
ABSTRACT
RB1CC1/FIP200 is an essential macroautophagy/autophagy protein that plays an important role in a variety of biological and disease processes through its canonical autophagy-dependent and -independent functions. However, it remains largely unknown whether post-translational modifications could regulate RB1CC1 and its associated autophagy functions. Here, we report acetylation of several lysine residues of RB1CC1 by acetyltransferase CREBBP (CREB binding protein), with K276 as the major CREBBP acetylation site. K276 is also identified as a ubiquitination site by mass spectrometry, and acetylation at this site reduces ubiquitination of RB1CC1 to inhibit its ubiquitin-dependent degradation. We also find that RB1CC1 contains an N-terminal intrinsically disordered region (IDR) capable of forming liquid-liquid phase separation (LLPS) in vitro, which may drive formation of RB1CC1 puncta with LLPS properties in cells independent of SQSTM1/p62 and other autophagy receptors CALCOCO2/NDP52, NBR1, TAX1BP1 and OPTN. Mutational analysis shows that both K276 acetylation and the N-terminal IDR containing it are important for maintaining canonical autophagy function of RB1CC1 in breast cancer cells. Our findings demonstrate regulation of RB1CC1 by a new post-translational mechanism and suggest potential therapeutic application of inducing RB1CC1 degradation through blocking K276 acetylation in the treatment of cancer and other diseases.
Abbreviations: Baf-A1: bafilomycin A1; CREBBP/CBP: CREB binding protein; CHX: cycloheximide; EP300/p300: E1A binding protein p300; FRAP: fluorescence recovery after photobleaching; HADCs: histone deacetylases; IDR: intrinsically disordered region; LLPS: liquid-liquid phase separation; KAT2A/GCN5: lysine acetyltransferase 2A; KAT2B/PCAF: lysine acetyltransferase 2B; KAT5/TIP60: lysine acetyltransferase 5; KAT8/MOF: lysine acetyltransferase 8; NAM: nicotinamide; PAS: phagophore assembly site; PEG-8000: polyethylene glycol 8000; RB1CC1/FIP200: RB1 inducible coiled-coil 1; TSA: trichostatin A.
Introduction
Macroautophagy/autophagy (termed autophagy thereafter) is a highly conserved cellular process for degradation of bulk cytoplasmic materials, including protein aggregates and damaged organelles, to maintain cellular homeostasis in response to starvation and other stress conditions [Citation1–3]. As an essential cellular process, dysfunction of autophagy contributes to a variety of diseases such as cancer, cardiovascular disorders, and neurodegenerative diseases [Citation4–10]. The whole process of autophagy can be generally divided into four stages: autophagy initiation, autophagosome formation, autophagosome maturation, and lysosomal degradation [Citation11,Citation12]. The sequential coordination of these stages ensures effective autophagic degradation and continuous autophagy flux.
RB1CC1/FIP200 (RB1 inducible coiled-coil 1) is a component of the ULK1 (unc-51 like autophagy activating kinase 1)-ATG13-RB1CC1-ATG101 complex essential for the induction of autophagy initiation [Citation13]. Residues in the N-terminal region of RB1CC1 that resembles yeast Atg17 have been shown to mediate its interaction with ATG13 for bulk autophagy [Citation13–16]. More recent studies also suggest that the C-terminal region of RB1CC1 that shares homology with yeast Atg11 contributes to its scaffolding functions in selective autophagy by interacting with multiple selective autophagy cargo receptors including SQSTM1/p62 [Citation17–22]. By generation and analysis of conditional knockout of RB1CC1 in different tissues and disease models, our previous studies demonstrated important roles of RB1CC1 in several different subtypes of breast cancer through regulating various downstream pathways, as well as in the maintenance of neural stem cell self-renewal and differentiation [Citation23–31]. We also took a rigorous genetic approach to generate RB1CC1-4A mutant knock-in allele that blocks its interaction with ATG13 to block its bulk autophagy function specifically. Using this unique model, we showed that whereas RB1CC1-mediated bulk autophagy is important for post-natal survival, breast cancer development and progression, and neuronal cell survival, ablation of both its bulk autophagy and selective autophagy functions by RB1CC1 deletion leads to several distinct phenotypes implicating specific roles of RB1CC1 scaffold role in selective autophagy (by itself or in combination with bulk autophagy). These roles include supporting embryogenesis by protecting embryonic cells from TNF/TNFα-induced apoptosis [Citation14], promoting maintenance and differentiation of postnatal neural stem cells [Citation32,Citation33], and suppressing the activation of TBK1 (TANK binding kinase 1)-IFN (interferon) signaling and expression of multiple chemokines in breast cancer and other cells [Citation14,Citation31,Citation34,Citation35]. Despite the significant progress in understanding the multitude of functions of RB1CC1 in various biological and disease processes, our current knowledge of how RB1CC1 protein is regulated is very limited, except for previous reports on phosphorylation of RB1CC1 by ULK1and ULK2 but with unknown consequences [Citation16].
Protein acetylation is a post-translational modification mediated by various protein acetyltransferases, such as CREBBP (CREB binding protein) and the related EP300/p300 (E1A binding protein p300) [Citation36]. CREBBP and EP300/p300 interact with a variety of transcriptional activators and are best known for their acetyltransferase activity for histones such as H3K27ac to increase transcriptional activities [Citation37,Citation38]. They both can also catalyze acetylation of lysine in many non-histone proteins to regulate a variety of cellular functions [Citation39–41]. Increasing evidence suggests the potential roles of non-histone protein acetylation in the regulation of autophagy. Several autophagy-related proteins, including ATG5, ATG7, Atg8-family proteins, and ATG12, can be acetylated by EP300/p300 to regulate autophagy under conditions of limited nutrient availability [Citation42]. Acetylation of another autophagy protein, PIK3C3/VPS34 (phosphatidylinositol 3-kinase catalytic subunit type 3) by EP300/p300, has been shown to affect its lipid kinase activity and promote the initiation of canonical and non-canonical autophagy [Citation43]. On the other hand, acetylation of another key autophagy protein, BECN1 (beclin 1) by EP300/p300, was found to inhibit autophagosome maturation and endocytic trafficking by promoting the recruitment of RUBCN/rubicon to the BECN1-PIK3C3/VPS34 complex [Citation44]. Despite the shared functions of CREBBP and EP300/p300 in catalyzing H3K27ac and their large degree of sequence homology with similar structural domains, less is known about the potential role of CREBBP in the regulation of autophagy. Moreover, acetylation of RB1CC1 and its impact on autophagy and function in cancer cells has not been examined.
Phase separation of fluids is a physicochemical process by which molecules separate into a dense phase and a dilute phase. Phase-separated biomolecular condensates, which include the nucleolus, nuclear speckles, stress granules, and others, provide a mechanism to compartmentalize and concentrate biochemical reactions within a cell [Citation45–47]. Biomolecular condensates produced by liquid-liquid phase separation (LLPS) allow rapid movement of components into and within the dense phase and exhibit properties of liquid droplets, such as fusion and fission [Citation48]. Although the molecular determinants underlying LLPS in cells still remain unclear, several proteins harboring intrinsically disordered regions (IDRs) have been shown to phase separate under various solution conditions [Citation49]. A recent study has shown that the phagophore assembly site (PAS) is a liquid-like condensate of Atg proteins in yeast, including Atg17 (paralog of mammalian RB1CC1) [Citation50], suggesting that RB1CC1 could participate in LLPS as a new regulatory mechanism to affect its function in mammalian cell autophagy.
In this study, we identify K276 in the N terminus of RB1CC1 as an acetylation site by CREBBP, which could compete with ubiquitination at this site to increase RB1CC1 stability and its associated autophagy activity. We also found that RB1CC1 could form puncta with LLPS properties mediated by an IDR in the N-terminus containing K276 and that this IDR and K276 acetylation are both important for maintaining canonical autophagy function of RB1CC1 in breast cancer cells. These findings reveal a previously unknown regulatory mechanism of RB1CC1 and its autophagy functions, suggesting a potential new approach to inhibit autophagy by inducing RB1CC1 degradation through reduced CREBBP acetylation at this site.
Results
Acetylation of RB1CC1 by CREBBP (CREB binding protein) at lysine 276
To explore potential acetylation of RB1CC1, HEK293T cells were transfected with a plasmid encoding HA-RB1CC1 along with vectors expressing several different acetyltransferases individually, including EP300/p300, CREBBP, KAT2A/GCN5 (lysine acetyltransferase 2A), KAT2B/PCAF (lysine acetyltransferase 2B), KAT8/MOF (lysine acetyltransferase 8), and KAT5/Tip60 (lysine acetyltransferase 5). We found that ectopic expression of CREBBP, but not other acetyltransferases, significantly increased acetylation of RB1CC1, as measured by a pan-specific anti–acetylated lysine antibody (). Similar results were also observed in human breast cancer MCF7 cells; ectopic expression of wild-type CREBBP, but not an enzymatically inactive CREBBPL1435A,D1436A (CREBBP[LD]) mutant, increased the level of RB1CC1 acetylation (). We also detected acetylation of endogenous RB1CC1 (), which could be further elevated after incubation of MCF7 cells with a combination of nicotinamide (NAM), an inhibitor of class III histone deacetylases (HDACs) (SIRT family deacetylases) [Citation51] and trichostatin A (TSA), class I and II HDAC blockers [Citation52]. The same treatment significantly increased H3K27 acetylation in these cells (Figure S1), demonstrating the effectiveness of deacetylation inhibitors. Moreover, we found that endogenous CREBBP and RB1CC1 were associated in MCF7 cells by co-immunoprecipitation using either antibody (). These results suggest that RB1CC1 is subjected to CREBBP-mediated acetylation in breast cancer and other cells.
Figure 1. RB1CC1 is acetylated by CREBBP and associates with the acetyltransferase. (A) HEK293T cells were co-transfected with expression vector for HA-RB1CC1 and vectors encoding MYC-tagged KAT2A/GCN5 (lysine acetyltransferase 2A) or Flag-tagged EP300/p300, CREBBP, KAT2B/PCAF (lysine acetyltransferase 2B), KAT8/MOF (lysine acetyltransferase 8) or KAT5/Tip60 (lysine acetyltransferase 5), as indicated. Cell lysates were immunoprecipitated by anti-HA and analyzed for acetylation of HA-RB1CC1 by immunoblotting with a pan-acetyl lysine antibody (AcK) or anti-HA antibody in top two panels. Cell lysates were also analyzed directly by immunoblotting with various antibodies, as indicated for bottom 3 panels. (B) MCF7 cells were co-transfected with expression vector for HA-RB1CC1 and vectors encoding Flag-tagged CREBBP or CREBBP[LD] mutant, as indicated. Cell lysates were immunoprecipitated by anti-HA and analyzed for acetylation of HA-RB1CC1 by immunoblotting with anti-AcK or anti-HA antibody in top two panels. Cell lysates were also analyzed directly by immunoblotting with various antibodies, as indicated for bottom 3 panels. (C) MCF7 cells were treated with or without a combination of inhibitors for HDACs (all 3 classes) nicotinamide (NAM, 5 mM for 4 h) and trichostatin A (TSA, 0.5 μM for 16 h). Cell lysates were immunoprecipitated by control immunoglobulin G (IgG) or anti-RB1CC1 antibodies and then analyzed by immunoblotting with different antibodies, as indicated. (D and E) Cell lysates from MCF7 cells were immunoprecipitated by control immunoglobulin G (IgG) (D and E), anti-CREBBP (D), or anti-RB1CC1 (E) antibodies. The immunoprecipitates (top two panels) or cell lysates (bottom two panels) were then analyzed by immunoblotting with the indicated antibodies.
![Figure 1. RB1CC1 is acetylated by CREBBP and associates with the acetyltransferase. (A) HEK293T cells were co-transfected with expression vector for HA-RB1CC1 and vectors encoding MYC-tagged KAT2A/GCN5 (lysine acetyltransferase 2A) or Flag-tagged EP300/p300, CREBBP, KAT2B/PCAF (lysine acetyltransferase 2B), KAT8/MOF (lysine acetyltransferase 8) or KAT5/Tip60 (lysine acetyltransferase 5), as indicated. Cell lysates were immunoprecipitated by anti-HA and analyzed for acetylation of HA-RB1CC1 by immunoblotting with a pan-acetyl lysine antibody (AcK) or anti-HA antibody in top two panels. Cell lysates were also analyzed directly by immunoblotting with various antibodies, as indicated for bottom 3 panels. (B) MCF7 cells were co-transfected with expression vector for HA-RB1CC1 and vectors encoding Flag-tagged CREBBP or CREBBP[LD] mutant, as indicated. Cell lysates were immunoprecipitated by anti-HA and analyzed for acetylation of HA-RB1CC1 by immunoblotting with anti-AcK or anti-HA antibody in top two panels. Cell lysates were also analyzed directly by immunoblotting with various antibodies, as indicated for bottom 3 panels. (C) MCF7 cells were treated with or without a combination of inhibitors for HDACs (all 3 classes) nicotinamide (NAM, 5 mM for 4 h) and trichostatin A (TSA, 0.5 μM for 16 h). Cell lysates were immunoprecipitated by control immunoglobulin G (IgG) or anti-RB1CC1 antibodies and then analyzed by immunoblotting with different antibodies, as indicated. (D and E) Cell lysates from MCF7 cells were immunoprecipitated by control immunoglobulin G (IgG) (D and E), anti-CREBBP (D), or anti-RB1CC1 (E) antibodies. The immunoprecipitates (top two panels) or cell lysates (bottom two panels) were then analyzed by immunoblotting with the indicated antibodies.](/cms/asset/af8deb3a-c168-47ac-9337-38ec4f42ee54/kaup_a_2148432_f0001_b.gif)
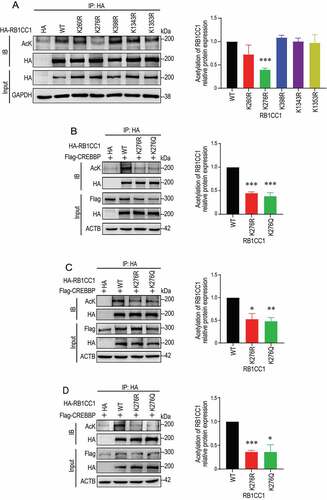
We next sought to determine the specific lysine residues in RB1CC1 that are acetylated by CREBBP. Mass spectrometry analysis showed that 22 lysines in RB1CC1 were possibly acetylated by CREBBP in cells. We focused on the top 5 high-confidence sites based on the number of Peptide-Spectrum Match (PSMs) (Figure S2). Each of these five putative acetylation sites was mutated to arginine from lysine (K to R, non-acetylatable mutant) and then examined for its effect on acetylation levels of RB1CC1. We found that RB1CC1K276R mutant, but not the other four mutants, significantly reduced the total level of RB1CC1 acetylation by CREBBP (). Inspection of the original spectra containing the peptide fragment including K276 is consistent with acetylation at this site in RB1CC1, which is also highly conserved across different species (Figure S3). Mutation of K276 to either non-acetylatable arginine (RB1CC1K276R) or acetylmimetic glutamine (RB1CC1K276Q) also markedly reduced the RB1CC1 acetylation in MCF7 cells (), as well as BT474 and SKBR3 cells, two other subtypes of breast cancer cells (). Together, these results identify K276 in the N-terminus of RB1CC1 as an acetylation site by CREBBP.
Figure 2. CREBBP promotes acetylation of RB1CC1 at K276 in HEK293T and breast cancer cells. (A) HEK293T cells were transfected with expression vectors encoding HA-RB1CC1 or several HA-tagged RB1CC1 mutants, as indicated. Cell lysates were immunoprecipitated by anti-HA antibodies and analyzed for acetylation of HA-RB1CC1 or different mutants by immunoblotting with anti-AcK or anti-HA antibody in top two panels. Cell lysates were also analyzed directly by immunoblotting with various antibodies, as indicated for bottom 2 panels. The graph on the right shows relative levels of acetylated RB1CC1/total RB1CC1 protein normalized to wild-type HA-RB1CC1 transfection samples (means ± SEM; n = 3 biologically independent samples). (B and D) MCF7 (B), BT474 (C), or SKBR3 (D) human breast cancer cells were co-transfected with expression vector for HA-RB1CC1 and vectors encoding Flag-CREBBP, as indicated. Cell lysates were immunoprecipitated by anti-HA and analyzed for acetylation of HA-RB1CC1 by immunoblotting with anti-AcK or anti-HA antibody in top two panels. Cell lysates were also analyzed directly by immunoblotting with various antibodies, as indicated for bottom 3 panels. The graphs on the right show relative levels of acetylated RB1CC1:total RB1CC1 protein normalized to wild-type HA-RB1CC1 transfection samples (means ± SEM; n = 3 biologically independent samples). *P < 0.05; **P < 0.01; ***P < 0.001.
Acetylation at K276 stabilizes RB1CC1 by inhibiting ubiquitin-dependent degradation
To investigate the importance of RB1CC1 acetylation at K276 by CREBBP, we first examined whether CREBBP may affect RB1CC1 degradation/stability in breast cancer cells. We found that wild-type CREBBP, but not its inactive mutant CREBBP[LD], increased stability of co-transfected HA-RB1CC1 protein in MCF7 cells (), as well as in BT474 cells (). Ectopic expression of CREBBP, but not CREBBP[LD] mutant, also increased stability of endogenous RB1CC1 in these two cells ( and S4). Conversely, upon the silencing of CREBBP, the levels and stability of endogenous RB1CC1 were reduced in MCF7 cells (, S5). Moreover, treatment of these cells with the CREBBP inhibitor A485 also reduced steady state RB1CC1 levels (i.e., no blockade of new protein synthesis by cycloheximide [CHX]) in a time-dependent manner (, S6A). Together, these results suggested that RB1CC1 acetylation by CREBBP may reduce RB1CC1 degradation to increase its levels in breast cancer cells.
Figure 3. RB1CC1 acetylation by CREBBP enhances RB1CC1 stability. (A and D) MCF7 (A and B) or BT474 (C and D) cells were co-transfected with expression vector for HA-RB1CC1 and Flag-tagged CREBBP or CREBBP[LD]. They were treated with CHX for various times, as indicated, to block new protein synthesis. Cell lysates were then analyzed by immunoblotting with different antibodies, as indicated (A and C). Panels B and D show quantification of relative levels of RB1CC1 signals normalized to GAPDH levels in A and C, respectively (means ± SEM; n = 3 biologically independent samples). (E and F) MCF7 cells were transfected with expression vector for Flag-tagged CREBBP or CREBBP[LD], and then treated with CHX for various times, as indicated. Cell lysates were then analyzed by immunoblotting with different antibodies, as indicated (E). Panel F shows quantification of relative levels of RB1CC1 signals normalized to GAPDH levels in E (means ± SEM; n = 3 biologically independent samples). (G and H) MCF7 cells were transfected with expression vector for shRNA for CREBBP and then treated with CHX for various times, as indicated. Cell lysates were then analyzed by immunoblotting with different antibodies, as indicated. Panel H shows quantification of relative levels of RB1CC1 signals normalized to GAPDH levels in G (means ± SEM; n = 3 biologically independent samples). (I) MCF7 cells were treated with CREBBP inhibitor A485 (10 μM) for various times, as indicated. Cell lysates were then analyzed by immunoblotting with different antibodies, as indicated (top). The right graph shows quantification of relative levels of RB1CC1 signals normalized to GAPDH levels (means ± SEM; n = 3 biologically independent samples).
![Figure 3. RB1CC1 acetylation by CREBBP enhances RB1CC1 stability. (A and D) MCF7 (A and B) or BT474 (C and D) cells were co-transfected with expression vector for HA-RB1CC1 and Flag-tagged CREBBP or CREBBP[LD]. They were treated with CHX for various times, as indicated, to block new protein synthesis. Cell lysates were then analyzed by immunoblotting with different antibodies, as indicated (A and C). Panels B and D show quantification of relative levels of RB1CC1 signals normalized to GAPDH levels in A and C, respectively (means ± SEM; n = 3 biologically independent samples). (E and F) MCF7 cells were transfected with expression vector for Flag-tagged CREBBP or CREBBP[LD], and then treated with CHX for various times, as indicated. Cell lysates were then analyzed by immunoblotting with different antibodies, as indicated (E). Panel F shows quantification of relative levels of RB1CC1 signals normalized to GAPDH levels in E (means ± SEM; n = 3 biologically independent samples). (G and H) MCF7 cells were transfected with expression vector for shRNA for CREBBP and then treated with CHX for various times, as indicated. Cell lysates were then analyzed by immunoblotting with different antibodies, as indicated. Panel H shows quantification of relative levels of RB1CC1 signals normalized to GAPDH levels in G (means ± SEM; n = 3 biologically independent samples). (I) MCF7 cells were treated with CREBBP inhibitor A485 (10 μM) for various times, as indicated. Cell lysates were then analyzed by immunoblotting with different antibodies, as indicated (top). The right graph shows quantification of relative levels of RB1CC1 signals normalized to GAPDH levels (means ± SEM; n = 3 biologically independent samples).](/cms/asset/a919eb91-b39d-4ca1-8ec9-a1af29c727b4/kaup_a_2148432_f0003_oc.jpg)
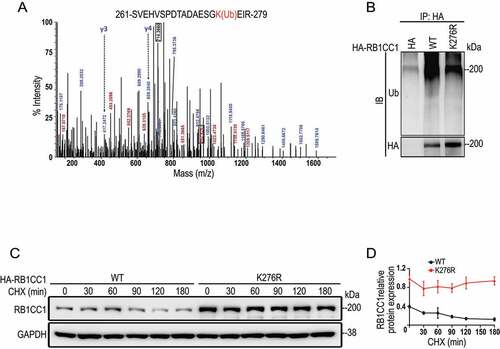
The ubiquitination induced degradation in proteasomes and autophagic pathways for lysosomal degradation are two major cellular mechanisms for protein degradation [Citation53]. Consistent with a role for CREBBP-mediated acetylation to stabilize RB1CC1, treatment of MCF7 cells with a combination of NAM [Citation51] and TSA [Citation52] increased levels of RB1CC1 in these breast cancer cells (). Further, autophagy inhibition by bafilomycin A1 (Baf-A1) did not affect RB1CC1 levels in the presence or absence of HDAC inhibitors (, see S1 for effectiveness of Baf-A1). In contrast, proteasome inhibitor MG132 increased RB1CC1 to comparable levels with or without HDAC inhibitors in these cells (, S6B), suggesting that RB1CC1 acetylation may affect its stability through modification of its ubiquitination and proteosome degradation. To test this directly, we examined the effects of CREBBP on the ubiquitination levels of RB1CC1. We found that wild-type CREBBP, but not its inactive CREBBP[LD] mutant, significantly decreased the ubiquitination level of RB1CC1 (). Conversely, CREBBP knockdown by shRNA increased the levels of RB1CC1 ubiquitination (). Likewise, treatment of the cells with the CREBBP inhibitor A485 also upregulated RB1CC1 ubiquitination levels (). Indeed, reexamination of the mass spectrum data revealed that lysine 276 was also a ubiquitination site in RB1CC1 (). In support of K276 as a ubiquitination site in RB1CC1, we found that RB1CC1K276R mutant exhibited decreased overall level of ubiquitination compared to wild-type RB1CC1 in breast cancer cells (). Lastly, RB1CC1K276R mutant showed increased levels and reduced degradation compared to wild-type RB1CC1 (). Together, these results indicate that acetylation of RB1CC1 at K276 by CREBBP promotes RB1CC1’s stability, likely by competing to prevent ubiquitination at this site.
Figure 4. Acetylation of RB1CC1 increases its stability by inhibiting ubiquitin-dependent degradation. (A and B) MCF7 cells were treated with autophagy inhibitor Baf-A1 (200 nM for 4 h) (A) or proteasome inhibitor MG132 (10 μM for 6 h) (B) in the presence or absence of a combination of inhibitors for HDACs (all 3 classes) NAM (5 mM for 4 h) and TSA (0.5 μM for 16 h). Cell lysates were then analyzed by immunoblotting with different antibodies, as indicated. The right graph shows quantification of relative levels of RB1CC1 signals normalized to un-treated samples (means ± SEM; n = 3 biologically independent samples). *P < 0.05; **P < 0.01. (C) MCF7 cells were co-transfected with expression vector for HA-RB1CC1, GFP-tagged ubiquitin, and vectors encoding Flag-tagged CREBBP or CREBBP[LD], as indicated. Following treatment of cells with 10 μM MG132 for 6 h, cell lysates were immunoprecipitated by anti-HA and analyzed by immunoblotting with a ubiquitin antibody (Ub) or anti-HA antibody. (D) MCF7 cells were co-transfected with expression vector for HA-RB1CC1, GFP-tagged ubiquitin, and vectors encoding 2 different shRNA for CREBBP, as indicated. Following treatment of cells with 10 μM MG132 for 6 h, cell lysates were immunoprecipitated by anti-HA and analyzed by immunoblotting with a ubiquitin antibody (Ub) or anti-HA antibody. Cell lysates were also analyzed directly by antibodies for CREBBP and GAPDH (bottom two panels). (E) MCF7 cells were co-transfected with expression vector for HA-RB1CC1 and GFP-tagged ubiquitin. Following treatment of cells with 10 μM MG132 for 6 h with or without CREBBP inhibitor A485 (10 μM) for 2 h, cell lysates were immunoprecipitated by anti-HA and analyzed by immunoblotting with a ubiquitin antibody (Ub) or anti-HA antibody.
![Figure 4. Acetylation of RB1CC1 increases its stability by inhibiting ubiquitin-dependent degradation. (A and B) MCF7 cells were treated with autophagy inhibitor Baf-A1 (200 nM for 4 h) (A) or proteasome inhibitor MG132 (10 μM for 6 h) (B) in the presence or absence of a combination of inhibitors for HDACs (all 3 classes) NAM (5 mM for 4 h) and TSA (0.5 μM for 16 h). Cell lysates were then analyzed by immunoblotting with different antibodies, as indicated. The right graph shows quantification of relative levels of RB1CC1 signals normalized to un-treated samples (means ± SEM; n = 3 biologically independent samples). *P < 0.05; **P < 0.01. (C) MCF7 cells were co-transfected with expression vector for HA-RB1CC1, GFP-tagged ubiquitin, and vectors encoding Flag-tagged CREBBP or CREBBP[LD], as indicated. Following treatment of cells with 10 μM MG132 for 6 h, cell lysates were immunoprecipitated by anti-HA and analyzed by immunoblotting with a ubiquitin antibody (Ub) or anti-HA antibody. (D) MCF7 cells were co-transfected with expression vector for HA-RB1CC1, GFP-tagged ubiquitin, and vectors encoding 2 different shRNA for CREBBP, as indicated. Following treatment of cells with 10 μM MG132 for 6 h, cell lysates were immunoprecipitated by anti-HA and analyzed by immunoblotting with a ubiquitin antibody (Ub) or anti-HA antibody. Cell lysates were also analyzed directly by antibodies for CREBBP and GAPDH (bottom two panels). (E) MCF7 cells were co-transfected with expression vector for HA-RB1CC1 and GFP-tagged ubiquitin. Following treatment of cells with 10 μM MG132 for 6 h with or without CREBBP inhibitor A485 (10 μM) for 2 h, cell lysates were immunoprecipitated by anti-HA and analyzed by immunoblotting with a ubiquitin antibody (Ub) or anti-HA antibody.](/cms/asset/83de0378-ae37-4e7d-ad7f-2d287d092c24/kaup_a_2148432_f0004_oc.jpg)
Figure 5. K276 is a ubiquitination site in RB1CC1 that regulates its ubiquitin-dependent degradation. (A) Mass spectrometry of RB1CC1 fragments containing ubiquitinated K276 (marked in red) in HEK293T cells. (B) RB1CC1-null MCF7 cells were co-transfected with expression vectors for HA-tagged RB1CC1 or RB1CC1K276R mutant and vector encoding GFP-tagged ubiquitin, as indicated. Following treatment of cells with 10 μM MG132 for 6 h, cell lysates were immunoprecipitated by anti-HA and analyzed by immunoblotting with a ubiquitin antibody (Ub) or anti-HA antibody. (C and D) RB1CC1-null MCF7 cells were transfected with expression vectors for HA-RB1CC1 or HA- RB1CC1K276R mutant. They were then treated with CHX for various times, as indicated. Cell lysates were analyzed by immunoblotting with different antibodies, as indicated (C). Panel D shows quantification of relative levels of RB1CC1 signals normalized to GAPDH levels in C (means ± SEM; n = 3 biologically independent samples).
Puncta formation of RB1CC1 mediated by their intrinsically disordered regions with liquid-liquid phase separation properties
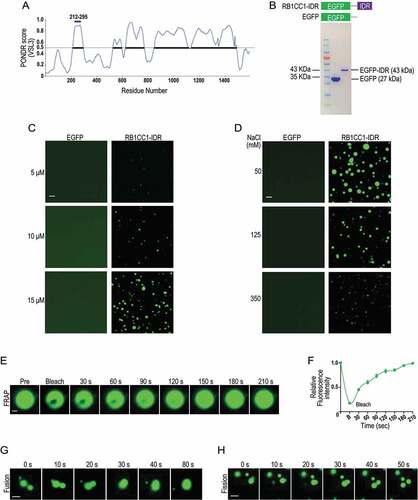
RB1CC1 is a large protein with multiple domains [Citation54], and inspection of the protein using Predictor of Natural Disordered Regions (PONDR) score revealed that K276 is located within IDR in the N-terminal region (residues 212–295) (). IDRs possess high numbers of proline, glutamine, and serine residues as acidic and basic segments and can mediate protein participation in LLPS [Citation55–58]. We, therefore, prepared a fusion recombinant protein EGFP-IDR (i.e., EGFP fused with the IDR of RB1CC1) to examine its possible formation of LLPS in vitro (). Similar to other purified IDRs involved in LLPS formation that also form droplets in vitro [Citation56,Citation59], purified recombinant EGFP-IDR, but not EGFP control, formed droplets in a concentration-dependent manner in the presence of 10% polyethylene glycol 8000 (PEG-8000) (). Moreover, the size distributions or opacity of RB1CC1-IDR droplets shifted toward smaller droplets with increasing NaCl concentration (from 50 mM to 350 mM) (), as in other LLPS [Citation45,Citation60]. Lastly, fluorescence recovery after photobleaching (FRAP) studies verified the dynamics of droplets formed by EGFP-IDR () that reflect high mobility or rapid exchange kinetics of the labels as a hallmark of LLPS [Citation45–47]. Fusion () and fission () events were also observed, supporting that these were liquid droplets. Together, these results suggest that the IDR region in the N-terminus could potentially mediate RB1CC1 participation in LLPS, although future studies using full length recombinant RB1CC1 will be needed to confirm the ability of RB1CC1 to form LLPS in vitro.
Figure 6. RB1CC1 N-terminal IDR containing K276 can form LLPS in vitro. (A) Schematic showing PONDR VSL3 score for intrinsic disorder of amino acid sequences of RB1CC1 based on the program VL3-BA. The horizontal bar in purple indicates the N-terminal IDR containing K276. (B and D) Purified EGFP fusion protein containing RB1CC1 N-terminal IDR (RB1CC1-IDR) or EGFP alone (B) were incubated at different concentrations with 125 mM NaCl (C) or at 20 μM with various NaCl concentration (D) in Buffer D containing 10% PEG-8000, as described in the Methods. Representative images of droplet formation of the fusion protein, as viewed under a fluorescence microscope. Scale bar: 2 μm. (E) Fluorescence intensity recovery of a droplet formed by RB1CC1-IDR in vitro after photobleaching. Scale bar: 0.5 μm. (F) Quantification of FRAP data for images in E. Bleaching event occurs at t = 0 s. Data plotted as means ± SEM (n = 3). (G and H) Fusion (G) and fission (H) of droplets formed by RB1CC1-IDR during phase separation process in vitro. Scale bar: 2 μm.
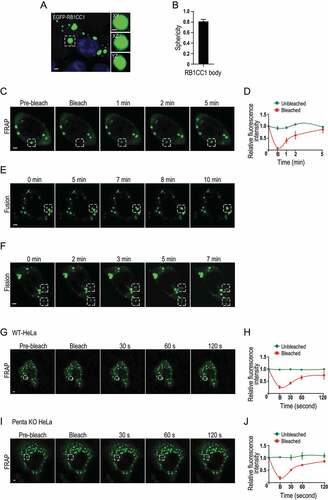
We next prepared an expression vector encoding RB1CC1 fused to EGFP (designated as EGFP-RB1CC1) and transfected it into MCF7 breast cancer cells to test for the potential of RB1CC1 participation in LLPS in cells. We found that EGFP-RB1CC1 formed spherical bodies in the cells (). FRAP studies in live cells showed that the fluorescence signal recovered within one minute after photobleaching of RB1CC1 bodies (), indicating that RB1CC1 can exchange rapidly among RB1CC1 bodies or between a RB1CC1 body and the surrounding cytosol. Further, we observed fusion of proximal RB1CC1 bodies (), as well as fission for other RB1CC1 bodies () within a 5–10 min time frame, both of which are other signature characteristics of LLPS [Citation45–47]. Recent studies suggested RB1CC1 interacts with autophagy receptors such as SQSTM1 which can form LLPS [Citation61], raising the possibility that RB1CC1 is recruited to LLPS formed by SQSTM1 and/or other autophagy receptors. To test this possibility, we performed similar studies in HeLa cells depleted of TAX1BP1 (Tax1 binding protein 1), CALCOCO2/NDP52 (calcium binding and coiled-coil domain 2), NBR1 (NBR1 autophagy cargo receptor), OPTN (optineurin) and SQSTM1 (penta KO cells) [Citation62]. We found that RB1CC1 was able to form LLPS in both wild-type HeLa cells () and penta KO cells (). Together, these results suggest that RB1CC1 could form puncta with LLPS properties in breast cancer cells independent of SQSTM1 and the other autophagy receptors CALCOCO2/NDP52, NBR1, TAX1BP1 and OPTN.
Figure 7. RB1CC1 participates in phase separation in cells. (A and F) MCF7 cells were transfected with expression vector encoding EGFP-RB1CC1. (A and B) Shows formation of RB1CC1 bodies, as viewed under a fluorescence microscope. Scale bar: 2 μm. Insets on the right shows 3D image of a RB1CC1 body at XY, XZ, and YZ planes. Scale bar: 1 μm. Panel B shows a plot of the sphericity of RB1CC1 bodies (n = 20). Data plotted as means ± SEM. (C) Fluorescence intensity recovery of a RB1CC1 body (boxed by dotted lines) after photobleaching. Scale bar: 2 μm. (D) Quantification of FRAP data for images in C. Bleaching event occurs at t = 0 s. Data plotted as means ± SEM (n = 3). (E and F) Fusion (E) and fission (F) of RB1CC1 bodies in MCF7 cells. Scale bar: 2 μm. (G and J) Wild-type HeLa cells (G and H) or penta KO cells (I and J) were transfected with expression vector encoding EGFP-RB1CC1. Panels G and I show fluorescence intensity recovery of a RB1CC1 body (boxed by dotted lines) after photobleaching for HeLa and penta KO cells, respectively. Panels H and J show quantification of FRAP data for images in G and I, respectively. Bleaching event occurs at t = 0 s. Data plotted as means ± SEM (n = 3).
Acetylation of RB1CC1 at K276 in its N-terminal intrinsically disordered region by CREBBP is important for autophagosome formation
To evaluate the physiological effects of acetylation of RB1CC1 IDR, we first generated a RB1CC1 mutant lacking its N-terminal IDR (designated as EGFP-ΔIDR) and examined its ability to form LLPS and rescue autophagy function of RB1CC1 KO MCF7 cells. As expected, EGFP-ΔIDR showed significantly decreased ability to form LLPS compared to EGPF-RB1CC1 (), consistent with that the N-terminal IDR is important for LLPS formed by RB1CC1. Furthermore, we found that the EGFP-ΔIDR mutant showed reduced ability to restore autophagy activity relative to wild-type RB1CC1 when expressed in RB1CC1-null MCF7 cells (), suggesting that RB1CC1 IDR is required for optimal autophagy functions. In contrast to ΔIDR mutant, RB1CC1K276R mutant of RB1CC1 showed similar activity as wild-type RB1CC1 in FRAP assays (Figure S7), suggesting that acetylation at K276 might not affect RB1CC1 puncta formation. We then examined the effect of K276 acetylation directly using in vitro assays. We found that direct incubation of purified EGFP-RB1CC1-IDR with a recombinant CREBBP catalytic domain protein and acetyl-CoA increased RB1CC1 puncta formation (Figure S8A), suggesting that K276 acetylation may increase the amount of RB1CC1 and/or its autophagy activities. Indeed, analysis of pellets from the MCF7 cell cytoplasm extracts showed that CREBBP reduced ubiquitination levels of EGFP-RB1CC1-IDR (Figure S8B), consistent with its decreased degradation to allow more protein accumulation. They also suggested that the lack of effect of RB1CC1K276R mutant in vivo could be due to that it also blocked ubiquitination of RB1CC1 at this site (so that the effect on blocking acetylation to influence ubiquitination and degradation could not be seen). Consistent with these in vitro findings, co-expression of EGFP-RB1CC1 with mCherry-CREBBP, but not inactive mCherry-CREBBP[LD] mutant, increased the size and numbers of EGFP-RB1CC1 puncta in MCF7 cells with CREBBP knockdown (). Similarly, ectopic expression of Flag-CREBBP exhibited greater ability to increase autophagy activity than Flag-CREBBP[LD] mutant, as measured by LC3-II levels in wild-type MCF7 cells (). Autophagy flux assays also showed that Flag-CREBBP expression slightly increased autophagy flux in wild-type MCF7 cells whereas Flag-CREBBP[LD] mutant reduced it (). Lastly, we further showed that A485 treatment of RB1CC1 KO MCF7 cells with ectopic expression of wild-type RB1CC1, but not RB1CC1K276R mutant, reduced RB1CC1 levels and starvation induced autophagy flux (), as well as the size and numbers of their puncta (). Together, these results suggest that acetylation of K276 in RB1CC1-IDR by CREBBP promotes RB1CC1 puncta formation and autophagosome formation.
Figure 8. Acetylation of K276 in N-terminal IDR of RB1CC1 is important for autophagosome formation. (A and B) MCF7 cells were transfected with expression vector encoding EGFP-RB1CC1 and EGFP-ΔIDR as indicated. (A) Fluorescence intensity recovery of a RB1CC1 body (boxed by dotted lines) after photobleaching. Scale bar: 2 μm. (B) Quantification of FRAP data for images in A. Bleaching event occurs at t = 0 s. Data plotted as means ± SEM (n = 3). (C and D) RB1CC1-null MCF7 cells were transfected with expression vectors for EGFP-tagged RB1CC1 or its mutant ΔIDR. (C) Cell lysates from these cells cultured under normal or starvation (HBSS) conditions were analyzed by immunoblotting with different antibodies, as indicated. (D) These cells were subjected to autophagy flux assay with Baf-A1 and lysates were analyzed under various conditions, as indicated. (E and F) MCF7 cells with CREBBP knockdown were co-transfected with expression vector for EGFP-RB1CC1 and mCherry-tagged CREBBP or CREBBP[LD] mutant, as indicated. Quantification of the puncta size (E) and number per cell (F) of RB1CC1 bodies are shown (n > 10 cells per sample) as mean ± SEM from three independent experiments. *P < 0.05; ***P < 0.001. (G and H) MCF7 cells were transfected with expression vectors for Flag-tagged CREBBP or CREBBP[LD] mutant. (G) Cell lysates from these cells cultured under normal or starvation (HBSS) conditions were analyzed by immunoblotting with different antibodies, as indicated. (H) These cells were subjected to autophagy flux assay with Baf-A1 and lysates were analyzed under various conditions, as indicated. (I and L) RB1CC1-null MCF7 cells were transfected with expression vectors for EGFP-tagged RB1CC1 or RB1CC1K276R mutant. (I) Following treatment of cells with 10 μM A485 treatment for 2 h, cell lysates were prepared and analyzed by immunoblotting with different antibodies, as indicated. (J) Cells were subjected to autophagy flux assay with Baf-A1 in the presence or absence of 10 μM A485 treatment for 2 h nd lysates were analyzed under various conditions, as indicated. Panels K and L show quantification of the puncta size (K) and number per cell (L) of RB1CC1 bodies (n > 10 cells per sample) as mean ± SEM from three independent experiments. *P < 0.05.
![Figure 8. Acetylation of K276 in N-terminal IDR of RB1CC1 is important for autophagosome formation. (A and B) MCF7 cells were transfected with expression vector encoding EGFP-RB1CC1 and EGFP-ΔIDR as indicated. (A) Fluorescence intensity recovery of a RB1CC1 body (boxed by dotted lines) after photobleaching. Scale bar: 2 μm. (B) Quantification of FRAP data for images in A. Bleaching event occurs at t = 0 s. Data plotted as means ± SEM (n = 3). (C and D) RB1CC1-null MCF7 cells were transfected with expression vectors for EGFP-tagged RB1CC1 or its mutant ΔIDR. (C) Cell lysates from these cells cultured under normal or starvation (HBSS) conditions were analyzed by immunoblotting with different antibodies, as indicated. (D) These cells were subjected to autophagy flux assay with Baf-A1 and lysates were analyzed under various conditions, as indicated. (E and F) MCF7 cells with CREBBP knockdown were co-transfected with expression vector for EGFP-RB1CC1 and mCherry-tagged CREBBP or CREBBP[LD] mutant, as indicated. Quantification of the puncta size (E) and number per cell (F) of RB1CC1 bodies are shown (n > 10 cells per sample) as mean ± SEM from three independent experiments. *P < 0.05; ***P < 0.001. (G and H) MCF7 cells were transfected with expression vectors for Flag-tagged CREBBP or CREBBP[LD] mutant. (G) Cell lysates from these cells cultured under normal or starvation (HBSS) conditions were analyzed by immunoblotting with different antibodies, as indicated. (H) These cells were subjected to autophagy flux assay with Baf-A1 and lysates were analyzed under various conditions, as indicated. (I and L) RB1CC1-null MCF7 cells were transfected with expression vectors for EGFP-tagged RB1CC1 or RB1CC1K276R mutant. (I) Following treatment of cells with 10 μM A485 treatment for 2 h, cell lysates were prepared and analyzed by immunoblotting with different antibodies, as indicated. (J) Cells were subjected to autophagy flux assay with Baf-A1 in the presence or absence of 10 μM A485 treatment for 2 h nd lysates were analyzed under various conditions, as indicated. Panels K and L show quantification of the puncta size (K) and number per cell (L) of RB1CC1 bodies (n > 10 cells per sample) as mean ± SEM from three independent experiments. *P < 0.05.](/cms/asset/724eca5b-6b37-4822-9c7d-6ed85b808992/kaup_a_2148432_f0008_oc.jpg)
Discussion
As a key component of the autophagy induction complex, RB1CC1 plays essential roles in multiple biological and disease processes, including promoting breast cancer development, progression, and resistance to current immunotherapies [Citation23–31,Citation34]. In this study, we show regulation of RB1CC1 protein stability and its autophagy functions by CREBBP-mediated acetylation of RB1CC1 at K276. We also identify a N-terminal IDR containing K276 that could mediate formation of RB1CC1 puncta with LLPS properties. These findings provide insight into the regulatory mechanism of a key autophagy protein and also suggest potential new ways to modulate autophagy for therapeutic applications.
Protein acetylation is a reversible modification by counteracting acetyltransferases and deacetylases [Citation63]. Previous studies have shown that Sirt1 interacts with several essential components of the autophagy machinery, including autophagy genes ATG5, ATG7, and those encoding Atg8-family proteins [Citation64]. Deacetylation of LC3 by Sirt1 in the nucleus help its redistribution toward the cytoplasm for autophagy initiation under starvation conditions [Citation65]. HDAC6 (histone deacetylase 6) is another deacetylase capable of binding to ubiquitin and implicated in the regulation of autophagy by targeting protein aggregates and damaged mitochondria, as well as controlling the fusion of autophagosomes to lysosomes [Citation66]. It will be interesting to determine whether these or other deacetylases act on RB1CC1 to regulate its stability and autophagy functions.
Whereas histone acetylation is well-established in regulating gene transcription, non-histone protein acetylation by CREBBP or other acetyltransferases could influence a variety of protein functions and activities [Citation67]. Our results suggest that RB1CC1 acetylation at K276 by CREBBP opposed ubiquitination at the same site, leading to reduced degradation of RB1CC1 by ubiquitin-proteosome and increased autophagy function in various breast cancer cells. Although full understanding of the involved molecular mechanisms requires further study, this observation could potentially be exploited to decrease cellular levels of RB1CC1 by inhibiting CREBBP activities. Given the recently identified functions of RB1CC1 independent of its role in the ULK1-ATG13-RB1CC1 complex for the induction of canonical autophagy (e.g., suppressing T-cell infiltration to reduce efficacy of immunotherapy by PD1 blockade [Citation34],), such a strategy potentially would be superior to current approaches using autophagy inhibitors for cancer treatment, as it would abolish both canonical autophagy-dependent and -independent functions of RB1CC1.
Given the well-characterized important role of CREBBP and EP300/p300-mediated histone acetylation in many cancers, several inhibitors for CREBBP-EP300/p300 have been developed as potential new anti-cancer treatments [Citation68,Citation69]. For example, the CREBBP-EP300/p300 bromodomain inhibitor CCS1477, identified by CellCentric, is currently in phase 1b/2a clinical evaluation for the treatment of hematological malignancies and advanced drug-resistant prostate cancer. Our findings raise the interesting possibility that CREBBP inhibitors like CCS1477 could also reduce RB1CC1 levels to contribute to its anti-cancer effects at least in some cancer types, based on previous studies of RB1CC1 in breast and other cancers through both its canonical autophagy-dependent and -independent functions [Citation34]. Nevertheless, more mechanistic understanding of the relative contributions of CREBBP-EP300/p300-mediated acetylation of histones versus non-histone proteins including RB1CC1 to the development, progression, and therapeutic resistance in different cancers will be necessary in better application of inhibitors for cancer treatments. In contrast to the potential benefits of CREBBP-EP300/p300 inhibitors with broad roles affecting both histone and non-histone protein acetylation, such mechanistic understanding may also allow us to design more specific inhibitors targeting a more restricted set of substrates by CREBBP-EP300/p300-mediated acetylation to reduce potential side effects of the inhibitors. In this regard, it is interesting to note that CREBBP, but not the closely related EP300/p300, induced acetylation of K276 of RB1CC1 in our studies. This could provide a model to dissect the potential differences in these two acetyltransferases for future development of more specific inhibitors. Likewise, it will be interesting to determine whether CREBBP may act on other autophagy-related proteins and how this may contribute to cancer development, including those previously shown to be acetylated by EP300/p300 like ATG5 and PIK3C3/VPS34 [Citation42,Citation43].
As in recent studies for other supra-molecular complexes, the PAS forms LLPS in yeast, with Atg13 phosphorylation by TORC1 playing a key role to promote the process [Citation50]. Our findings suggest that RB1CC1 in the ULK1-ATG13-RB1CC1 of mammalian cells might also contribute to LLPS formation, although the relative contributions of these two proteins will require future investigations. One caveat is that our in vitro studies only used a small fragment of RB1CC1 containing the N-terminal IDR (see ). As IDRs can easily undergo LLPS by addition of PEG, these results simply show that attaching IDR to EGFP accelerates its LLPS upon exposure to PEG. Therefore, future studies using full-length RB1CC1 and preferably without PEG will be required to demonstrate that RB1CC1 possesses the activity to undergo LLPS. Nevertheless, our studies in several cells including in penta KO HeLa cells are consistent with the idea that RB1CC1 can form LLPS independent of SQSTM1, CALCOCO2/NDP52, NBR1, TAX1BP1 and OPTN in mammalian cells. Protein acetylation has been shown to alter the phase separation threshold of IDRs [Citation70], therefore, it will also be interesting to determine whether acetylation of RB1CC1 at K276 located in the N-terminal IDR affect its LLPS in vitro and puncta formation in cells. Interestingly, we found that CREBBP inhibitor A485 reduced RB1CC1 puncta formation in breast cancer cells, suggesting a potential effect on LLPS by RB1CC1 possibly in addition to inducing RB1CC1 degradation to contribute to the decreased autophagy in these cells. Collectively, our studies suggest that blocking CREBBP-mediated acetylation of RB1CC1 offers a new therapeutic strategy to induce RB1CC1 degradation with implications for the treatment of breast cancer.
Materials and methods
Antibodies and reagents
Antibodies used in this study were pan acetylated-lysine (AcK; Cell Signaling Technology, 9441), HA (Cell Signaling Technology, 3724), Flag (Cell Signaling Technology, 8146S), MYC (Cell Signaling Technology, 2278S), RB1CC1/FIP200 (Cell Signaling Technology, 12,436), GAPDH (Cell Signaling Technology, 2118), CREBBP/CBP (Cell Signaling Technology, 7389), ACTB/actin (Sigma-Aldrich, A5441), TUBA/alpha-tubulin (Sigma-Aldrich, T9026), H3 (Cell Signaling Technology, 4499), acetyl-histone H3 (Lys27; Cell Signaling Technology, 8173), TP53/p53 (Santa Cruz Biotechnology, sc-126), LC3B (Cell Signaling Technology, 3868) and ubiquitin (Cell Signaling Technology, 3936). Anti-HA-agarose antibody was purchased from Sigma-Aldrich (A2095). Cycloheximide (C7698), A-485 (SML2192), bafilomycin A1 (B1793), MG132 (M7449) and isopropyl-β-D-thiogalactopyranoside (IPTG; I6758) were purchased from Sigma-Aldrich. NAM (S1899) and TSA (S1045) were purchased from Selleck.
Plasmid constructs and shRNA
Plasmids pcDNA3β-FLAG-CREBBP-HA (32,908), pcDNA3β-FLAG-CREBBP[LD]-HA (32,906) were purchased from Addgene (provided by Tso-Pang Yao). Flag-EP300/p300, MYC-KAT2A/GCN5, Flag-KAT2B/PCAF and Flag-KAT8/MOF were kind gifts from L. Cao (China Medical University, Shenyang, China). Plasmids encoding Flag-tagged human KAT5/Tip60 (Flag-KAT5/Tip60), HA-tagged human RB1CC1 (HA-RB1CC1) and EGFP-tagged human RB1CC1 (EGFP-RB1CC1) were constructed using Vazyme ClonExpress II One Step Cloning Kit (Vazyme, C112-01). Plasmids encoding specific point mutations of RB1CC1 and a deletion of its N-terminal IDR (ΔIDR) in these two vectors were constructed using New England Biolabs mutagenesis kit according to manufacturer’s instructions (New England Biolabs, E0552S). shRNAs for CREBBP knockdown were purchased from Sigma-Aldrich and Dharmacon (TRCN0000367481, RHS4430-200,236,268).
Cell culture and transfections
HEK293T, MCF7 and RB1CC1-null MCF7 cells (the parental cells were purchased from ATCC [HTB-22], and the knockout cells was produced in our lab) were cultured in high-glucose Dulbecco’s modified Eagle’s medium (DMEM; Gibco, 11,995–065), supplemented with 10% fetal bovine serum (Gibco, A3160602), penicillin (100 U), and streptomycin (100 μg/ml). BT474 and SKBR3 cells (ATCC) were cultured in RPMI 1640 medium (Gibco, 11,875–119) with the same supplements. RB1CC1-null MCF7 cells were generated by CRISPR-Cas9-mediated knockout of RB1CC1 in MCF7 cells and described previously [Citation71]. HeLa penta KO cells were provided as a kind gift by Dr. Richard Youle at NIH and were maintained as described previously [Citation62]. Plasmids were transfected into various cells at 60% confluence using Lipofectamine 3000 reagent (Thermo Fisher Scientific, L3000015) according to the manufacturer’s instructions. Cells were harvested at 24–72 h post transfection for different analyses. For autophagy flux assays, cells were starved in EBSS (Gibco, 24,010–043) for 6 h with or without Baf-A1for the last 4 h before lysis for western blotting with various antibodies.
Immunoprecipitation and immunoblotting
Cells were washed with PBS buffer (Gibco, 14,190–250), and lysed by RIPA buffer (Sigma-Aldrich, C2978) with Halt Protease Inhibitor Cocktail (Thermo Fisher Scientific, PI78425). Protein concentrations were determined using BCA assay (Thermo Fisher Scientific, 23,225). For immunoprecipitation analysis, cell lysates were mixed with anti-HA affinity beads (Sigma-Aldrich, A2095) at 4°C overnight or incubated with other appropriate antibodies followed by the protein A/G-Agarose beads (Santa Cruz Biotechnology, sc-2002) overnight at 4°C. The protein-antibody complexes were collected by centrifugation, and then washed three times with cold lysis buffer at 4°C. The samples were boiled in 2 × loading buffer for 10 min, and the supernatants were subjected to SDS-PAGE. Immunoblotting was performed as previously described [Citation72]. In experiments for the detection of ubiquitinated RB1CC1, cells were lysed in denaturing lysis buffer (1% SDS, 50 mM Tris, pH 7.5, 0.5 mM EDTA, 1 mM DTT) and boiled for 5 min. They were then diluted 10-fold in Tris-HCl buffer (pH 7.5) and used for immunoprecipitation by anti-HA affinity beads for HA-tagged RB1CC1, as outlined above.
Mass spectrometry analysis
For mass spectrometry analysis, 10-cm dishes of HEK293T cells were co-transfected with expression vectors encoding HA-RB1CC1 and Flag-CREBBP or Flag vector control. 36 h after transfection, cells were lysed with lysis buffer (137 mM NaCl, 50 mM tris-HCl, pH 7.6, 10% glycerol, 10 mM NaF, 1 mM EDTA, 0.1 mM Na3VO4, 1% NP-40 [Fluka, 74,385]) containing 5 μM TSA and 20 mM NAM. Cell lysates were immunoprecipitated with the anti-HA affinity beads overnight at 4°C. The immunoprecipitate was resolved by 10% SDS-PAGE and acetylated protein fragments were detected by mass spectrometry analysis, as described previously [Citation72,Citation73].
Recombinant protein expression and purification
cDNA encoding the N-terminal IDR of RB1CC1 was inserted into a modified version of pET28a-EGFP-6× HIS (Addgene, 61,603; deposited by Maarten Merkx) to generate an expression vector encoding recombinant IDR with both EGFP and His tags (designated as EGFP-IDR). This plasmid or the vector control were transformed in E. coli BL21 (DE3) cells and cultured with 1 mM IPTG (Sigma, I6758) at 18°C overnight. Bacterial cells were collected and stored frozen at −80°C. To prepare for the recombinant proteins, bacterial cells were re-suspended in Buffer A (50 mM Tris-HCl, pH 7.5, 500 mM NaCl) containing 10 mM imidazole, protease inhibitors cocktail (Thermo Fisher Scientific) for 30 min on ice, followed by sonication. The lysate was centrifuged at 12,000 g for 30 min at 4°C and was incubated with Ni-NTA agarose (Invitrogen, R901-15) pre-equilibrated with 10 × volumes of Buffer A at 4°C for 1.5 h. The Ni-NTA agarose was packed in a column and washed with 15 volumes of Buffer A containing 10 mM imidazole. Recombinant protein was eluted with Buffer A containing different concentrations of imidazole. Fractions containing EGFP-IDR recombinant proteins as determined by SDS-PAGE followed by Coomassie blue staining were combined and dialyzed against Buffer D (500 mM NaCl, 50 mM Tris-HCl, pH 7.5, 10% glycerol, 1 mM DTT). The purity of the protein was verified by SDS-PAGE and Coomassie blue staining.
In vitro droplet assay
Recombinant EGFP-IDR or EGFP control proteins were concentrated and desalted to an appropriate protein concentration and 125 mM NaCl using Amicon Ultra centrifugal filters (50 K or 10 K MWCO; Millipore, UFC901024). The samples were then added to solutions at varying concentrations with indicated NaCl and molecular crowder concentrations (10% PEG-8000 [Sigma, 25,322–68-3] or 10% Ficoll-400 [Millipore, 26,873–85-8]) in Buffer D (50 mM Tris-HCl, pH 7.5, 10% glycerol, 1 mM DTT) as indicated in the figure legends. For droplet FRAP and time-lapse imaging experiments, phase separation assay was carried out on glass-bottomed 35 mm dishes. The droplets were observed using on a Zeiss LSM 710 confocal laser scanning microscope with a 60 × oil immersion objective.
Immunofluorescence assay
Cells were washed three times with PBS and then fixed with ice-cold 4% paraformaldehyde for 15 min and permeabilized with 0.5% Triton X-100 (Sigma, 9036–19-5) in PBS for 15 min before blocking with 2% BSA (Thermo Fisher Scientific, BP9703) for 1 h at room temperature. Then appropriate primary antibodies were incubated at 4°C overnight. After 3 washes with PBS, cells were incubated with FITC or Texas red–conjugated secondary antibodies (Jackson, 111–075-003, 115–097-003) for 1 h at room temperature. Cells were washed 3 times with PBS and stained with DAPI at room temperature for 15 min. All images were generated on Zeiss LSM 710 confocal laser scanning microscope.
Fluorescence recovery after photobleaching (FRAP)
FRAP experiments were performed on a Zeiss LSM 710 confocal laser scanning microscope with a 60 × oil immersion objective. RB1CC1 bodies (in vivo) or EGFP-IDR droplets (in vitro) were bleached using 100% laser power at 480 nm. Recovery was recorded for the indicated time. Background intensity was subtracted and fluorescence intensity was measured using Fiji Is Just ImageJ (FIJI).
In vitro ubiquitination assay for recombinant RB1CC1-IDR
MCF7 cells were resuspended in cold hypotonic buffer (1 M Tris-HCl, pH 7.4, 2.5 M KCl, 1 M MgCl2) then centrifuge at 4°C. Repeat the above process twice and break up the cell membrane using a Dounce homogenizer (Bellco, 1984–10,002). Check the cells under the microscope after trypan blue staining. Transfer the fragmented cells to a new tube followed by centrifugation at 3000 g, 15 min. Carefully remove the top layer and transfer the supernatant into a new tube. The cytoplasm extracts were tested on SDS-PAGE gels using cytoplasm marker antibodies. For droplet ubiquitination assay within the in vitro acetylation reaction, 40 μg of the HA-tagged RB1CC1-IDR recombinant protein and MCF7 cell cytoplasm extracts was incubated with 2.5 μg of human recombinant CREBBP catalytic domain (Enzo, BML-SE452) and 2 mM acetyl-CoA (Sigma-Aldrich, A2056) for 3 h at 37°C. The resulting mixture was analyzed by western blotting by anti-HA to detect ubiquitinated RB1CC1-IDR protein.
Supplemental Material
Download MS Word (767.6 KB)Acknowledgments
We would like to thank Dr. Liu Cao of China Medical University, Shenyang, China for Flag-EP300/p300, Myc-KAT2A/GCN5, Flag-KAT2B/PCAF and Flag-KAT8/MOF plasmids, Dr. Ken Greis and the UC Proteomics Laboratory for mass spectrometry data collection and analyses, Dr. Richard Youle at NIH for the kind gift of HeLa penta KO cells. We appreciate the help from Lindsey Huether for graphics support and preparation of figures. We are grateful to members of the Guan lab for their critical comments and suggestions in the preparation of this manuscript. The work in this study is funded by NIH grants R01CA163493, R01NS094144 and R01HL073394 to J.-L. Guan.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/15548627.2022.2148432
Additional information
Funding
References
- Mizushima N. Autophagy: process and function. Genes Dev. 2007;21(22):2861–2873.
- Klionsky DJ. Autophagy: from phenomenology to molecular understanding in less than a decade. Nat Rev Mol Cell Biol. 2007;8(11):931–937.
- Yang Z, Klionsky DJ. Eaten alive: a history of macroautophagy. Nat Cell Biol. 2010;12(9):814–822.
- Mizushima N, Levine B. Autophagy in mammalian development and differentiation. Nat Cell Biol. 2010;12(9):823–830.
- Mizushima N, Komatsu M. Autophagy: renovation of Cells and Tissues. Cell. 2011;147(4):728–741.
- Rubinsztein DC, Marino G, Kroemer G. Autophagy and aging. Cell. 2011;146(5):682–695.
- White E. Deconvoluting the context-dependent role for autophagy in cancer. Nat Rev Cancer. 2012;12(6):401–410.
- Kenific CM, Debnath J. Cellular and metabolic functions for autophagy in cancer cells. Trends Cell Biol. 2015;25(1):37–45.
- White E. The role for autophagy in cancer. J Clin Invest. 2015;125(1):42–46.
- Mizushima N. A brief history of autophagy from cell biology to physiology and disease. Nat Cell Biol. 2018;20(5):521–527.
- Feng Y, He D, Yao Z, et al. The machinery of macroautophagy. Cell Res. 2014;24(1):24–41.
- Lamb CA, Dooley HC, Tooze SA. Endocytosis and autophagy: shared machinery for degradation. Bioessays. 2013;35(1):34–45.
- Hara T, Takamura A, Kishi C, et al. FIP200, a ULK-interacting protein, is required for autophagosome formation in mammalian cells. J Cell Biol. 2008;181(3):497–510.
- Chen S, Wang C, Yeo S, et al. Distinct roles of autophagy-dependent and -independent functions of FIP200 revealed by generation and analysis of a mutant knock-in mouse model. Genes Dev. 2016;30(7):856–869.
- Ganley IG, Lam DH, Wang J, et al. ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J Biol Chem. 2009;284(18):12297–12305.
- Jung CH, Jun CB, Ro S-H, et al. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol Biol Cell. 2009;20(7):1992–2003.
- Ravenhill BJ, Boyle KB, von Muhlinen N, et al. The cargo receptor NDP52 initiates selective autophagy by recruiting the ULK complex to cytosol-invading bacteria. Mol Cell. 2019;74(2):320–329.e6.
- Vargas JNS, Wang C, Bunker E, et al. Spatiotemporal control of ULK1 activation by NDP52 and TBK1 during selective autophagy. Mol Cell. 2019;74(2):347–362.e6.
- Ohnstad AE, Delgado JM, North BJ, et al. Receptor-mediated clustering of FIP200 bypasses the role of LC3 lipidation in autophagy. EMBO J. 2020;39(24):e104948.
- Turco E, Savova A, Gere F, et al. Reconstitution defines the roles of p62, NBR1 and TAX1BP1 in ubiquitin condensate formation and autophagy initiation. Nat Commun. 2021;12(1):5212.
- Smith MD, Harley ME, Kemp AJ, et al. CCPG1 is a non-canonical autophagy cargo receptor essential for ER-phagy and pancreatic ER proteostasis. Dev Cell. 2018;44(2):217–232 e11.
- Zhou Z, Liu J, Fu T, et al. Phosphorylation regulates the binding of autophagy receptors to FIP200 Claw domain for selective autophagy initiation. Nat Commun. 2021;12(1):1570.
- Gan B, Peng X, Nagy T., et al. Role of FIP200 in cardiac and liver development and its regulation of TNFalpha and TSC-mTOR signaling pathways. J Cell Biol. 2006;175(1):121–133.
- Wei H, Gan B, Wu X, et al. Inactivation of FIP200 leads to inflammatory skin disorder, but not tumorigenesis, in conditional knock-out mouse models. J Biol Chem. 2009;284(9):6004–6013.
- Wei H, Wei S, Gan B, et al. Suppression of autophagy by FIP200 deletion inhibits mammary tumorigenesis. Genes Dev. 2011;25(14):1510–1527.
- Liang CC, Wang C, Peng X, et al. Neural-specific deletion of FIP200 leads to cerebellar degeneration caused by increased neuronal death and axon degeneration. J Biol Chem. 2010;285(5):3499–3509.
- Liu F, Lee JY, Wei H., et al. FIP200 is required for the cell-autonomous maintenance of fetal hematopoietic stem cells. Blood. 2010;116(23):4806–4814.
- Wang C, Liang -C-C, Bian ZC, et al. FIP200 is required for maintenance and differentiation of postnatal neural stem cells. Nat Neurosci. 2013;16(5):532–542.
- Liu F, Fang F, Yuan H, et al. Suppression of autophagy by FIP200 deletion leads to osteopenia in mice through the inhibition of osteoblast terminal differentiation. J Bone Miner Res. 2013;28(11):2414–2430.
- Wei H, Wang C, Croce CM, et al. p62/SQSTM1 synergizes with autophagy for tumor growth in vivo. Genes Dev. 2014;28(11):1204–1216.
- Wang C, Haas MA, Yang F, et al. Autophagic lipid metabolism sustains mTORC1 activity in TSC-deficient neural stem cells. Nat Metab. 2019;1(11):1127–1140.
- Wang C, Chen S, Yeo S, et al. Elevated p62/SQSTM1 determines the fate of autophagy-deficient neural stem cells by increasing superoxide. J Cell Biol. 2016;212(5):545–560.
- Liu H, Wang C, Yi F, et al. Non-canonical function of FIP200 is required for neural stem cell maintenance and differentiation by limiting TBK1 activation and p62 aggregate formation. Sci Rep. 2021;11(1):23907.
- Okamoto T, Yeo SK, Hao M, et al. FIP200 suppresses immune checkpoint therapy responses in breast cancers by limiting AZI2/TBK1/IRF signaling independent of its canonical autophagy function. Cancer Res. 2020;80(17):3580–3592.
- Wang C, Yeo S, Haas MA, et al. Autophagy gene FIP200 in neural progenitors non-cell autonomously controls differentiation by regulating microglia. J Cell Biol. 2017;216(8):2581–2596.
- Dancy BM, Cole PA. Protein lysine acetylation by p300/CBP. Chem Rev. 2015;115(6):2419–2452.
- Parker SC, Stitzel ML, Taylor DL, et al. Chromatin stretch enhancer states drive cell-specific gene regulation and harbor human disease risk variants. Proc Natl Acad Sci U S A. 2013;110(44):17921–17926.
- Hnisz D, Abraham B, Lee T, et al. Super-enhancers in the control of cell identity and disease. Cell. 2013;155(4):934–947.
- Gu W, Roeder RG. Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell. 1997;90(4):595–606.
- Wang Z, Yang X, Liu C, et al. Acetylation of PHF5A modulates stress responses and colorectal carcinogenesis through alternative splicing-mediated upregulation of KDM3A. Mol Cell. 2019;74(6):1250–1263 e6.
- Dai X, et al. Acetylation-dependent regulation of BRAF oncogenic function. Cell Rep. 2022;38(3):110250.
- Lee IH, Finkel T. Regulation of autophagy by the p300 acetyltransferase. J Biol Chem. 2009;284(10):6322–6328.
- Su H, Yang F, Wang Q, et al. VPS34 acetylation controls its lipid kinase activity and the initiation of canonical and non-canonical autophagy. Mol Cell. 2017;67(6):907–921 e7.
- Sun T, Li X, Zhang P, et al. Acetylation of beclin 1 inhibits autophagosome maturation and promotes tumour growth. Nat Commun. 2015;6:7215.
- Hyman AA, Weber CA, Julicher F. Liquid-liquid phase separation in biology. Annu Rev Cell Dev Biol. 2014;30(1):39–58.
- Banani SF, Lee HO, Hyman AA, et al. Biomolecular condensates: organizers of cellular biochemistry. Nat Rev Mol Cell Biol. 2017;18(5):285–298.
- Shin Y, Brangwynne CP. Liquid phase condensation in cell physiology and disease. Science. 2017;357(6357):6357.
- Brangwynne CP, Eckmann CR, Courson DS, et al. Germline P granules are liquid droplets that localize by controlled dissolution/condensation. Science. 2009;324(5935):1729–1732.
- Tompa P. Intrinsically disordered proteins: a 10-year recap. Trends Biochem Sci. 2012;37(12):509–516.
- Fujioka Y, Alam JM, Noshiro D, et al. Phase separation organizes the site of autophagosome formation. Nature. 2020;578(7794):301–305.
- Avalos JL, Bever KM, Wolberger C. Mechanism of sirtuin inhibition by nicotinamide: altering the NAD(+) cosubstrate specificity of a Sir2 enzyme. Mol Cell. 2005;17(6):855–868.
- Furumai R, Komatsu Y, Nishino N, et al. Potent histone deacetylase inhibitors built from trichostatin A and cyclic tetrapeptide antibiotics including trapoxin. Proc Natl Acad Sci U S A. 2001;98(1):87–92.
- Varshavsky A. The ubiquitin system, autophagy, and regulated protein degradation. Annu Rev Biochem. 2017;86(1):123–128.
- Gan B, Guan JL. FIP200, a key signaling node to coordinately regulate various cellular processes. Cell Signal. 2008;20(5):787–794.
- Elbaum-Garfinkle S, Kim Y, Szczepaniak K, et al. The disordered P granule protein LAF-1 drives phase separation into droplets with tunable viscosity and dynamics. Proc Natl Acad Sci U S A. 2015;112(23):7189–7194.
- Nott TJ, Petsalaki E, Farber P, et al. Phase transition of a disordered nuage protein generates environmentally responsive membraneless organelles. Mol Cell. 2015;57(5):936–947.
- Li P, Banjade S, Cheng H-C, et al. Phase transitions in the assembly of multivalent signalling proteins. Nature. 2012;483(7389):336–340.
- Patel A, Lee H, Jawerth L, et al. A liquid-to-solid phase transition of the ALS protein FUS accelerated by disease mutation. Cell. 2015;162(5):1066–1077.
- Pak CW, et al. Sequence determinants of intracellular phase separation by complex coacervation of a disordered protein. Mol Cell. 2016;63(1):72–85.
- Sabari BR, Dall’Agnese A, Boija A, et al. Coactivator condensation at super-enhancers links phase separation and gene control. Science. 2018;361(6400):6400.
- Turco E, et al. FIP200 claw domain binding to p62 promotes autophagosome formation at ubiquitin condensates. Mol Cell. 2019;74(2):330–346 e11.
- Lazarou M, Sliter DA, Kane LA, et al. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature. 2015;524(7565):309–314.
- Drazic A, Myklebust LM, Ree R, et al. The world of protein acetylation. Biochim Biophys Acta. 2016;1864(10):1372–1401.
- Lee IH, Cao L, Mostoslavsky R, et al. A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc Natl Acad Sci U S A. 2008;105(9):3374–3379.
- Huang R, Xu Y, Wan W, et al. Deacetylation of nuclear LC3 drives autophagy initiation under starvation. Mol Cell. 2015;57(3):456–466.
- Lee JY, Koga H, Kawaguchi Y, et al. HDAC6 controls autophagosome maturation essential for ubiquitin-selective quality-control autophagy. EMBO J. 2010;29(5):969–980.
- Narita T, Weinert BT, Choudhary C. Functions and mechanisms of non-histone protein acetylation. Nat Rev Mol Cell Biol. 2019;20(3):156–174.
- Lasko LM, Jakob CG, Edalji RP, et al. Discovery of a selective catalytic p300/CBP inhibitor that targets lineage-specific tumours. Nature. 2017;550(7674):128–132.
- Welti J, Sharp A, Brooks N, et al. Targeting the p300/CBP axis in lethal prostate cancer. Cancer Discov. 2021;11(5):1118–1137.
- Saito M, Hess D, Eglinger J, et al. Acetylation of intrinsically disordered regions regulates phase separation. Nat Chem Biol. 2019;15(1):51–61.
- Hao M, Yeo SK, Turner K, et al. Autophagy blockade limits HER2+ breast cancer tumorigenesis by perturbing HER2 trafficking and promoting release via small extracellular vesicles. Dev Cell. 2021;56(3):341–355 e5.
- Yi F, Zhang Y, Wang Z., et al. The deacetylation-phosphorylation regulation of SIRT2-SMC1A axis as a mechanism of antimitotic catastrophe in early tumorigenesis. Sci Adv. 2021;7(9). DOI:10.1126/sciadv.abe5518.
- Chutipongtanate S, Greis KD. Multiplex biomarker screening assay for urinary extracellular vesicles study: a targeted label-free proteomic approach. Sci Rep. 2018;8(1):15039.