
ABSTRACT
Lysosomes are essential catabolic organelles responsible for the degradation of biomacromolecules into low-molecular-weight materials for subsequent reuse. Neuronal ceroid lipofuscinoses (NCLs) are a group of fatal neurodegenerative lysosomal storage disorders characterized by the intracellular accumulation of lipoprotein aggregates (called ceroid lipofuscin) in neurons and other tissues. Mutations in KCTD7, which encodes a substrate-binding adaptor for the CUL3-RING E3 (CRL3) ubiquitin ligase complex, are categorized as a unique NCL subtype. However, the molecular mechanisms underlying the KCTD7-mutated NCLs remain unclear. In our recent study, we showed that KCTD7 deficiency leads to the accumulation of lysosomal storage deposits owing to lysosomal dysfunction and macroautophagic/autophagic defects. We identified CLN5 as an authentic substrate of CRL3-KCTD7 E3s. Wild-type KCTD7 targets CLN5 for ubiquitination and proteasomal degradation, whereas NCL patient-derived KCTD7 mutations disrupt the interaction between KCTD7-CUL3 or KCTD7-CLN5 and ultimately lead to excessive CLN5 accumulation in the endoplasmic reticulum. Accumulated CLN5 disrupts the interaction between CLN6-CLN8 and lysosomal enzymes, leading to impaired ER-to-Golgi trafficking of lysosomal enzymes. Thus, our findings indicate that KCTD7 is a key player in maintaining lysosomal and autophagic homeostasis and demonstrate that KCTD7 and CLN5, two NCL causative genes, are biochemically linked and function in a common neurodegenerative pathway.
Lysosomes are major degradative organelles that contain a variety of acid hydrolases that are capable of breaking down all types of biological polymers, such as DNA, RNA, proteins, carbohydrates, and lipids. The physiological importance of lysosomal homeostasis has been illustrated by the discovery of over 70 rare inherited metabolic diseases, called lysosomal storage diseases (LSDs). LSDs are characterized by lysosomal dysfunction associated with the accumulation of non-degraded materials in lysosomes, resulting in stress and dysfunction in cells, tissues, and organs. Neuronal ceroid lipofuscinoses (NCLs) constitute a broad class of fatal LSDs, also known as Batten disease. At the cellular level, NCLs show excessive accumulation of autofluorescent lipoprotein aggregates (called ceroid lipofuscin) in neurons and other cell types outside the central nervous system. Severe neurodegeneration is a prominent manifestation of most NCLs, and other clinical symptoms include epileptic seizures, vision loss, progressive motor/cognitive decline, and eventual premature death.
NCLs result from mutations in at least thirteen CLN genes. Usually, causative gene mutations in NCLs result in defects in lysosomal enzyme activity or impaired trafficking of lysosomal enzymes to perturb lysosomal homeostasis. Almost all NCL gene mutations lead to extensive autophagic defects, indicating that autophagy plays an indispensable role in the development and progression of NCLs. Among these, mutations in KCTD7, a gene encoding a substrate-binding adaptor of CRL3 E3s, are categorized as subtype NCL14. However, the physiological substrates of the CRL3-KCTD7 complex have not yet been identified, impeding the establishment of potential therapeutic interventions.
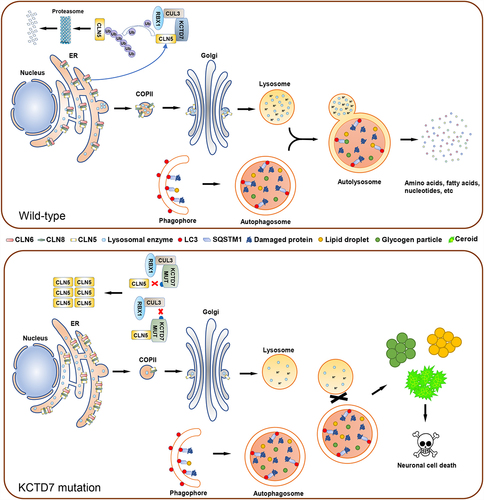
In a recent study, we attempted to delineate the molecular mechanisms underlying KCTD7- mutated NCLs [Citation1]. First, we generated KCTD7 knockout cells using CRISPR-Cas9-mediated gene editing. Transmission electron microscopy and immunofluorescence analysis demonstrated that KCTD7 deficiency in cells leads to the accumulation of various lysosomal storage materials such as lipid droplets, fingerprint-like profiles, granular osmiophilic deposits, and glycogen particles, reminiscent of observations in KCTD7-mutated patient biopsies, indicating that the disease-specific subcellular structures of KCTD7-mutated NCLs can be reproduced in cultured cell lines. NCLs are characterized by lysosomal dysfunction. Although KCTD7-deficient cells show a gross morphology similar to that of the parental cells, lysosomal acidification and enzymatic composition/activity are severely impaired. Moreover, KCTD7 deficiency leads to drastic lipidomic changes, autophagic defects, mTORC1 inactivation, unfolded protein response activation, and increased susceptibility to spermine-induced lysosome-dependent cell death, which have been described in other NCL subtypes.
As KCTD7 is a substate-binding adaptor of CRL3 E3s, we suspected that KCTD7 may control the ubiquitination and degradation of one or more substrates that are critical for lysosomal homeostasis. To explore this possibility, we combined CRISPR-Cas9–mediated endogenous gene tagging and affinity purification followed by mass spectrometry to identify potential KCTD7-interacting partners. CLN5 is ranked as a high-confidence interactor on the interaction hit list. Given that CLN5 mutations lead to another NCL subtype, we investigated any possible link between KCTD7 mutation-induced NCLs and KCTD7-CLN5 interaction. Although CLN5 has been reported to regulate lysosomal enzyme trafficking, little is known about the mechanisms through which CLN5 functions are regulated. We demonstrated that KCTD7 directly interacts with CLN5 and reduces CLN5 protein stability by inducing the ubiquitin-proteasomal degradation of CLN5, establishing that CLN5 is an authentic substrate for CRL3-KCTD7 E3s.
More than 30 patient-derived KCTD7 mutations have been identified in previous studies. We found that KCTD7 mutants with mutations in the BTB domain show reduced CUL3-binding ability, whereas KCTD7 mutants with mutations in the region between amino acids 139 and 289 (which harbors a CLN5-interacting domain) show reduced CLN5-binding ability. Patient-derived KCTD7 mutants are defective in promoting KCTD7 ubiquitination and degradation and fail to reverse lysosomal defects caused by KCTD7 deficiency. At first glance, it seems counterintuitive that both CLN5 accumulation induced by KCTD7 deficiency and CLN5 loss-of-function mutations independently lead to different NCL subtypes. If CLN5 accumulation induced by KCTD7 deficiency causes this disease, CLN5 overexpression could be expected to elicit similar NCL phenotypes. Indeed, CLN5 overexpression also leads to a reduction in lysosomal enzymatic composition/activity and autophagic defects. Moreover, CLN5 knockdown (KD) largely reverses KCTD7 deficiency-induced reductions in lysosomal enzymatic composition/activity and autophagic defects. These results indicated that the dosage balance of CLN5 plays a vital role in lysosomal homeostasis.
CLN5 is generally recognized as a lysosomal protein but has also been reported to be localized in the ER, Golgi, and endo/lysosomes. In KCTD7-deficient cells, CLN5 protein levels in the lysosomal fraction markedly decrease, while CLN5 protein levels in the ER fraction markedly increase. We proposed that the abnormally accumulated CLN5 may impair the trafficking of lysosomal enzymes from the ER to the Golgi based on the finding that KCTD7 deficiency induces CLN5 accumulation and retention in the ER. Indeed, retention using selective hooks (RUSH) cargo sorting assays show that KCTD7 deficiency leads to a significant delay in the ER-to-Golgi trafficking of lysosomal enzymes, which is largely reversed by CLN5 KD. Moreover, the ER-to-Golgi trafficking of lysosomal enzymes is delayed in cells with CLN5 KD or overexpression, suggesting that proper ER-to-Golgi trafficking is sensitive to CLN5 protein levels. Two ER-resident transmembrane proteins, CLN6 and CLN8, form a complex called the ER-to-Golgi relaying of enzymes of the lysosomal system (EGRESS) complex because of its role in recruiting lysosomal enzymes for trafficking to the Golgi. CLN5 has been reported to interact with CLN6-CLN8 and lysosomal enzymes. We found that the interactions between CLN6-CLN8 and lysosomal enzymes are markedly reduced in KCTD7-deficient cells, and this effect is largely reversed by CLN5 KD, indicating that abnormal CLN5 accumulation disrupts CLN6-CLN8-mediated recruitment of lysosomal enzymes, thus impairing the ER-to-Golgi trafficking of lysosomal enzymes.
Taken together, our findings that the NCL phenotypes caused by KCTD7 mutations are largely rescued by reducing CLN5 protein levels and that CLN5 overexpression phenocopies KCTD7 deficiency indicate that KCTD7 affects a common lysosomal enzyme trafficking pathway by modulating CLN5 protein levels (). Currently, there is no curative treatment for KCTD7-mutated NCLs, and our findings suggest that reducing CLN5 expression may represent a potential target for effective treatment.
Figure 1. Schematic diagram depicting a model in which NCL-associated KCTD7 mutations lead to lysosomal dysfunction and autophagic defects in NCL14 patients.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
Reference
- Wang Y, Cao X, Liu P, et al. KCTD7 mutations impair the trafficking of lysosomal enzymes through CLN5 accumulation to cause neuronal ceroid lipofuscinoses. Sci Adv. 2022 Aug 5;8(31):eabm5578.