
ABSTRACT
The Golgi apparatus regulates the process of modification and subcellular localization of macromolecules, including proteins and lipids. Aberrant protein sorting caused by defects in the Golgi leads to various diseases in mammals. However, the role of the Golgi apparatus in organismal longevity remained largely unknown. By employing a quantitative proteomic approach, we demonstrated that MON-2, an evolutionarily conserved Arf-GEF protein implicated in Golgi-to-endosome trafficking, promotes longevity via upregulating macroautophagy/autophagy in C. elegans. Our data using cultured mammalian cells indicate that MON2 translocates from the Golgi to the endosome under starvation conditions, subsequently increasing autophagic flux by binding LGG-1/GABARAPL2. Thus, Golgi-to-endosome trafficking appears to be an evolutionarily conserved process for the upregulation of autophagy, which contributes to organismal longevity.
Macroautophagy (hereafter referred to as autophagy) is a large-scale cellular recycling process responsible for degradation of damaged or dysfunctional cytoplasmic components via the autophagosomal-lysosomal pathway. Autophagy plays multiple fundamental roles in physiological processes, including development and immunity. Autophagy is also associated with aging and age-related disorders, and is required for longevity in various species.
C. elegans is an excellent model organism to study aging and longevity because of its relatively short lifespan, genetic tractability, and evolutionarily conserved aging-regulating processes, including autophagy. The Golgi apparatus is essential for the transport of macromolecules among organelles, including autophagosomes. However, evidence for the role of the Golgi complex in the regulation of organismal aging has been scarce.
Because C. elegans has been underappreciated as a model employing biochemical approaches, in particular for aging research, we performed unbiased quantitative proteomic experiments to identify novel factors that contribute to longevity [Citation1]. Specifically, we sought to identify proteins whose levels were altered by a longevity-promoting condition, reduced mitochondrial respiration. We subsequently performed a large-scale RNAi-based lifespan screen targeting genes that encode proteins identified from our proteomic analysis. Among four proteins that are required for the longevity, we further focused our research on MON-2 (), an evolutionarily conserved Arf-GEF protein localized to the trans-Golgi network and implicated in Golgi-to-endosome trafficking. We chose MON-2 as the focus of our research mainly because the role of Golgi proteins in the regulation of aging remained largely unknown. We showed that mon-2 contributes to longevity caused by reduced insulin/insulin-like growth factor 1 signaling, and by dietary restriction regimens, in addition to reduced mitochondrial respiration. We then found that PAD-1/DOP1, which forms a complex with Mon2 in yeast, sorting nexin SNX-3/SNX3 and TBC-3/TBC1D22A, all of which contribute to vesicular trafficking between the Golgi and endosomes, as MON-2 does, are also required for C. elegans longevity. These data suggest that MON-2-regulated Golgi-to-endosome trafficking is required for longevity in C. elegans.
Figure 1. Upregulation of autophagy by MON-2/MON2 is conserved between C. elegans and mammalian cells. Reduced mitochondrial respiration or dietary restriction, and starvation appear to increase MON-2/MON2 levels in the Golgi in C. elegans and mammalian cells, respectively. This leads to the translocation of MON-2/MON2 from the Golgi to endosomes, leading to increases in autophagosome formation by MON-2/MON2 binding LGG-1/GABARAPL2, a key autophagy factor, on the phagophore. Consequently, enhanced MON-2/MON2-mediated autophagy leads to increased longevity in C. elegans.
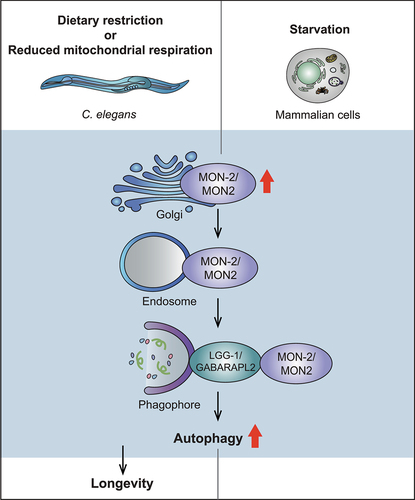
How does MON-2 contribute to lifespan extension by multiple interventions? By assessing the contribution of various established factors that affect aging, we found that autophagy mediates MON-2-regulated longevity. We showed that MON-2 is required for enhanced autophagosome formation and autophagy activation conferred by reduced mitochondrial respiration and dietary restriction. We then found that mammalian MON2 upregulates autophagy under starvation, likely by translocating from the Golgi to phagophores via endosomes, and by physically interacting with GABARAPL2, a homolog of key autophagy-regulating factor Atg8/LGG-1, in cultured human cells. Thus, the role of MON-2 in the regulation of autophagy appears to be conserved between C. elegans and mammals.
Collectively, our research combining unbiased proteomics and molecular genetic approaches revealed new roles of MON-2 and Golgi-to-endosome trafficking in lifespan regulation. It is of note that we reported the first mass spectrometry analysis revealing the proteomic changes caused by two extensively studied longevity-promoting mutations in mitochondrial respiration genes, clk-1 and isp-1, in C. elegans. We uncovered the essential role of the Golgi protein MON-2/MON2 in enhancing autophagosome formation in both C. elegans and cultured mammalian cells. Considering the evolutionarily conserved nature of MON-2 and the role of autophagy in aging, it is tempting to speculate that MON2 may also contribute to longevity in mammals.
While we demonstrated the role of MON-2 as a lifespan- and autophagy-regulating factor, many questions need to be answered in future studies. Although our data are consistent with the possibility that proper trafficking of autophagy-related factors by MON-2 enhances autophagosome formation, we have not provided direct evidence linking MON-2-mediated membrane trafficking to autophagy. In addition, factors that are responsible for upregulation of MON-2 are required to be identified and characterized. It will also be important to test whether upregulation of autophagy by MON-2 affects organismal aging in mammals. Further investigation concerning these issues will open up an exciting new avenue in the research fields of autophagy and aging.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
Reference
- Jung Y, Artan M, Kim N, et al. MON-2, a Golgi protein, mediates autophagy-dependent longevity in Caenorhabditis elegans. Sci Adv. 2021;8156:1–16.