
ABSTRACT
RHOA (ras homolog family member A) is a small G-protein that regulates a range of cellular processes including cell growth and survival. RHOA is a proximal downstream effector of G protein-coupled receptors coupling to GNA12/Gα12-GNA13/Gα13 proteins, and is activated in response to stretch and oxidative stress, functioning as a stress-response molecule. It has been demonstrated that RHOA signaling provides cardioprotection through inhibition of mitochondrial death pathways. Mitochondrial integrity is preserved not only by inhibition of mitochondrial death pathways but also by mitochondrial quality control mechanisms including mitophagy. One of the most well-established mechanisms of mitophagy is the mitochondrial membrane depolarization-dependent PINK1-PRKN/Parkin pathway. However, depolarization of the mitochondrial membrane potential is a late-stage event that occurs just before cell death, and additional intracellular mechanisms that enhance the PINK1-PRKN pathway have not been fully determined. We recently discovered that RHOA activation engages a unique mechanism to regulate PINK1 protein stability without inducing mitochondrial membrane depolarization, leading to increased mitophagy and protection against ischemia in cardiomyocytes. Our results suggest regulation of RHOA signaling as a potential strategy to enhance protective mitophagy against stress without compromising mitochondrial functions.
Preservation of the integrity of mitochondria is essential for maintaining cellular homeostasis, as damaged mitochondria trigger mitochondrial death pathways. Thus, there exists a process for the selective elimination of damaged mitochondria by autophagy (mitophagy). In homeostatic conditions, PINK1, a mitochondrial serine/threonine kinase, is constitutively cleaved and released from mitochondria to the cytosol where it undergoes proteasomal degradation. Dissipation of the mitochondrial membrane potential induced by stress leads to inhibition of PINK1 cleavage and accumulation of full-length PINK1 at the outer mitochondrial membrane, recruiting PRKN, an E3 ubiquitin ligase, to mitochondria. Subsequently, PRKN ubiquitinates and tags damaged mitochondria for selective removal. Loss of mitochondrial membrane potential occurs as a relatively late event during the process of cell death, thus additional intracellular signaling mechanisms that enhance PINK1-PRKN-mediated mitophagy at earlier stages would prolong cell survival during stress. However, such signaling mechanisms have not been fully elucidated.
Examining the role of RHOA in cardioprotection, we found that either expression of active RHOA or activation of endogenous RHOA with the G protein-coupled receptor ligand sphingosine-1-phosphate (S1P) leads to accumulation of exogenously expressed PINK1 at mitochondria in cardiomyocytes [Citation1]. We further confirmed that endogenous PINK1 is increased at mitochondria in response to activation of RHOA in cardiomyocytes, and that this leads to PRKN recruitment to mitochondria and increased mitophagy (). Furthermore, AAV9-mediated RHOA expression potentiates increases in mitochondrial PINK1, PRKN and ubiquitinated proteins induced by myocardial infarction in mouse hearts and decreases the infarct size. The effect of RHOA signaling on PINK1 is evident in human astrocytoma and rat hepatocyte cell lines, indicating that RHOA regulation of PINK1 accumulation is neither a cell-type nor a species-specific signaling event.
Figure 1. RHOA activation, without inducing depolarization of mitochondrial membrane potential, stabilizes PINK1 protein, induces mitophagy and provides protection against ischemic stress.
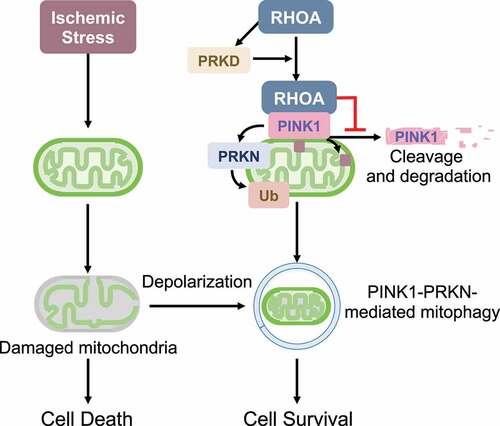
Notably, mitochondrial membrane potential is unaffected by RHOA activation, indicating that RHOA-mediated PINK1 accumulation at mitochondria is independent of the canonical mitochondrial membrane potential depolarization pathway. Although RHOA signaling regulates gene transcription, no significant differences in Pink1 mRNA levels are observed in control versus RHOA-expressing cardiomyocytes. Instead, we found that RHOA decreases the formation of cleaved PINK1 and increases PINK1 protein stability. Our experiments show that PRKD (protein kinase D), a downstream RHOA effector, is required but not sufficient for RHOA-mediated PINK1 accumulation on mitochondria. Interestingly, we observed that activated RHOA translocates to mitochondria and this is the step in which PRKD plays a role. Through co-immunoprecipitation studies, we further demonstrated that RHOA interacts with PINK1 at mitochondria. We speculate that formation of a RHOA-PINK1 molecular complex impedes the cleavage of PINK1. However, it remains to be elucidated how PRKD regulates the mitochondrial localization of RHOA and how RHOA inhibits PINK1 cleavage at mitochondria.
Activation of RHOA has been demonstrated to provide cardioprotection through inhibition of mitochondrial death pathways, and our study reveals an additional novel mechanism for RHOA-mediated protection involving PINK1-dependent mitophagy. The contribution of mitophagy to RHOA-mediated cardioprotection is supported by the demonstration that the protection provided by RHOA activation is significantly inhibited by PINK1 knockdown in cardiomyocytes. Although it is still controversial, mitochondrial fission facilitates mitophagic degradation through segregation of damaged mitochondria, and our previous study demonstrated that RHOA has an ability to induce mitochondrial fission through regulation of DNM1 L/DRP1 (dynamin 1-like), contributing to RHOA-mediated cellular protection. Together, these observations suggest that mitochondria are a convergence point for RHOA-mediated cellular protection and that RHOA is a unique signaling molecule that inhibits the mitochondrial death pathway as well as enhances removal of compromised mitochondria during stress, ensuring cellular protection.
Mitochondria are essential organelles producing cellular energy and regulating cell fate, and mitochondrial dysfunction is commonly observed in many diseases including heart failure, neurodegenerative and inflammatory disease. Developing drugs capable of enhancing mitophagy is of significant interest, and our study suggests that RHOA signaling pathways can serve as druggable targets to facilitate protective mitophagy through stabilization of PINK1 without concomitantly diminishing mitochondrial membrane potential and function. However, further studies in vivo are required to elucidate the relevance of RHOA signaling-mediated, PINK1-dependent mitophagy in protection against stress.
Acknowledgements
This work was supported by National Institutes of Health (NIH) grants R01HL145459, and American Heart Association grant 19TPA34910011 to SM.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
Reference
- Tu M, Tan VP, Yu JD, et al. RhoA signaling increases mitophagy and protects cardiomyocytes against ischemia by stabilizing PINK1 protein and recruiting Parkin to mitochondria. Cell Death Diff. 2022 Epub 2022 June 27. DOI:10.1038/s41418-022-01032-w.