
ABSTRACT
NLRP3 (NLR family pyrin domain containing 3) inflammasome is a potent mediator of inflammation due to its ability to produce the pro-inflammatory cytokines IL1B (interleukin 1 beta) and IL18 in response to numerous danger signals and pathogens. Mitophagy, a selective form of autophagy, restricts NLRP3 inflammasome activation by limiting the mitochondrial-derived danger signals. Here, we demonstrated that the adaptor protein APPL1 together with its interaction partner RAB5 in early endosomes negatively regulate NLRP3 inflammasome activation via induction of mitophagy in macrophages. Hematopoietic-deletion of Appl1 exacerbates systemic NLRP3 inflammasome activation in rodent models under obese or septic conditions. Our study identified a new regulatory network between early endosomes and mitochondria in control of NLRP3 inflammasome activation.
The NLRP3 inflammasome senses a wide array of pathogen- and danger/damage-associated molecular patterns (PAMPs and DAMPs) and initiates immune responses. Activated NLRP3 oligomerizes and forms the multiprotein inflammasome together with the adaptor protein PYCARD/ASC (PYD and CARD domain containing) and CASP1 (caspase 1), leading to maturation of the pro-inflammatory cytokines IL1B and IL18. Uncontrolled or unresolved activation of the NLRP3 inflammasome triggers inflammatory diseases, including obesity-related type 2 diabetes and vascular complications as well as sepsis.
Mitophagy removes dysfunctional mitochondria, and therefore is essential for cellular homeostasis. This process is a concerted event involving multiple organelles, including endosomes, endoplasmic reticulum, proteasomes and lysosomes, at different stages. Disruption of this highly coordinated pathway results in uncontrolled NLRP3 inflammasome activation. In particular, defective mitophagy increases mitochondrial danger signals, including mitochondrial reactive oxygen species (mtROS) and oxidized mitochondrial DNA (mtDNA), which are the key triggering and amplifying signals for the NLRP3 inflammasome.
The endosomal protein APPL1 mediates ADIPOQ/adiponectin and INS (insulin) signaling, and its dysfunction contributes to obesity and type 2 diabetes; both feature chronic low-grade inflammation. Our latest work uncovered a new role of APPL1 in controlling early endosome-mediated mitophagy, which restricts NLRP3 inflammasome activation in primary macrophages [Citation1]. Bone marrow-derived macrophages (BMDMs) from appl1 knockout (KO) mice exhibit an overactivation of the NLRP3 inflammasome in response to ATP, nigericin, monosodium urate crystals and palmitic acid, when compared to those BMDMs isolated from wild-type (WT) littermates. Conversely, APPL1 does not regulate NLRC4, AIM2 and non-canonical inflammasome activation in BMDMs and has no impact on M1 macrophage polarization.
How does APPL1 restrict NLRP3 inflammasome activation? It seems that APPL1 does not regulate the priming step of the NLRP3 inflammasome. The mitochondrial danger signal is one of the signals for the activation step of the NLRP3 inflammasome. Upon stimulation with NLRP3 agonists, APPL1 deficiency induces mitochondria damage, leading to a dramatic increase of mtROS generation and cytosolic oxidized mtDNA. Inhibition of mtROS production or removal of mtDNA largely dampens APPL1 deficiency-induced NLRP3 inflammasome activation, suggesting that mitochondrial danger signals are the primary inducers of NLRP3 inflammation activation.
Why are excessive damaged mitochondria and its associated danger signals accumulated in appl1-KO macrophages? Recent studies indicate that early endosomes or endosome-related proteins participate in mitophagy. We hypothesized that APPL1 as an endosomal-resident protein and an interaction partner of RAB5 (a small GTPase that mediates early endosome trafficking and functions) might control mitophagy. Indeed, APPL1 deficiency impairs mitochondrial clearance after NLRP3 inflammasome activation, evidenced by the reduced degradation rate of mitochondrial proteins and reduced RAB5+-labeled mitochondria. By using the fluorescent mitophagy reporter mito-Keima, we further showed that mitophagic flux is substantially decreased in appl1-KO BMDMs in response to the NLRP3 agonists. Likewise, confocal imaging also showed an increased colocalization of LC3+ puncta with mitochondria but a diminished number of LAMP+ (a lysosome marker) and MitoTracker+ puncta in APPL1-deficient BMDMs, indicating a defective mitophagic response. Furthermore, recruitment of the autophagy receptor SQSTM1/p62 to the damaged mitochondria is also greatly abrogated in APPL1-deficient BMDMs compared to the WT controls upon NLRP3 agonist stimulation. Collectively, these findings hint that the APPL1 deficiency leads to impairment of mitophagic flux in macrophages.
APPL1 is a highly dynamic early endosomal protein that translocates between different organelles under various stress conditions. We then tested whether APPL1 is recruited to the mitochondria during NLRP3 inflammasome activation. Immunoblotting and confocal imaging demonstrated that APPL1 can be found in the mitochondrial fraction and MitoTracker+ puncta under NLRP3 inflammasome activation conditions. The trafficking of APPL1 to the mitochondria is concurrent with the recruitment of RAB5. Both APPL1 and RAB5 are localized in mitophagosomes, but are rarely detected in the mitochondria-containing autolysosomes. Wild-type APPL1, but not the APPL1 mutants with defective RAB5 binding ability or endosomal localizing ability, is recruited to the damaged mitochondria upon NLRP3 stimulation.
RAB5+ early endosomes deliver mitochondria to lysosomes for clearance. Next, we explored the role of APPL1+ and RAB5+ endosomes in control of mitophagy and inflammasome activation. Trafficking of the RAB5+ endosome to the damaged mitochondria and mitophagosome is impaired in appl1-KO BMDMs, whereas replenishment of wild-type APPL1 expression not only rescues RAB5+ endosome-mediated mitochondrial clearance but also reduces NLRP3 inflammasome activation. However, replenishment with APPL1 mutants lacking RAB5 binding ability or endosomal localizing ability does not exert the similar rescue effects. Constitutive activation of RAB5 rescues the defective mitophagic response and NLRP3 inflammasome hyperactivation in appl1-KO BMDMs, indicating that RAB5 is a downstream mediator of APPL1. Hematopoietic deletion of Appl1 exaggerates obesity-induced adipose tissue inflammation and septic responses in animal models, which in particular are reflected by overproduction of IL1B and IL18.
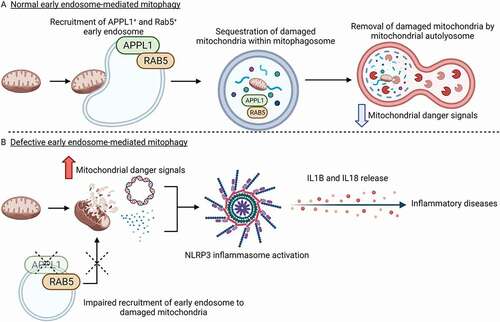
Taken together, our work shows a crucial role of the early endosome controlled by the APPL1-RAB5 axis in restricting NLRP3 inflammasome activation by clearing the mitochondrial danger signals via mitophagy ()
Figure 1. The APPL1-RAB5 axis controls early endosome-mediated mitochondrial clearance and NLRP3 inflammasome activation. (A) In macrophages with the functional APPL1-RAB5 axis, the early endosomes are recruited to damaged mitochondria in response to NLRP3 inflammasome activator. APPL1+ and RAB5+ endosomes aid the delivery of damaged mitochondria to the mitophagy machinery for its degradation, thereby preventing NLRP3 inflammasome overactivation by mitochondrial-derived danger signals such as mtDNA and mtROS. (B) Defective APPL1-mediated mitophagy impairs the recruitment of RAB5 to mitochondria and the downstream mitochondrial clearance, leading to accumulation of damaged mitochondria and its associated danger signals. Elevated mitochondrial-derived danger signals trigger the hyperactivation of the NLRP3 inflammasome for the production of IL1B and IL18, thereby exacerbating inflammatory diseases such as type 2 diabetes and sepsis. (Created with BioRender.com).
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
Reference
- Wu KKL, Long KK, Lin HG, et al. The APPL1-Rab5 axis restricts NLRP3 inflammasome activation through early endosomal-dependent mitophagy in macrophages. Nat Commun. 2021;12. DOI:https://doi.org/10.1038/s41467-021-26987-1.