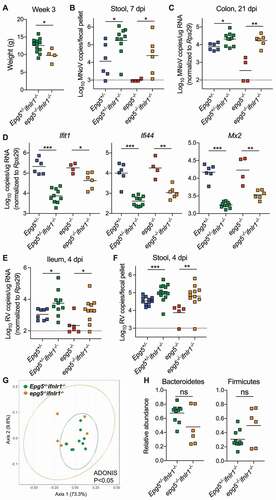
To further investigate the relationship between the microbiota and inflammatory phenotypes in epg5−/- mice, we generated germ-free (GF) Epg5+/- and epg5−/- mice by rederiving embryos into GF Swiss-Webster dams. GF mice were either maintained as GF until 9 weeks of age, or colonized at weaning at 3 weeks of age by fecal transplant (FT) from specific-pathogen free (SPF) Epg5 mice (). We confirmed that the mice were GF or had been successfully recolonized by analysis of 16S rRNA gene copies in stool at 9 weeks of age (). Growth retardation has previously been reported for epg5−/- mice [Citation7]; we measured weight of SPF and GF mice at 3 weeks of age prior to colonization, and found that epg5−/- were significantly smaller than Epg5+/- littermates regardless of the presence or absence of the microbiota (). We next evaluated whether the microbiota played any role in SQSTM1 accumulation in the colonic epithelium observed in SPF epg5−/- mice (), and found this accumulation was also present in GF and recolonized (GF+FT) epg5−/- mice (). We also found that GF and recolonized epg5−/- mice exhibited increased expression of colonic ISGs compared to Epg5+/- littermates (). Similarly, we found that ex vivo in the absence of microbial products, colonoids derived from epg5−/- but not Epg5+/- mice showed elevated levels of ISG Ifit1 (). Our results thus indicate that the microbiota is not responsible for the growth retardation or colonic IFN-associated inflammatory signature in epg5−/- mice.
Figure 4. The microbiota does not regulate the basal inflammatory state of the epg5−/- colon. (A) Epg5+/- and epg5−/- embryos were rederived into germ-free (GF) Swiss-Webster dams, then were either maintained as GF until 9 weeks of age, or colonized at weaning at 3 weeks of age by fecal transplant (FT) from specific-pathogen free (SPF) Epg5+/- mice. (B) 16S rRNA gene copies per fecal pellet from Epg5+/- and epg5−/- GF, or colonized (GF+FT) littermates as detected by qPCR of the V4 hypervariable region of the 16S rRNA gene. (C) Weights of age-matched SPF and GF Epg5+/- and epg5−/- mice were collected. N = 5–18 mice per group. (D) Histological analysis of Epg5+/- and epg5−/- SPF, GF, or GF+FT littermate colons, stained for epithelial cell marker CTNNB1 and autophagy substrate SQSTM1 with DAPI counterstaining. Representative photomicrographs are shown. Scale bars are 50 μm. (E) QPCR was used to analyze expression of canonical ISGs Ifit1, Ifi44, and Oas1a in colon from Epg5+/- and epg5−/- SPF, GF, or GF+FT littermates at 9 weeks of age. N = 5–8 mice per group. (F) QPCR was used to analyze expression of Ifit1 in colonoids derived from Epg5+/+ and epg5−/- SPF colons. N = 9–10 colonoids per group pooled from two independent experiments. Statistical analyses performed using Mann-Whitney test.
The microbiota does not regulate the inflammatory state of the epg5−/- lung
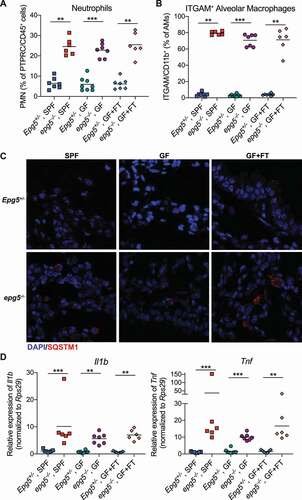
It has been previously reported that epg5−/- mice are resistant to influenza infection due to elevated basal lung inflammation [Citation18]. We thus assessed the role of the microbiota in inflammatory lung phenotypes previously observed in epg5−/- mice. Consistent with findings in SPF epg5−/- mice, GF and recolonized epg5−/- mice exhibited histologic lung abnormalities including the appearance of lymphoid aggregates, large macrophage-like cells, and infiltration by polymorphonuclear leukocytes (Figure S5A). Flow cytometric analysis confirmed inflammatory phenotypes previously observed in epg5−/- lungs, including increased neutrophils and upregulation of ITGAM/CD11b on alveolar macrophages, regardless of the presence or absence of the microbiota (, Figure S5B) [Citation18]. Similarly, the microbiota did not regulate the dysregulated SQSTM1 accumulation observed in epg5−/- lungs by histological analysis (). Finally, expression of cytokines Il1b and Tnf was significantly upregulated in GF and recolonized epg5−/- lungs, but not colons, compared to Epg5+/- littermates, consistent with our prior report in SPF epg5−/- mice (, Figure S6A) [Citation18]. Cumulatively, these results indicate that the microbiota is dispensable for the dysregulated autophagy, immune cell infiltration, and inflammatory cytokine milieu of the lungs in mice lacking Epg5.
Figure 5. The microbiota does not control inflammatory lung phenotypes in epg5−/- mice. (A,B) Flow cytometric analysis was performed on lung tissue from Epg5+/- and epg5−/- SPF, GF, or GF+FT littermates, and the percentage of neutrophils (PTPRC/CD45+ Ly6G+ ITGAM/CD11b+) (A) as well as the ITGAM/CD11b-positivity of alveolar macrophages (pre-gated on PTPRC/CD45+ SIGLECF+ ITGAX/CD11c+) (B) was assessed. (C) Histological analysis of Epg5+/- and epg5−/- SPF, GF, or GF+FT littermate lungs, stained for SQSTM1 with DAPI counterstaining. Representative photomicrographs are shown. (D) QPCR was used to analyze expression of Il1b and Tnf in lungs from these mice. N = 6–8 mice per group over two independent experiments. Statistical analyses performed using Mann-Whitney test.
The colonic antiviral inflammatory signature in epg5−/- mice is driven by IFNL signaling
Upon finding that the microbiota played a negligible role in the basal inflammatory state in epg5−/- mice, we furthered our investigation into the specific signaling molecules that may be driving these intestinal antiviral effects by interrogating the role of IFNL signaling. Expression of Ifnlr1 in the colons of SPF and GF was equivalent between epg5−/- and Epg5+/- littermates (Figure S6A). We generated epg5−/- ifnlr1−/- and Epg5+/- ifnlr1−/- mice, which lack IFNL signaling. While disruption of IFNL signaling did not affect the growth retardation observed in epg5−/- mice (), we found that MNoV susceptibility was restored in epg5−/- ifnlr1−/- mice compared to epg5−/- mice, as measured by stool viral shedding at 7 dpi and colon MNoV levels at 21 dpi (). We also observed significantly decreased expression of canonical ISGs Ifit1, Ifi44, and Mx2 in post-infection colons of epg5−/-ifnlr1−/- mice compared to epg5−/- mice (). Disruption of IFNL signaling in epg5−/- mice also restored susceptibility to RVEC (, but had no effect on the accumulation of SQSTM1 in intestinal epithelial cells (Figure S6B). We also assessed whether, in the absence of IFNL signaling, the dysbiosis observed in epg5−/- mice would still be present, by analyzing 16S rRNA gene sequencing from pre-infection stools of epg5−/-ifnlr1−/- and Epg5+/- ifnlr1−/- mice. We found that, similar to the epg5−/- mouse microbiota compared to that of littermate controls (), the epg5−/- ifnlr1−/- microbiota was significantly different from that of Epg5+/-ifnlr1−/- mice (). Though not statistically significant, relative abundances of Bacteroidetes and Firmicutes in epg5−/-ifnlr1−/- samples also followed similar trends as observed in epg5−/- samples (). Disruption of IFNL signaling similarly did not affect the histologic lung abnormalities observed in epg5−/- mice (Figure S6C). We also assessed previously-harvested lungs from epg5−/- sqstm1−/- mice and a single mouse deficient for both Epg5 and Sting1 in parallel, and observed that neither autophagic cargo protein SQSTM1 nor cytosolic DNA sensor STING1 appear to contribute to histologic lung abnormalities in epg5−/- mice (Figure S6C). In sum, these data support that IFNL signaling is the major driver of dysregulated ISG expression and antiviral signaling, but not of microbial dysbiosis, in the intestine.
Figure 6. Antiviral signaling, but not dysbiosis, in epg5−/- mice is driven by IFNL signaling.
Discussion
Autophagy is an essential cellular pathway for processing intracellular pathogens and cellular materials during periods of stress and plays critical roles at barrier sites in the body such as the intestinal and pulmonary mucosa. Here, we discovered a novel interaction between autophagy gene Epg5 and the intestinal antiviral response, which is predominantly mediated by elevated IFNL signaling. Epg5 deficiency resulted in complete resistance to multiple enteric viruses including norovirus and rotavirus in mice, and genetic studies leveraging epg5−/-ifnlr1−/- mice revealed that these intestinal hyperinflammation and antiviral resistance phenotypes are dependent on the IFNL-ISG pathway. Loss of Epg5 is also associated with dysbiosis of commensal microbiota in the intestine. However, strikingly, GF mouse studies indicated that the hyperinflammation in the intestine and lung occurs without contribution from the dysbiotic microbiota.
This study provides an unexpected observation which uncouples the traditional “stimulus and response” link between the microbiota and inflammation. In recent years, several studies have shown that sensing of microbial metabolites or the microbiota itself is a primary driver of inflammatory responses in the intestine and other tissues [Citation17,Citation20,Citation24]. Susceptible animals such as autophagy-deficient mice are thought to be hypersensitive to these microbial stimuli, resulting in elevated cytokine production and immune cell infiltration in the tissue. As shown, Epg5-deficient mice exhibited hyperinflammation at two mucosal sites, lung and intestine, as well as dysbiosis of the bacterial microbiota. Gene expression analysis in the intestine revealed a strong signature of ISG expression, which is predominantly IFNL-mediated. Elevated IFNL appeared to be derived predominantly from the ileum, while STAT1 activation and antiviral ISG activity were present throughout the intestine. epg5−/- ifnlr1−/- mice lost this increased expression of ISGs, but still maintained dysbiosis of the microbiota. Conversely, GF epg5−/- mice developed hyperinflammation at comparable levels to conventional epg5−/- in both the lung and intestine, which is independent of reconstitution of the commensal microbiota. Although inflammatory signatures in the lung and intestine exhibit different features (e.g., increased TNF and IL1B in the lung, and increased IFNL and ISGs in the intestine), none of these phenotypes required any stimulus from microbiota, indicating that hyperinflammation and microbial dysbiosis are completely independent in these animals. We also found that neither STING1, which is involved in induction of both inflammation and autophagy [Citation37,Citation38], nor SQSTM1, which may also contribute to regulation of inflammatory pathways [Citation39,Citation40], play any role in epg5−/–associated inflammatory lung phenotypes. Thus, our study in the epg5−/- mouse model broadens the conventional view of immune responses and their regulation by autophagy genes.
It has been shown in multiple studies that deficiency of autophagy genes is associated with hyperinflammation in multiple organs (e.g., lung, intestine and peritoneum) via elevation of different proinflammatory cytokines including type I IFNs, IFNG, TNF and IL1B [Citation14,Citation15,Citation18,Citation20–22,Citation24]. In a recent study, defects in autophagy genes in intestinal epithelial cells are reported to induce type I IFN responses, driven by the commensal microbiota [Citation24]. However, immune regulation of type III IFN signaling by the autophagy pathway has not previously been studied. Data in this study revealed a novel role for autophagy gene Epg5 in regulating intestinal antiviral responses via IFNL, which is a specialized antiviral cytokine at barrier sites and a previously unrecognized element to be controlled by an autophagy gene. As lung inflammation and intestinal IFNL responses are microbiota-independent, it is an open question as to what stimuli trigger hyperinflammation. Epg5-deficient intestines in GF and conventional conditions exhibited accumulated autophagic flux marker SQSTM1, particularly at the tips of epithelial villi, suggesting enterocytes are under autophagic stress and may be responsible for increased IFNL production. However, our data indicates that IFNL signaling itself does not contribute to SQSTM1 accumulation or to inflammatory lung phenotypes. Given the rapid turnover of the epithelial cell layer, dead cells, released mitochondrial DNA, or damaged DNA could serve as drivers of inflammation. Further studies will be needed to elucidate the upstream stimuli, sensing and regulation pathways to better understand the autophagy-mediated regulation of IFNL responses.
Although hyperinflammation is not directly driven by dysbiosis of the commensal microbiota in epg5−/- mice, key features of the microbiota in epg5−/- mice reveal important insights. Microbiota changes, including increased bacterial load, loss of Akkermansia, and increased abundance of genera in the Clostridiaceae family in epg5−/- mice () are consistent with prior reports of dysbiosis in mice lacking Atg5 or Atg7 in intestinal epithelial cells [Citation16,Citation36]. Our study thus supports the broad observation that dysregulated autophagy in the intestine contributes to dysregulation of microbial communities [Citation41].
Materials and methods
Ethics statement
All mice were used under regulations stipulated by Washington University Institutional Animal Care and Use Committee and on protocols approved by the Washington University Animal Studies Committee.
Mouse lines
C57BL/6 J mice were originally purchased from Jackson Laboratories (Jackson Laboratories, 000664) and bred and housed in Washington University in Saint Louis animal facilities under specific pathogen free, including MNoV-free, conditions. Crosses of Epg5+/− mice [Citation7] were used to generate experimental animals and littermates were used for all experiments. ifnlr1−/- mice, generated from a cross between Ifnlr1f/f (originally from Ifnlr1tm1a(EUCOMM)Wtsi ES cells) and a Deleter-Cre line as previously described [Citation30], were bred to Epg5+/− mice. sqstm1−/- mice [Citation42] and sting1−/- mice [Citation43] were also bred to Epg5+/− mice. Only a single epg5−/-sting1−/- mouse was obtained from six litters (28 mice) born from Epg5+/−sting1−/- intercrosses. Atg7f/f [Citation44] and atg7f/f-Vil-Cre [Citation14], as well as Atg16l1f/f [Citation11] and atg16l1f/f-Vil-Cre [Citation45], mice were generated by crossing Cre-positive and Cre-negative mice.
For rederivation of GF Epg5+/- and epg5−/- mice, sperm was harvested from epg5−/- males and eggs from Epg5+/- females for in vitro fertilization. The resulting embryos were then transferred into GF pseudopregnant Swiss Webster recipient females, and Epg5+/- and epg5−/- mice were born at approximately equal ratios. GF and recolonized mice were housed in the Washington University Gnotobiotic Core Facility until 9 weeks of age. Confirmation of a GF environment was performed by bi-weekly wet mount observation, anaerobic and aerobic culture, fungal culture, and monthly 16S rRNA gene PCR (Charles River) and shotgun sequencing (Transnetyx) of fecal samples.
Generation of viral stocks
Stocks of MNoV strains CR6 and CW3 were generated from molecular clones as previously described [Citation25]. Briefly, plasmid encoding the viral genomes was transfected into 293 T cells to generate infectious virus, which was subsequently passaged on BV2 (RRID:CVCL_0182) cells (obtained from the Virgin Lab at Washington University School of Medicine). After two passages, BV2 cultures were frozen and thawed to liberate virions. Cultures then were cleared of cellular debris and virus was concentrated by ultracentrifugation through a 30% sucrose cushion. Titers of virus stocks were determined by plaque assay on BV2 cells [Citation46].
For production of murine RV stocks, four-day-old suckling CD-1 mice (Charles River, Crl:CD1[ICR]) were infected orally with 400 diarrheal dose 50 (5 uL of a 1:10 dilution in PBS [Gibco, 14190–144] + Evan’s blue) from a stock of murine RV strain EC-RV received from the Estes Lab at Baylor College of Medicine. Three days post-infection, mice were sacrificed and the entire small intestine and colon with contents were harvested and pooled for weighing. A 20% homogenate was prepared in homogenization media of DMEM (Gibco, 11995–040) supplemented with 1% penicillin-streptomycin (Gibco, 15140–122) by three rounds of homogenization using a Mini-Beadbeater-24 (BioSpec Products, 112011) on ice (homogenize for 1 min, then allow to settle for 30 s). Homogenate was pooled on ice, aliquoted, and then stored at −80°C until use. The shedding dose 50 (SD50) of the stock was determined by orally infecting groups of five adult mice (6–8 weeks old), of both BALB/c and C57BL/6 genotypes, with 100 ul of 10-fold serial dilutions, ranging from 10−5 to 10−9 for BALB/c and 100 to 10−5 for C57BL/6, and analyzing fecal pellets for RV genome copies by qPCR.
Mouse treatments and infections
For intestinal transit time assays, mice were gavaged orally with 500 uL of 8% Evans blue dye (MP Biomedicals, 314–13-6). Fecal pellets were collected at 2, 4, 6, 9, 24, and 48 h, weighed and resuspended at 100 mg/mL in PBS. “Evan’s Blue Score” was determined by assessing the level of blue color in the feces: slightly blue = 1, light blue = 2, moderate blue = 3, dark blue = 4, intense blue = 5, as previously described [Citation47,Citation48].
For MNoV infections, mice were inoculated with virus at 6–8 weeks of age by the oral route in a volume of 25 μl; a dose of 106 plaque-forming units (pfu) of the indicated virus was used for all experiments. For RV infections, 6–8 week-old mice received 105 50% shedding doses (SD50) of RV in 100 μl preceded by 100 μl 1.33% (w:v) sodium bicarbonate (Fisher Scientific, BP328-500) by oral gavage.
Cells, infections, and IFN treatment
Primary bone marrow-derived macrophages were prepared as described previously [Citation11]. Briefly, bone marrow was incubated in culture medium (DMEM supplemented with 10% FBS, 5% defined equine serum [Invitrogen, 16050–114], 10% CMG14-12 supernatant [Citation49], 1% MEM nonessential amino acids [Gibco, 11140050], 1 mM sodium pyruvate, 2 mM l-glutamine) for 7 days, after which adherent cells were harvested and replated on fresh culture medium. For in vitro infections, cells were plated at a density of 1 × 105 cells/well in 24-well dishes. Infections were performed at day 11 of in vitro culture after 24 h of treatment with 100 U of recombinant IFNG (R&D Systems, 485-MI-100/CF). After 30 min of absorption at 4°C, cells were washed and IFNG added back into treated cultures. Cells were harvested by freezing at −80°C.
Colonoids were generated from crypts isolated from colons. Briefly, adult mice were euthanized to collect colon tissue, which was then cut into 2- to 4-mm pieces and washed with ice cold PBS. The tissue fragments were treated with EDTA solution to isolate intestinal crypts. The crypts were resuspended in Matrigel (Corning, 15585729) and seeded into culture plates. The colonoids were grown in basal culture media (Advanced DMEM/F12 [Gibco, 12634–010] supplemented with 2 mM GlutaMax [Invitrogen, 35050–061], 10 mM HEPES, pH 7.5). For differentiation, the colonoids were cultured in differentiation media consisting of basal culture media with (1X) N2 supplement (Invitrogen, 17502048), (1X) B27 supplement (Invitrogen, 17504044), and 1 mM N-acetylcysteine (Sigma Aldrich, A9165), 50 ng/mL murine recombinant EGF (Invitrogen, PMG8041), 100 ng/mL murine recombinant NOG (noggin; PeproTech, AF-250-38), and 1 mg/mL human recombinant RSPO1 (R-spondin 1; R&D Systems, 4645-RS-025/CF).
Lung and colon histological analysis
Lungs were inflated and colons were flushed with PBS, then both tissues were fixed with 10% formalin, embedded in paraffin, cut into 5-μm sections, and adhered to charged slides. Slides were stained with H&E or left unstained. For IHC, lung sections were deparaffinized in CitriSolv (Fisher, 1601) and rehydrated in graded ethanol. Heat-mediated antigen retrival was performed with citrate-based Antigen Unmasking Solution (Vector labs, H3300). Sections were blocked and incubated with primary antibodies overnight at 4°C, and detected by Alexa Fluor 488- (Jackson ImmunoResearch Laboratories, 111–545-003) and 555- (Invitrogen, A21435) conjugated secondary antibodies. Slides were mounted using DAPI-containing mounting media (Vector labs, H-1000). Images were obtained with immunofluorescence microscopy (BX-51; Olympus) and analyzed with SPOT Advanced software (Diagnostic Instruments). Antibodies used: SQSTM1/p62 (Progen, GP-P62 C), CTNNB1 (Sigma, HPA029160), MKI67/Ki67 (eBioscience, SolA15), DCLK1 (Abcam, ab31704).
Analysis of intestinal tissues by western blotting and ELISA
Tissues were processed by mechanical homogenization in RIPA buffer (Beyotime, P0013K) containing PMSF (protease inhibitor; Beyotime, ST506). Pierce™ BCA Protein Assay Kit (Thermo, 23227) was utilized to quantify protein amounts. Following SDS-PAGE to resolve samples, transfer to PVDF membrane (GE, 10600023) was performed. Membranes were then subjected to a 60 min incubation at room temperature with blocking buffer containing PBS or TBST (TBS [Servicebio, G0015] + 0.01% Tween-20 [Sangon Biotech, A600560]) and 5% nonfat dry milk, followed by incubation overnight at 4°C with either a 1:1000 dilution of anti-LC3 (Cell Signaling Technology, 83506), anti-TBK1/NAK (Cell Signaling Technology, 3504), anti-phospho-TBK1/NAK (Ser172; Cell Signaling Technology, 5483), a 1:750 dilution of anti-phospho-STAT1 (Tyr701; Invitrogen, 7649 T), or a 1:5000 dilution of anti-GAPDH (Bio-Rad, MCA4739P) or anti-ACTB/β-actin (HuaBio, EM21002). This was followed by PBST (PBS and 0.05% Tween-20) and treatment with 1:5000 horseradish peroxidase: goat anti-mouse IgG (Cell Signaling Technology, 7076) at room temperature for 60 min. The Sensitive Enhanced Chemiluminescence D kit (Proteintech, B2202103) or ECL Western Blotting Substrate (Pierce, 32106) was utilized to detect specific reactive proteins on the membrane.
For IFNL ELISA, serum or 30–50 mg of ileum or colon homogenate were collected and IFNL2 and IFNL3 levels were analyzed by ELISA as described previously (R&D Systems, DY1789B05) [Citation50].
RNA extraction and quantitative reverse transcription-PCR
As previously described [Citation30], RNA was isolated from stool using a ZR-96 Viral RNA kit (Zymo Research, D7023). RNA from tissues or cells was isolated using TRI Reagent with a Direct-zol-96 RNA kit (Zymo Research, R2056) according to the manufacturer’s protocol. Five μl of RNA from stool or tissue was used for cDNA synthesis with the ImProm-II reverse transcriptase system (Promega, A3800). MNoV TaqMan assays were performed, using a standard curve for determination of absolute viral genome copies, as described previously [Citation51]. RV genome copies were detected using custom NSP3-specific primer pairs (5ʹ-CTTCTGGACTACGACAGACTATTT-3ʹ, 5ʹ-GATGTTTGATCGGTTCGTTGTG-3ʹ) with a (5ʹ-AGCGCTGCAACTTAGACTACGCAT-3ʹ) 5′ 6-carboxyfluorescein (FAM) dye label, 3′ nonfluorescent quencher (NFQ), and minor groove binder (MGB) from Integrated DNA Technologies. PrimeTime qPCR assays for Ifna4 (Mm.PT.58.7678281.g), Ifnb (Mm.PT.58.30132453.g), Ifng (Mm.PT.58.30096391), Ifit1 (Mm.PT.58.32674307), Ifi44 (Mm.PT.58.12162024), and Oas1a (Mm.PT.58.30459792) all from Integrated DNA Technologies, and a Taqman assay for Ifnl2 and Ifnl3 (Mm04204156_gH) from Thermo Fisher Scientific were performed using the same protocol. Quantitative PCR for housekeeping gene Rps29 was also performed as described and used to normalize absolute values of other analytes [Citation30].
RNA-Seq
For RNA-Seq, mRNA was extracted from colon tissue lysate by means of oligo-dT beads (Invitrogen, 61002). For cDNA synthesis, we used custom oligo-dT primer with a barcode and adaptor-linker sequence (CCTACACGACGCTCTTCCGATCT-XXXXXXXX-T15). After first-strand synthesis, samples were pooled together based on Actb qPCR values and RNA-DNA hybrid was degraded with consecutive acid-alkali treatment. Then, a second sequencing linker (AGATCGGAAGAGCACACGTCTG) was ligated with T4 ligase (New England BioLabs, M0202) followed by solid phase reversible immobilization/SPRI clean-up. The mixture then was PCR enriched 12 cycles and solid phase reversible immobilization purified to yield final strand-specific RNA-seq libraries. Libraries were sequenced using a HiSeq 2500 (Illumina) using 50 bp X 25 bp pair-end sequencing. Second mate was used for sample demultiplexing. Reads were aligned to the GRCm38.p2 assembly of the mouse genome using STAR aligner. Additional options were “- outFilterMultimapNmax 15 – outFilterMismatchNmax 6-quantMode TranscriptomeSAM”. Aligned reads were quantified using quant3p script (github.com/ctlab/quant3p) to account for specifics of 3ʹ sequencing: higher dependency on good 3ʹ annotation and lower level of sequence specificity close to 3ʹ end. RefSeq genome annotation was used. Using MACS2 peak caller, putative exons were identified (peaks in rna-seq alignment not overlapping with known exons), then assigned to a gene located within 5000 bp and added into the annotation. For on average 1 million reads per sample that mapped to multiple genome positions there were only one exonic alignment. For these reads multimapper flags were removed for read counting purposes. Actual read counting was carried out by ht-seq with enriched genome annotation and alignment with fixed multimapper flags. DESeq2 was used for differential gene expression analysis which was used as ranked list in preranked GSEA analysis to identify pathway enrichments [Citation52]. An IFNL-specific gene set was generated from the top differentially expressed genes after treatment with recombinant IFNL [Citation53]. RNA-Seq data have been uploaded to the European Nucleotide Archive (accession no. PRJEB42341).
16S rDNA qPCR and Illumina sequencing and analysis
Phenol:chloroform-extracted DNA from fecal pellets was used for both 16S rRNA gene qPCR and sequencing. SYBR green qPCR for 16S was performed with 515 F (5ʹ-GTGCCAGCMGCCGCGGTAA-3ʹ) and 805 R (5ʹ-GACTACCAGGGTATCTAATCC-3ʹ) primers to detect the V4 hypervariable region of the 16S rRNA gene.
For 16S sequencing, primer selection and PCRs were performed as described previously [Citation54]. Briefly, each sample was amplified in triplicate with Golay-barcoded primers specific for the V4 region (F515/R806), combined, and confirmed by gel electrophoresis. PCR reactions contained 18.8 μL RNase/DNase-free water, 2.5 μL 10X High Fidelity PCR Buffer (Invitrogen, 11304–102), 0.5 μL 10 mM dNTPs, 1 μL 50 mM MgSO4, 0.5 μL each of the forward and reverse primers (10 μM final concentration), 0.1 μL Platinum High Fidelity Taq (Invitrogen, 11304–102) and 1.0 μL genomic DNA. Reactions were held at 94°C for 2 min to denature the DNA, with amplification proceeding for 26 cycles at 94°C for 15 s, 50°C for 30 s, and 68°C for 30 s; a final extension of 2 min at 68°C was added to ensure complete amplification. Amplicons were pooled and purified with 0.6x Agencourt AMPure XP beads (Beckman-Coulter, A63882) according to the manufacturer’s instructions. The final pooled samples, along with aliquots of the three sequencing primers, were sent to the DNA Sequencing Innovation Lab (Washington University School of Medicine) for sequencing by the 2 X 250bp protocol with the Illumina MiSeq platform.
Read quality control and the resolution of amplicon sequence variants were performed with the dada2 R package [Citation55]. Amplicon sequence variants that were not assigned to the kingdom Bacteria were filtered out. The remaining reads were assigned taxonomy using the Ribosomal Database Project (RDP trainset 16/release 11.5) 16S rRNA gene sequence database [Citation56]. Ecological analyses, such as alpha-diversity (richness, Faith’s phylogenetic diversity) and beta-diversity analyses (weighted UniFrac distances), were performed using PhyloSeq and additional R packages [Citation57], and differentially abundant taxa between sample groups were identified by performing pairwise comparisons using LEfSe [Citation58]. 16S sequencing data have been uploaded to the European Nucleotide Archive (accession no. PRJEB42341).
Flow cytometric analysis of lung and intestinal cells
Single-cell suspensions were made from minced lung tissue and subjected to collagenase (Liberase Thermolysin Medium; Sigma-Aldrich, 5401119001), hyaluronidase (Sigma-Aldrich, H4272), and DNase I (grade II; Sigma-Aldrich, 10104159001) digestion, then treated with ACK buffer to remove red blood cells. Cells were suspended in PBS with 0.1% sodium azide and 1% BSA (Fisher Scientific, BP9706100), blocked with anti-FCGR2B/FcγRII-FCGR3/FcγRIII (BioLegend, 101320), and labeled with specific antibodies. Gating strategies were the same as previously reported [Citation18]. Briefly, antibodies used included: PTPRC/CD45 (APC-Fire750; BioLegend, 147714), SIGLECF (BV605; BD Pharmingen, 747673), FCGR1/CD64 (APC; BioLegend, 139306), LY6G (PerCP; BioLegend, 127654), ITGAX/CD11c (FITC; BioLegend, 117306), and ITGAM/CD11b (BV421; BioLegend, 101236). We used flow cytometry to identify specific cell populations in mouse lungs using the following gates: alveolar macrophages as PTPRC/CD45+ FCGR1/CD64+ SIGLECF+ ITGAX/CD11c+ (in Epg5+/- mice, ITGAM/CD11b expression was low; in epg5−/- mice, expression of ITGAM/CD11b was predominantly high), and neutrophils as PTPRC/CD45+ LY6G+ ITGAM/CD11b+. Flow cytometric analysis was performed on an LSRFortessa (BD Biosciences).
Intraepithelial or lamina propria lymphocytes were isolated from the small and large intestines as described previously [Citation59]. Briefly, small or large intestines were collected and opened longitudinally washed with cold PBS to remove fecal content. The intestines were shaken in HBSS containing 5 mM EDTA and 0.5% FBS for 20 min at 37°C. Detached epithelial cells were filtered with a 70um cell strainer and saved for Percoll (Sigma-Aldrich, P1644-500ML) gradient separation. After removing the epithelial cells, the remaining portions of the intestines were cut into small pieces and incubated with RPMI 1640 (Gibco, 11875093) containing 4% FBS, 1 mg/ml collagenase D (Roche Diagnostics, 11088858001), 0.5 mg/ml dispase (Roche Diagnostics, 10269638001) and 40 µg/ml DNase I (Roche Diagnostics, 10104159001) for 1 h at 37°C in a shaking water bath. The digested tissues were washed and filtered with a 70-um cell strainer. The detached epithelial and digested lamina propria cells were resuspended in 5 ml of 40% Percoll, overlaid on 2.5 ml of 80% Percoll in a 15-ml Falcon tube. Percoll gradient separation was performed by centrifugation at 780 g for 20 min at 25°C. The interface cells were collected, washed with cold PBS and then used immediately for experiments as intraepithelial or lamina propria lymphocytes.
After dead cell labeling with Zombie Aqua™ Fixable Viability Kit (BioLegend, 423102), the cells were stained with PTPRC/CD45 (APC-CY7, BV421, and BV711; BioLegend, 103116, 103134, 103147), CD3 (BUV 395; BD Biosciences, 563565), CD4 (PE; BioLegend, 100408), KLRB1C/NK-1.1 (APC-CY7; BioLegend, 108724), PTPRC/B220 (AF700; BioLegend, 103231), CD8A (FITC; BioLegend, 100706), CD8B1 (PE-Cy7; BioLegend, 126616), TCRB/TCRβ (BV421; BioLegend, 109230), TCRG/TCRγ-TCRD/TCRδ (APC; BioLegend, 118116), CX3CR1 (FITC; BioLegend, 149020), LY6C (APC-CY7; BioLegend, 128025), ADGRE1/F4/80 (BV785; BioLegend, 123141), ITGAX/CD11c (PE-CY7; BioLegend, 117317), ITGAM/CD11b (PerCP-CY5.5; BioLegend, 101229), MHC-II (AF700; BioLegend, 107621), FCGR1/CD64 (PE; BioLegend, 139303), and IL2RA/CD25 (APC; BioLegend, 101910). The gating strategy to distinguish macrophages from dendritic cells in the gut was as previously described [Citation59]. For the analysis of regulatory T cells (Treg) from colon, isolated lymphocytes were permeabilized and stained with FOXP3 (FITC; eBioscience, 11–5773-82). Flow cytometric data were collected on a BD X-20 (BD Biosciences). Both lung and intestinal flow cytometric data were analyzed with FlowJo software (Tree Star Inc.).
Statistical analysis
Data were analyzed with Prism 7 software (GraphPad Software). In all graphs, three asterisks indicate a P value of <0.001, two asterisks indicate a P value of <0.01, one asterisk indicates a P value of <0.05, and ns indicates not significant (P > 0.05) as determined by Mann-Whitney test, one-way analysis of variance (ANOVA) or Kruskal-Wallis test, or two-way ANOVA with Tukey’s multiple-comparison test, as specified in the relevant figure legends. Differences in beta-diversity were determined using Permutational Multivariate Analysis of Variance (ADONIS).
Data and materials availability
The data from this study are tabulated in the main paper and supplementary materials. All reagents are available from M.T.B. under a material transfer agreement with Washington University. Sequencing data have been uploaded to the European Nucleotide Archive (accession no. PRJEB42341).
Supplemental Material
Download MS Word (7.6 MB)Acknowledgments
We acknowledge the Washington University Gnotobiotic Core Facility for assistance with germ-free mouse generation and recolonization services, as well as the Washington University Transgenic, Knockout and Micro-Injection Core for assistance with generation of mouse embryos. We also acknowledge the Pulmonary Morphology Core for assistance with lung histology and the Washington University Center for Cellular Imaging for assistance with microscopy.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
References
- Cullup T, Kho AL, Dionisi-Vici C, et al. Recessive mutations in EPG5 cause Vici syndrome, a multisystem disorder with defective autophagy. Nat Genet. 2013;45(1):83–87.
- Byrne S, Jansen L, U-King-Im JM, et al. EPG5-related Vici syndrome: a paradigm of neurodevelopmental disorders with defective autophagy. Brain. 2016;139(3):765–781.
- Alzahrani A, Alghamdi AA, Waggass R. A Saudi infant with Vici syndrome: case report and literature review. Open Access Maced J Med Sci. 2018;6(6):1081–1084.
- Murrow L, Debnath J. Autophagy as a stress-response and quality-control mechanism: implications for cell injury and human disease. Annu Rev Pathol. 2013;8:105–137.
- Cadwell K. Crosstalk between autophagy and inflammatory signalling pathways: balancing defence and homeostasis. Nat Rev Immunol. 2016;16:661–675.
- Wang Z, Miao G, Xue X, et al. The Vici syndrome protein EPG5 is a Rab7 effector that determines the fusion specificity of autophagosomes with late endosomes/lysosomes. Mol Cell. 2016;63:781–795.
- Zhao H, Zhao YG, Wang X, et al. Mice deficient in Epg5 exhibit selective neuronal vulnerability to degeneration. J Cell Biol. 2013;200:731–741.
- Zhao YG, Zhao H, Sun H, et al. Role of Epg5 in selective neurodegeneration and Vici syndrome. Autophagy. 2013;9:1258–1262.
- Tian Y, Li Z, Hu W, et al. C. elegans screen identifies autophagy genes specific to multicellular organisms. Cell. 2010;141:1042–1055.
- Deretic V. Autophagy as an innate immunity paradigm: expanding the scope and repertoire of pattern recognition receptors. Curr Opin Immunol. 2012;24(1):21–31.
- Hwang S, Maloney NS, Bruinsma MW, et al. Nondegradative role of Atg5-Atg12/ Atg16L1 autophagy protein complex in antiviral activity of interferon gamma. Cell Host Microbe. 2012;11(4):397–409.
- Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature. 2011;469(7330):323–335.
- Keller MD, Torres VJ, Cadwell K. Autophagy and microbial pathogenesis. Cell Death Differ. 2020;27(3):872–886.
- Cadwell K, Patel KK, Komatsu M, et al. A common role for Atg16L1, Atg5 and Atg7 in small intestinal paneth cells and Crohn disease. Autophagy. 2009;5(2):250–252.
- Cadwell K, Patel KK, Maloney NS, et al. Virus-plus-susceptibility gene interaction determines Crohn’s disease gene Atg16L1 phenotypes in intestine. Cell. 2010;141(7):1135–1145.
- Yang L, Liu C, Zhao W, et al. Impaired autophagy in intestinal epithelial cells alters gut microbiota and host immune responses. Appl Environ Microbiol. 2018;84(18):e00880–18.
- Levy J, Cacheux W, Bara MA, et al. Intestinal inhibition of Atg7 prevents tumour initiation through a microbiome-influenced immune response and suppresses tumour growth. Nat Cell Biol. 2015;17(8):1062–1073.
- Lu Q, Yokoyama CC, Williams JW, et al. Homeostatic control of innate lung inflammation by Vici syndrome gene Epg5 and additional autophagy genes promotes influenza pathogenesis. Cell Host Microbe. 2016;19(1):102–113.
- Abdel Fattah E, Bhattacharya A, Herron A, et al. Critical role for IL-18 in spontaneous lung inflammation caused by autophagy deficiency. J Immunol. 2015;194(11):5407–5416.
- Kanayama M, He YW, Shinohara ML. The lung is protected from spontaneous inflammation by autophagy in myeloid cells. J Immunol. 2015;194(11):5465–5471.
- Wang YT, Zaitsev K, Lu Q, et al. Select autophagy genes maintain quiescence of tissue-resident macrophages and increase susceptibility to Listeria monocytogenes. Nat Microbiol. 2020;5(2):272–281.
- Park S, Buck MD, Desai C, et al. Autophagy genes enhance murine gammaherpesvirus 68 reactivation from latency by preventing virus-induced systemic inflammation. Cell Host Microbe. 2016;19(1):91–101.
- Chu H, Khosravi A, Kusumawardhani IP, et al. Gene-microbiota interactions contribute to the pathogenesis of inflammatory bowel disease. Science. 2016;352(6289):1116–1120.
- Martin PK, Marchiando A, Xu R, et al. Autophagy proteins suppress protective type I interferon signalling in response to the murine gut microbiota. Nat Microbiol. 2018;3(10):1131–1141.
- Strong DW, Thackray LB, Smith TJ, et al. Protruding domain of capsid protein is necessary and sufficient to determine murine norovirus replication and pathogenesis in vivo. J Virol. 2012;86(6):2950–2958.
- Nice TJ, Strong DW, McCune BT, et al. A single-amino-acid change in murine norovirus NS1/2 is sufficient for colonic tropism and persistence. J Virol. 2013;87(1):327–334.
- Ezekowitz RA, Austyn J, Stahl PD, et al. Surface properties of bacillus Calmette-Guerin-activated mouse macrophages. Reduced expression of mannose-specific endocytosis, Fc receptors, and antigen F4/80 accompanies induction of Ia. J Exp Med. 1981;154(1):60–76.
- Ezekowitz RA, Gordon S. Down-regulation of mannosyl receptor-mediated endocytosis and antigen F4/80 in bacillus Calmette-Guerin-activated mouse macrophages. Role of T lymphocytes and lymphokines. J Exp Med. 1982;155(6):1623–1637.
- Wilen CB, Lee S, Hsieh LL, et al. Tropism for tuft cells determines immune promotion of norovirus pathogenesis. Science. 2018;360(6385):204–208.
- Baldridge MT, Lee S, Brown JJ, et al. Expression of Ifnlr1 on intestinal epithelial cells is critical to the antiviral effects of interferon lambda against norovirus and reovirus. J Virol. 2017;91(7). DOI:https://doi.org/10.1128/JVI.02079-16
- Nice TJ, Baldridge MT, McCune BT, et al. Interferon-lambda cures persistent murine norovirus infection in the absence of adaptive immunity. Science. 2015;347(6219):269–273.
- Grau KR, Zhu S, Peterson ST, et al. The intestinal regionalization of acute norovirus infection is regulated by the microbiota via bile acid-mediated priming of type III interferon. Nat Microbiol. 2020;5(1):84–92.
- Pott J, Mahlakoiv T, Mordstein M, et al. IFN-lambda determines the intestinal epithelial antiviral host defense. Proc Natl Acad Sci U S A. 2011;108(19):7944–7949.
- Lin JD, Feng N, Sen A, et al. Distinct roles of type I and type III interferons in intestinal immunity to homologous and heterologous rotavirus infections. PLoS Pathog. 2016;12(4):e1005600.
- Onoguchi K, Yoneyama M, Takemura A, et al. Viral infections activate types I and III interferon genes through a common mechanism. J Biol Chem. 2007;282(10):7576–7581.
- Tsuboi K, Nishitani M, Takakura A, et al. Autophagy protects against colitis by the maintenance of normal gut microflora and secretion of mucus. J Biol Chem. 2015;290(33):20511–20526.
- Wan D, Jiang W, Hao J. Research advances in how the cGAS-STING pathway controls the cellular inflammatory response. Front Immunol. 2020;11:615.
- Gonugunta VK, Sakai T, Pokatayev V, et al. Trafficking-mediated STING degradation requires sorting to acidified endolysosomes and can be targeted to enhance anti-tumor response. Cell Rep. 2017;21:3234–3242.
- Duran A, Linares JF, Galvez AS, et al. The signaling adaptor p62 is an important NF-kappaB mediator in tumorigenesis. Cancer Cell. 2008;13:343–354.
- Wu MY, Lu JH. Autophagy and macrophage functions: inflammatory response and phagocytosis. Cells. 2019;9(1):70.
- Larabi A, Barnich N, Nguyen HTT. New insights into the interplay between autophagy, gut microbiota and inflammatory responses in IBD. Autophagy. 2020;16:38–51.
- Okada K, Yanagawa T, Warabi E, et al. The alpha-glucosidase inhibitor acarbose prevents obesity and simple steatosis in sequestosome 1/A170/p62 deficient mice. Hepatol Res. 2009;39:490–500.
- Sauer JD, Sotelo-Troha K, von Moltke J, et al. The N-ethyl-N-nitrosourea-induced Goldenticket mouse mutant reveals an essential function of Sting in the in vivo interferon response to Listeria monocytogenes and cyclic dinucleotides. Infect Immun. 2011;79:688–694.
- Komatsu M, Waguri S, Ueno T, et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol. 2005;169:425–434.
- Matsuzawa-Ishimoto Y, Shono Y, Gomez LE, et al. Autophagy protein ATG16L1 prevents necroptosis in the intestinal epithelium. J Exp Med. 2017;214:3687–3705.
- Orchard RC, Wilen CB, Doench JG, et al. Discovery of a proteinaceous cellular receptor for a norovirus. Science. 2016;pii:aaf1220.
- Kuss SK, Etheredge CA, Pfeiffer JK. Multiple host barriers restrict poliovirus trafficking in mice. PLoS Pathog. 2008;4:e1000082.
- Kuss SK, Best GT, Etheredge CA, et al. Intestinal microbiota promote enteric virus replication and systemic pathogenesis. Science. 2011;334:249–252.
- Takeshita S, Kaji K, Kudo A. Identification and characterization of the new osteoclast progenitor with macrophage phenotypes being able to differentiate into mature osteoclasts. J Bone Miner Res. 2000;15:1477–1488.
- Peterson ST, Kennedy EA, Brigleb PH, et al. Disruption of type III interferon genes Ifnl2 and Ifnl3 recapitulates loss of the type III IFN receptor in the mucosal antiviral response. J Virol. 2019;93(22):e01073–19.
- Baert L, Wobus CE, Van Coillie E, et al. Detection of murine norovirus 1 by using plaque assay, transfection assay, and real-time reverse transcription-PCR before and after heat exposure. Appl Environ Microbiol. 2008;74:543–546.
- Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550.
- Lee S, Liu H, Wilen CB, et al. A secreted viral nonstructural protein determines intestinal norovirus pathogenesis. Cell Host Microbe. 2019;25:845–57 e5.
- Caporaso JG, Lauber CL, Walters WA, et al. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc Natl Acad Sci U S A. 2011;108(Suppl 1):4516–4522.
- Callahan BJ, McMurdie PJ, Rosen MJ, et al. DADA2: high-resolution sample inference from Illumina amplicon data. Nat Methods. 2016;13:581–583.
- Cole JR, Wang Q, Fish JA, et al. Ribosomal database project: data and tools for high throughput rRNA analysis. Nucleic Acids Res. 2014;42:D633–42.
- McMurdie PJ, Holmes S. phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One. 2013;8:e61217.
- Segata N, Izard J, Waldron L, et al. Metagenomic biomarker discovery and explanation. Genome Biol. 2011;12:R60.
- Tamoutounour S, Henri S, Lelouard H, et al. CD64 distinguishes macrophages from dendritic cells in the gut and reveals the Th1-inducing role of mesenteric lymph node macrophages during colitis. Eur J Immunol. 2012;42:3150–3166.