
ABSTRACT
Chemotherapy is an important treatment modality for osteosarcoma (OS), but the development of chemoresistance limits the therapeutic efficacy of OS and results in a poor prognosis. Thus, a better understanding of the mechanisms underlying chemoresistance in OS is essential. We previously demonstrated that COPS3/CSN3 (COP9 signalosome subunit 3) functions as an oncogene to promote OS cells lung metastasis, which is closely related to chemoresistance. Here, we showed that COPS3 was significantly upregulated in OS tissues with poor response to preoperative chemotherapy. Moreover, COPS3 depletion made OS cells more sensitive to cisplatin treatment in vitro and in vivo, implicating COPS3 as a driver of cisplatin resistance. Mechanistic investigations showed that COPS3 induced a cytoprotective macroautophagy/autophagy in response to cisplatin. Specifically, we identified FOXO3 as a critical target of COPS3, as high expression of COPS3 enhanced the nuclear abundance of FOXO3 and increased the expression of FOXO3-responsive genes, promoting autophagosome formation and maturation. In turn, FOXO3 regulated COPS3 levels by inhibiting ubiquitin-mediated degradation and attenuating SKP2-mediated COPS3 inhibition, cooperatively maintaining a high level of COPS3. In both COPS3-expressing OS cells and a murine xenograft model, inhibition of autophagy could also overcome resistance to cisplatin. Collectively, our results offer insights into the mechanisms of cisplatin resistance and suggest that targeting COPS3-mediated autophagy is a promising therapeutic strategy for overcoming the cisplatin resistance of OS.
Abbreviations: 3-MA: 3-methyladenine; BECN1: beclin 1; ChIP: chromatin immunoprecipitation; CHX: cycloheximide; COPS3/CSN3: COP9 signalosome subunit 3; CQ: chloroquine; DEGs: differentially expressed genes; FOXO3: forkhead box O3; GFP: green fluorescent protein; IC50: 50% inhibitory concentration; LAMP1: lysosomal associated membrane protein 1; MAP1LC3B/LC3B: microtubule associated protein 1 light chain 3 beta; MTOR: mechanistic target of rapamycin kinase; mRFP: monomeric red fluorescent protein; OS: osteosarcoma; PBS: phosphate-buffered saline; qRT-PCR: quantitative real-time PCR; RAB7: RAB7, member RAS oncogene family; RPS6KB1/p70S6K1: ribosomal protein S6 kinase B1; SEM: standard error of the mean; shRNA: short hairpin RNA; siRNA: small interfering RNA; SKP2: S-phase kinase associated protein 2; TEM: transmission electron microscopy; UPS: ubiquitin-proteasome system
Introduction
Osteosarcoma (OS) is the most common primary malignant tumor of the bone and primarily arises in children and adolescents, accounting for approximately 20% of all bone tumors and approximately 5% of pediatric tumors [Citation1]. Although curative surgery is the mainstay of OS treatment, the overall survival rate is only approximately 15–17% [Citation2]. In the early 1970s, chemotherapy was introduced as an adjuvant treatment [Citation3]. It was found that the histological response to preoperative chemotherapy is an important prognostic factor in OS [Citation4]. At present, the universal standard for evaluating the preoperative chemotherapy response is based on the Huvos grading system, where grade I denotes tumor necrosis of less than 50%; grade II, 50% to 89% tumor necrosis; grade III, 90% to 99% tumor necrosis; and grade IV, 100% tumor necrosis. Grades III and IV (≥ 90% necrosis) are considered to have a good response to chemotherapy, while grades I and II (< 90% necrosis) are considered to have a poor response to chemotherapy [Citation5]. Overall, the combination of surgical resection and systemic chemotherapy (cisplatin, doxorubicin, and high-dose methotrexate) resulted in a substantial improvement in the survival rate of patients with non-metastatic disease, with a survival rate of approximately 70% [Citation6]. However, for patients with metastatic or relapsed OS, survival has remained largely unchanged over the past 30 years, with an overall survival rate of approximately 20% [Citation7,Citation8]. Tumor metastasis is strongly associated with resistance to chemotherapy. However, several studies have shown that increasing the dose of first-line therapy or applying second-line chemotherapy in patients with a poor response to chemotherapy has very limited efficacy [Citation9,Citation10]; therefore, exploring the underlying mechanisms involved in OS chemoresistance is warranted.
OS is characterized by remarkable genomic instability and high heterogeneity. It is reported that several mutated tumor suppressor genes or altered candidate driver genes are associated with autophagy regulation, indicating that autophagy is closely related to the occurrence and progression of OS [Citation11–14]. Autophagy is an evolutionarily conserved catabolic process and can be induced by different signals and cellular stresses [Citation15]. In addition to the role of autophagy in OS proliferation and metastasis, several studies have demonstrated its role in chemoresistance [Citation16,Citation17]. Silencing of autophagy-related genes or applying the autophagy inhibitor chloroquine (CQ) enables OS cells to be more sensitive to chemotherapy. However, autophagy may also have an anti-tumor role in OS and lead to cell death [Citation18]. Given the context-dependent property of autophagy and the complexity of the OS genome, it is necessary to define the context of OS or identify new biomarkers that will be helpful in the therapeutic targeting of autophagy.
The COP9 signalosome is a protein complex consisting of eight to nine subunits (GPS1/CSN1/COPS1, COPS2 to COPS6, COPS7A, COPS7B and COPS8) and is found in plants and animals [Citation19–21]. A well-known function of the COP9 signalosome is the regulation of the catalytic dynamics of CUL (cullin)-based RING ligases (CRLs) and the coordination of CRL-mediated ubiquitination by utilizing its CUL/cullin deneddylation activity [Citation22,Citation23]. In addition, the COP9 signalosome has an emerging role in cancer. For example, COPS5 and COPS6 have been reported to be overexpressed in multiple tumors [Citation24]; COPS2 overexpression is associated with vascular endothelial growth factor production [Citation25]. Furthermore, low expression of COPS3 is associated with better prognosis in patients bearing tumors with a RAS signature [Citation26]. Moreover, several studies, including ours, have shown that COPS3 is highly amplified in OS, and we further demonstrated that COPS3 significantly correlates with the poor outcome of OS patients [Citation27–29].
In a recent study, we confirmed that COPS3 promotes OS cells lung metastasis [Citation30]. The occurrence of lung metastasis is partly due to resistance to chemotherapy. In the present study, we found that COPS3 was significantly upregulated in chemoresistant OS samples and that the depletion of COPS3 enabled OS cells to be more sensitive to cisplatin, indicating that COPS3 was a potential driver of cisplatin resistance. Mechanistically, we identified COPS3 as a regulator to induce cytoprotective autophagy against cisplatin. Here we reported that FOXO3 (forkhead box O3), a master transcription factor that regulates several autophagy genes, was a key target of COPS3. Our data also showed that COPS3 and FOXO3 formed a positive feedback loop, whereby COPS3 enhanced the nuclear abundance of FOXO3 and augmented its transcriptional activity. FOXO3 in turn maintained COPS3 levels through transcriptional regulation and post-translational modification, thereby inducing FOXO3-responsive gene expression and autophagy activation. Through both in vitro and in vivo experiments, we further validated that the inhibition of autophagy could prevent the resistance to cisplatin induced by COPS3. Together, our study offers insights into the mechanisms of cisplatin resistance in OS and provides a potential therapeutic opportunity to reverse cisplatin resistance.
Results
COPS3 expression reduces the efficacy of cisplatin in osteosarcoma cells
We have previously demonstrated that the oncogene COPS3 mediates the lung metastasis of OS cells in vitro and in vivo [Citation30]. The occurrence of lung metastasis is often accompanied by resistance to chemotherapy; therefore, we examined COPS3 expression in samples with different responses to preoperative chemotherapy. We found that the proportion of COPS3-positive cells and staining intensity in chemoresistant samples were significantly higher than that in chemoresponsive samples (), indicating that the abundance of COPS3 may be associated with chemoresistance. Cisplatin is one of the conventional drugs used in osteosarcoma. To determine whether COPS3 affects the anti-tumor efficacy of cisplatin, we constructed COPS3 knockdown cell models by transfecting COPS3 short hairpin RNA (shRNA) in 143B cells and treating cells with cisplatin at different concentrations for 24 h. The cell viability of COPS3 knockdown cells was significantly lower than that of control. We also overexpressed COPS3 in COPS3 knockdown cells and found that overexpression of COPS3 could rescue the decrease of cell viability caused by the knockdown of COPS3 (). Next, we transfected Saos-2 cells with lentivirus vector-expressing COPS3 or control lentivirus vector and examined cell viability in the two groups treated with cisplatin. We found that the cell viability of cells with high expression of COPS3 was higher than that of the control group. Simultaneously, we knocked down COPS3 in cells with overexpression of COPS3 and found that the cell viability decreased significantly after inhibition of COPS3 expression (). To further verify whether COPS3 influences the anti-tumor effects of cisplatin in vivo, we constructed an animal model by subcutaneous injection of COPS3 knockdown and control cells tagged with green fluorescent protein (GFP). The results showed that COPS3 knockdown rendered OS cells more sensitive to cisplatin treatment (), as evidenced by the reduced tumor volume () and tumor weight (). Collectively, these data suggest that COPS3 endows OS cells with cisplatin resistance, and COPS3 inhibition renders cells more sensitive to cisplatin.
Figure 1. COPS3 promotes cisplatin resistance in osteosarcoma. (A) Immunohistochemical images of COPS3 expression in human osteosarcoma tissues with different responses to preoperative chemotherapy (tumor necrosis ≥90%: chemosensitivity; tumor necrosis <90%: chemoresistance). Data are presented as mean ± SEM. *** p < 0.001. (B) 143B cells were transfected with the indicated vectors to construct cells with different COPS3 expression levels. The cells were treated with different concentrations of cisplatin for 24 h. CCK-8 assays were performed to examine cell viability. Data are presented as mean ± SEM of triplicates. *p < 0.05, *** p < 0.001, **** p < 0.0001. (C) Saos-2 cells were transfected with the indicated vectors to construct cells with different COPS3 expression levels. The cells were treated with different concentrations of cisplatin for 24 h. CCK-8 assays were performed to examine cell viability. Data are presented as mean ± SEM of triplicates. *p < 0.05, **p < 0.01,*** p < 0.001. (D-F) BALB/c nude mice (n = 4 mice per group) were injected with 143B cells that were transfected with either GFP-control or GFP-COPS3 shRNA. (D) Tumors excised from the different groups are shown. (E) Tumor growth curves of the different groups. The arrow indicates the time to start cisplatin administration. (F) Weight of the excised tumors in each group. Data are presented as mean ± SEM. *p < 0.05.
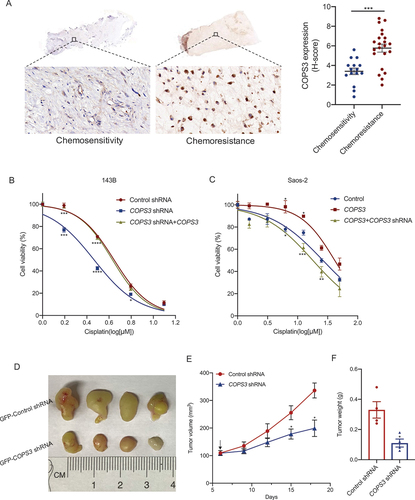
COPS3-induced autophagy is amplified in response to cisplatin
Autophagy is generally considered a cytoprotective mechanism against chemotherapy [Citation31,Citation32]. In a previous study, we noted the role of COPS3 in the regulation of autophagy [Citation30]. Therefore, we aimed to determine whether autophagy plays a role in cisplatin resistance, which is associated with COPS3 expression. Based on the cell viability curves mentioned above, we selected an optimal dose (3 μM for 143B, 5 μM for Saos-2) and time (24 h) of cisplatin treatment in subsequent studies. The expression of autophagic markers in the absence or presence of cisplatin treatment was examined, and further analyses were performed to evaluate the role of COPS3 in the induction of autophagy. There was basic autophagy activity in 143B cells, which themselves have high COPS3 expression without cisplatin treatment. After treatment with cisplatin, autophagy levels were increased in the cells. Further, BECN1 (beclin 1) expression and the conversion of MAP1LC3B/LC3B (microtubule associated protein 1 light chain 3 beta)-I to LC3B-II were significantly lower in COPS3 knockout cells than in the control cells ( and Fig. S1A-C), indicating that COPS3 is essential for autophagy activation. Analysis of the effect of COPS3 overexpression on autophagy activation showed that cells with high COPS3 expression had the highest levels of autophagy following cisplatin treatment ( and Fig. S1D). In addition, 143B and Saos-2 cells were infected with a GFP-monomeric red fluorescent protein (mRFP)-LC3 vector in which autophagosomes were represented by yellow puncta (GFP+ and mRFP+), while free red puncta (GFP− and mRFP+) indicated autolysosomes. Overall, the number of LC3 puncta in control 143B cells treated with cisplatin was higher than that in the absence of cisplatin. Further, the formation of LC3 puncta, especially the yellow puncta, was notably reduced in COPS3 knockout cells when compared with the control under the same conditions of cisplatin treatment (). Further, the treatment of cisplatin significantly increased LC3 puncta formation in Saos-2 cells overexpressing the COPS3 gene. However, there was no obvious change in control Saos-2 cells with low expression of COPS3 in the absence or presence of cisplatin treatment (), indicating that COPS3 is essential for cisplatin-stimulated autophagy activation. Transmission electron microscopy (TEM) further showed a low level of COPS3-induced autophagy in control 143B cells without cisplatin treatment because COPS3 knockdown diminished the formation of autophagic structures. Furthermore, few autophagic features were observed in control Saos-2 cells without cisplatin treatment, but after the cells were transfected with lentivirus vector-expressing COPS3, the autophagic structures were easily detected (). Importantly, this indicated that under normal survival conditions, there was a basic level of COPS3-mediated autophagy. While cisplatin treatment can increase the effect of COPS3-activated autophagy, as evidenced by an apparent increase in the number of autophagic vacuoles in high COPS3 expression cells following cisplatin treatment.
Figure 2. COPS3 is required for autophagy activation. (A) Western blotting analysis of BECN1 and LC3B in 143B cells transfected with COPS3 siRNAs with or without cisplatin treatment for 24 h (3 μM). (B) Western blotting analysis of BECN1 and LC3B in Saos-2 cells transfected with COPS3 overexpression vector with or without cisplatin treatment for 24 h (5 μM). (C) Representative confocal images of GFP-LC3 and mRFP-LC3 distribution in 143B cells transfected with COPS3 shRNA vector, with or without cisplatin treatment for 24 h (3 μM). (D) Representative confocal images of GFP-LC3 and mRFP-LC3 distribution in Saos-2 cells transfected with COPS3 overexpression vector with or without cisplatin treatment for 24 h (5 μM). (E) Representative images of the autophagic structures (red arrows) of 143B cells transfected with COPS3 shRNA vector with or without cisplatin treatment for 24 h (3 μM). The number of autophagic vacuoles per area was quantified (n = 5). Data are presented as mean ± SEM. *p < 0.05, **p < 0.01, *** p < 0.001. (F) Representative images of the autophagic structures (red arrows) of Saos-2 cells transfected with COPS3 overexpression vector with or without cisplatin treatment for 24 h (5 μM). The number of autophagic vacuoles per area was quantified (n = 5). Data are presented as mean ± SEM. *p < 0.05, **** p < 0.0001. (G) Western blotting analysis of changes induced by COPS3 silencing with siRNAs on MTOR, phosphorylated MTOR, RPS6KB1, and phosphorylated RPS6KB1 levels in 143B cells with cisplatin treatment for 24 h (3 μM). (H) Western blotting analysis of changes induced by COPS3 overexpression on MTOR, phosphorylated MTOR, RPS6KB1, and phosphorylated RPS6KB1 levels in Saos-2 cells with cisplatin treatment for 24 h (5 μM).
LC3, an autophagosome marker, is essential for autophagosome formation, which is an early stage of autophagy [Citation33]. Therefore, we concluded that COPS3 may be involved in the initial stage of autophagy following cisplatin treatment. The MTOR (mechanistic target of rapamycin kinase) pathway is a major negative regulator of autophagy at an early stage, and MTOR activation impairs autophagy [Citation34]. Thus, we investigated whether COPS3-mediated autophagy occurred via inhibition of the MTOR pathway. COPS3 downregulation resulted in increased levels of total MTOR, phosphorylated MTOR, and phosphorylated RPS6KB1/p70S6K1 (ribosomal protein S6 kinase B1) with cisplatin treatment in 143B cells ( and Fig. S1E-G). Similarly, COPS3 upregulation in Saos-2 cells inhibited MTOR pathway activity ( and Fig. S1H). Collectively, the data indicate that under normal conditions, OS cells have a low level of COPS3-dependent autophagy. However, after cisplatin treatment, COPS3-induced autophagy, which is in its initial stage, is amplified.
COPS3 promotes autophagosome and lysosome fusion by induction of RAB7
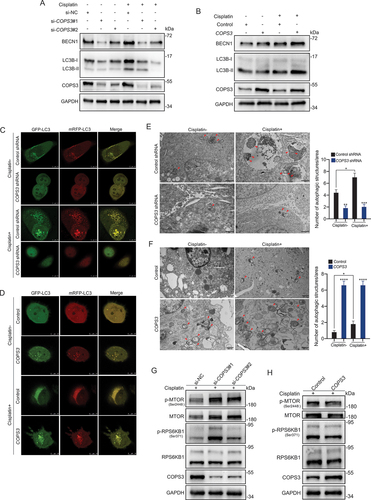
To gain a deeper insight into the role of COPS3 in the regulation of autophagy, RNA sequencing was performed in COPS3 knockout and control 143B cells after cisplatin treatment (). We found that COPS3 regulated multiple autophagic processes in OS, and the results of quantitative real-time PCR (qRT-PCR) revealed that several autophagy-related and lysosomal genes were significantly downregulated upon COPS3 knockdown ( and Fig. S2A). In addition, the results of gene set enrichment analysis demonstrated that the genes affected by COPS3 silencing were enriched in the MTORC1 pathway, indicating that the MTORC1 signaling cascade is the main pathway responsible for the autophagy regulation of COPS3 (Fig. S2B). Among differentially expressed genes (DEGs), RAB7 (RAB7, member RAS oncogene family) plays an indispensable role in the process of autophagosome maturation [Citation35] and can be inhibited by COPS3 silencing. In line with this, we confirmed that RAB7 protein levels were also positively regulated by COPS3 ( and Fig. S2C-F). To identify whether COPS3 promotes the fusion of the autophagosome and lysosome, immunofluorescence staining was performed. LC3 indicated that the autophagosomes and lysosomes were visualized by LAMP1 (lysosomal associated membrane protein 1), a lysosomal marker. Notably, the number of autolysosomes (LC3 and LAMP1 double-positive dots) was markedly decreased in the COPS3 knockout groups but not in the control group (). Similarly, the cells overexpressing COPS3 contained a higher density of autolysosomes than did the control cells (). These data indicate that COPS3 deficiency impairs the capability of the autophagosome to fuse with the lysosome by reducing RAB7 levels.
Figure 3. COPS3 promotes fusion of the autophagosome and the lysosome by induction of RAB7. (A) Clustered heatmap of DEGs acquired by RNA sequencing in 143B cells transfected with either control or COPS3 siRNA and treated with cisplatin for 24 h (3 μM). (B) Autophagy-related terms enriched by DEGs. (C) qRT-PCR analysis of the levels of autophagy-related and lysosomal genes in 143B cells transfected with COPS3 siRNA and treated with cisplatin for 24 h (3 μM). Data are presented as mean ± SEM of triplicates. *p < 0.05, **p < 0.01, *** p < 0.001. (D) Western blotting analysis of RAB7 levels in 143B cells transfected with COPS3 siRNAs and treated with cisplatin for 24 h (3 μM). (E) Western blotting analysis of RAB7 levels in Saos-2 cells transfected with COPS3 overexpression vector with or without cisplatin treatment for 24 h (5 μM). (F) Immunofluorescence analysis of the distribution of autophagosomes (LC3), lysosomes (LAMP1), and autolysosomes (LC3- and LAMP1-colocalized dots) in 143B cells transfected with COPS3 siRNAs and treated with cisplatin for 24 h (3 μM). The number of LC3 and LAMP1 double positive-dots per area was quantified (n = 5). Data are presented as mean ± SEM. **p < 0.01, *** p < 0.001. Scale bar: 10 μm. (G) Immunofluorescence analysis of the distribution of autophagosomes (LC3), lysosomes (LAMP1), and autolysosomes (LC3- and LAMP1-colocalized dots) in Saos-2 cells transfected with COPS3 overexpression vector and treated with cisplatin for 24 h (5 μM). The number of LC3 and LAMP1 double positive-dots per area was quantified (n = 5). Data are presented as mean ± SEM. *** p < 0.001. Scale bar: 10 μm.
COPS3-induced autophagy mediates cisplatin resistance
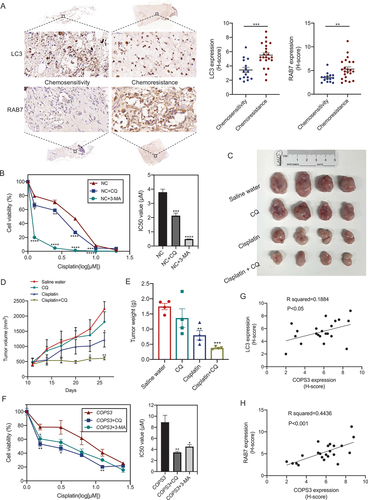
Autophagy can be either a pro-survival or a pro-death mechanism [Citation36]. Thus, to identify the role of autophagy in OS, autophagic levels in OS samples with different sensitivities to chemotherapy were investigated. The levels of LC3 and RAB7 were significantly upregulated in the chemotherapy-resistant group (), and thus, we speculated that autophagy may be associated with the development of chemoresistance. Then, 143B cells were treated with cisplatin and combined with the autophagy inhibitors CQ or 3-methyladenine (3-MA). Inhibition of autophagy enhanced the cytotoxicity of cisplatin in the cells (). The effect of autophagy on cisplatin chemoresistance was further investigated in 143B cell-grafted nude mice. Given the safety of autophagy inhibitors, we selected CQ for the inhibition of autophagy because it is approved for human use and is currently used in many clinical trials [Citation37]. On day 11 after tumor inoculation, tumor-bearing mice were treated with saline water, CQ, cisplatin, or cisplatin + CQ. After 15 days, the mice were sacrificed. We observed that tumor volume and tumor weight were lower in CQ-treated mice than in the saline water-treated mice, but the difference was not significant. Tumor volume and tumor weight were also lower in both the cisplatin- or cisplatin + CQ-treated mice than in the saline water-treated mice. Furthermore, the therapeutic effect of the combination therapy was superior to cisplatin monotherapy (), indicating that the inhibition of autophagy by CQ promoted cisplatin-induced apoptosis; therefore, autophagy serves as a pro-survival mechanism during cisplatin treatment in OS. COPS3-induced autophagy was amplified in response to cisplatin treatment. Next, the exact role of COPS3-induced autophagy in cell survival after cisplatin treatment was investigated using Saos-2 cells with few autophagic structures. Subsequent COPS3 overexpression in Saos-2 cells following cisplatin treatment resulted in the accumulation of autophagic vacuoles captured by TEM (Fig. S3), indicating autophagy activation. Combination treatment with cisplatin and autophagy inhibitors inhibited cell viability and decreased the 50% inhibitory concentration (IC50) (). These results reveal that COPS3-induced autophagy plays a protective role and leads to cisplatin resistance in OS. In addition, correlation analyses showed that the expression of LC3 and RAB7 was positively correlated with the expression of COPS3 (), indicating that autophagy was induced in COPS3-amplified chemoresistant OS and that targeting COPS3-mediated autophagy may reverse the chemoresistance of OS.
Figure 4. COPS3-induced autophagy mediates cisplatin resistance. (A) Immunohistochemical images of LC3 and RAB7 expression in human osteosarcoma tissues with different responses to preoperative chemotherapy (tumor necrosis ≥90%: chemosensitivity; tumor necrosis <90%: chemoresistance). Data are presented as mean ± SEM. **p < 0.01, *** p < 0.001. (B) Cell viability was measured using CCK-8 assay in 143B cells treated with cisplatin, cisplatin + CQ (10 μM), or cisplatin + 3-MA (5 mM) for 24 h. IC50 values of the different groups are statistically analyzed. Data are presented as mean ± SEM of triplicates. *p < 0.05, **p < 0.01, *** p < 0.001, **** p < 0.0001. (C-E) BALB/c nude mice (n = 4 mice per group) were injected with 143B cells and treated with saline water, CQ, cisplatin, or cisplatin + CQ. (C) Tumors excised from the different groups are shown. (D) Tumor growth curves of the different treatment groups. (E) Weight of the excised tumors in each group. Data are presented as mean ± SEM. *p < 0.05, **p < 0.01, *** p < 0.001. (F) Cell viability was measured using CCK-8 assay in Saos-2 cells transfected with COPS3 overexpression vector and treated with cisplatin, cisplatin + CQ (10 μM), or cisplatin + 3-MA (5 mM) for 24 h. The IC50 values of the different groups are statistically analyzed. Data are presented as mean ± SEM of triplicates. *p < 0.05, **p < 0.01. (G and H) The correlations between the levels of LC3, RAB7, and COPS3 expression in 22 chemoresistant OS samples are shown.
FOXO3 is required for COPS3-induced autophagy
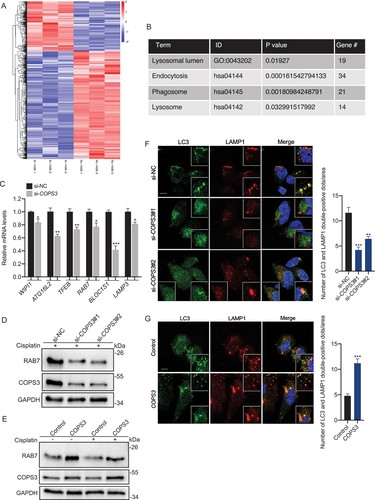
To explore the role of COPS3 in the transcriptional regulation of autophagy, differentially expressed transcription factors were determined via RNA sequencing analysis. Analysis of transcription factors related to autophagy indicated the most significant difference to be in FOXO3 ( and Table S1). FOXO3 is necessary and sufficient for autophagy induction [Citation38]. We found that FOXO3 protein levels were regulated by COPS3 ( and Fig. S4A). Analysis of the influence of COPS3 with or without cisplatin treatment on FOXO3 expression showed that although FOXO3 levels were upregulated after cisplatin treatment, it was still dependent on COPS3 ( and Fig. S4B-D). As a transcription factor, FOXO3 needs to be located in the nucleus to exert its effect. To confirm whether COPS3 regulates FOXO3 transcriptional activity, we first examined the distribution of FOXO3 in cytosolic and nuclear fractions with COPS3 knockout following cisplatin treatment. The results showed reduced nuclear levels of FOXO3, while cytosolic expression of FOXO3 remained unchanged after COPS3 knockout ( and Fig. S4E). Luciferase reporter assay driven by the FOXO promoter showed that COPS3 inhibition in 143B cells decreased FOXO promoter activity (). Consistently, induction of COPS3 in HEK-293T cells enhanced promoter activity (). Overall, COPS3 is a crucial regulator of FOXO3 transcriptional activity.
Figure 5. FOXO3 is required for COPS3-induced autophagy. (A) Differentially expressed transcription factors that may be associated with autophagy regulation. (B) Immunoblotting analysis of FOXO3 levels in 143B cells transfected with COPS3 siRNAs and treated with cisplatin for 24 h (3 μM). (C) Immunoblotting analysis of FOXO3 levels in 143B cells transfected with COPS3 siRNAs and treated with or without cisplatin for 24 h (3 μM). (D) 143B cells were transfected with COPS3 siRNAs and treated with cisplatin for 24 h (3 μM). Immunoblotting analysis of the distribution of FOXO3 protein in the cytoplasm and nucleus is shown. (E) Relative luciferase activity of the FOXO-Luc reporter construct in response to COPS3 knockdown with siRNA in 143B cells was examined. Data are presented as mean ± SEM of triplicates. *p < 0.05. (F) Relative luciferase activity of the FOXO-Luc reporter construct in response to COPS3 overexpression in HEK-293T cells was examined. Data are presented as mean ± SEM of triplicates. *p < 0.05. (G) ChIP assay for the interaction of FOXO3 and the LC3B promoter in 143B cells. (H) Immunoblotting analysis of LC3B levels in 143B cells transfected with FOXO3 overexpression vector. (I) ChIP assay for the interaction of FOXO3 and the RAB7 promoter in 143B cells. (J) Immunoblotting analysis of RAB7 levels in 143B cells transfected with FOXO3 overexpression vector. (K) Saos-2 cells were transfected with COPS3 overexpression vector. Immunoblotting analysis of LC3B and RAB7 levels in COPS3-overexpressing Saos-2 cells following knockdown of FOXO3 with siRNAs. (L) ChIP assays for the binding of FOXO3 to LC3B or the RAB7 promoter in 143B cells transfected with COPS3 siRNAs.
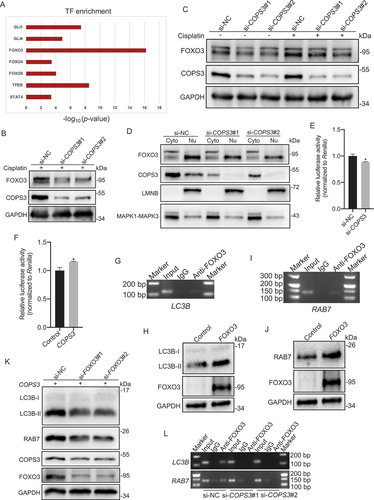
FOXO3 has been found to be involved in the transcriptional regulation of LC3 in skeletal muscle [Citation38]. Thus, we examined the ability of FOXO3 to bind to the LC3 promoter in 143B cells via chromatin immunoprecipitation (ChIP) assays. Endogenous FOXO3 was found to interact with the LC3B promoter (), and induction of FOXO3 expression increased LC3B protein level in transfected cells ( and Fig. S4F). Given that FOXO3 induces the expression of RAB7 [Citation39], further analysis was performed to determine whether FOXO3 was directly implicated in the transcriptional regulation of RAB7. Several FOXO3 binding sites in the promoter region of the RAB7 gene were then predicted using the JASPAR database (Table S2). We speculated that RAB7 might act as a downstream target gene, and the binding of FOXO3 to RAB7 was subsequently identified (). Furthermore, FOXO3 overexpression markedly induced RAB7 protein level, confirming that FOXO3 regulated RAB7 expression in 143B cells ( and Fig. S4G). To confirm whether FOXO3 is involved in COPS3-induced autophagy, COPS3 was overexpressed in Saos-2 cells, and the protein levels of LC3B and RAB7 were detected after FOXO3 knockdown. Autophagic levels were notably attenuated by FOXO3 inhibition, although COPS3 was overexpressed ( and Fig. S4H). Further, COPS3 knockout prevented the binding of FOXO3 to its target promoters, subsequently leading to the failure of target gene transcription induction (). These findings indicate that COPS3 results in FOXO3-dependent transcriptional activation of autophagy-related genes in the nucleus, which subsequently promotes the formation of autophagosomes and autolysosomes.
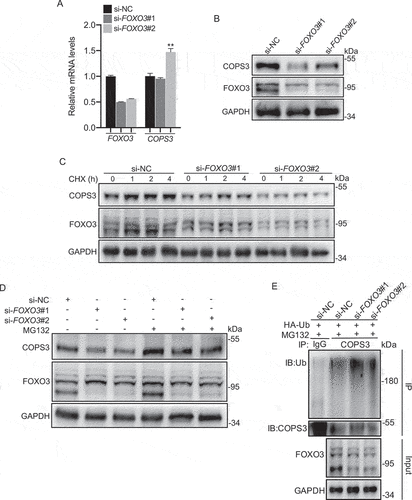
FOXO3 regulates the protein stability of COPS3
As shown in , FOXO3 inhibition with small interfering RNAs (siRNAs) decreased COPS3 protein levels in Saos-2 cells with COPS3 re-expression. Therefore, we speculated that FOXO3 might act as a positive regulator of COPS3. Investigation of the COPS3 mRNA levels in 143B cells after FOXO3 knockdown showed that the mRNA level of COPS3 was slightly altered by si-FOXO3#1 knockdown. However, si-FOXO3#2 increased mRNA level of COPS3 (). Detection of COPS3 protein levels in 143B cells showed that FOXO3 inhibition by siRNAs significantly decreased COPS3 levels in transfected cells ( and Fig. S5A). The discrepancy between the mRNA and protein levels indicates that COPS3 expression is more likely regulated by post-translational modifications rather than by transcription. Thus, we speculated that FOXO3 might influence the synthesis and/or degradation of the COPS3 protein. Accordingly, 143B cells were treated with cycloheximide (CHX), a protein synthesis inhibitor. FOXO3 knockout markedly shortened the half-life of COPS3 (). Moreover, treatment of si-FOXO3-transfected 143B cells with the 26S proteasome inhibitor MG132 inhibited COPS3 degradation ( and Fig. S5B), indicating that FOXO3 inhibits the 26S proteasome-dependent degradation of COPS3. To further explore whether the ubiquitin-proteasome system (UPS) is responsible for the degradation of COPS3, COPS3 ubiquitination was investigated. As expected, FOXO3 knockdown markedly enhanced COPS3 ubiquitination in 143B cells (). Collectively, these data suggest that FOXO3 promotes the protein stability of COPS3 by preventing COPS3 degradation mediated by the ubiquitin-proteasome pathway in OS.
Figure 6. FOXO3 depletion results in increased COPS3 ubiquitination. (A) qRT-PCR analysis of COPS3 levels in 143B cells transfected with FOXO3 siRNAs. Data are presented as mean ± SEM of triplicates. **p < 0.01. (B) Immunoblotting analysis of COPS3 levels in 143B cells transfected with FOXO3 siRNAs. (C) 143B cells infected with FOXO3 siRNAs were subjected to a CHX (50 μg/ml) experiment. (D) COPS3 expression levels in the absence or presence of MG132 (20 μM, 6 h) were analyzed in 143B cells transfected with FOXO3 siRNAs. (E) 143B cells expressing HA-Ubiquitin (Ub) were infected with FOXO3 siRNAs and treated with MG132 (20 μM) for 6 h. Immunoblotting analysis of COPS3 ubiquitination with anti-COPS3 antibody.
The FOXO3-SKP2 axis is involved in COPS3 transcriptional regulation
Based on the findings that FOXO3 inhibited ubiquitin-mediated COPS3 degradation, we then sought to identify which E3 ubiquitin ligase is responsible for COPS3 ubiquitination. FOXO3 is an important transcription factor in the repression of SKP2 (S-phase kinase associated protein 2), an F-box protein subunit of the SCF E3 ubiquitin ligase complex [Citation40]. Thus, we examined the SKP2 levels regulated by FOXO3 in 143B cells. Decreased levels of FOXO3 resulted in an increase in both mRNA and protein levels of SKP2 ( and Fig. S6A). Furthermore, detection of COPS3 levels after SKP2 knockdown showed an increase in COPS3 mRNA () and protein levels ( and Fig. S6B) in 143B cells. These results support the notion that increased COPS3 levels regulated by SKP2 resulted from the transcriptional level rather than the post-translational modification level. Collectively, these findings indicate that SKP2 functions as a new regulator in the repression of COPS3.
Figure 7. The FOXO3-SKP2 axis is involved in the transcriptional regulation of COPS3. (A) qRT-PCR analysis of SKP2 levels in 143B cells transfected with FOXO3 siRNAs. Data are presented as mean ± SEM of triplicates. *p < 0.05, **p < 0.01. (B) Immunoblotting analysis of SKP2 levels in 143B cells transfected with FOXO3 siRNAs. (C) qRT-PCR analysis of COPS3 levels in 143B cells transfected with SKP2 siRNAs. Data are presented as mean ± SEM of triplicates. *p < 0.05, *** p < 0.001. (D) Immunoblotting analysis of COPS3 levels in 143B cells transfected with SKP2 siRNAs. (E) Immunoblotting analysis of COPS3 levels in 143B cells transfected with FOXO3 siRNAs and SKP2 siRNA. (F) qRT-PCR analysis of FOXO3 levels in 143B cells transfected with SKP2 siRNAs. Data are presented as mean ± SEM of triplicates. **p < 0.01, *** p < 0.001. (G) Immunoblotting analysis of FOXO3 levels in 143B cells transfected with SKP2 siRNAs. (H) 143B cells infected with SKP2 siRNAs were subjected to a CHX (50 μg/ml) experiment.
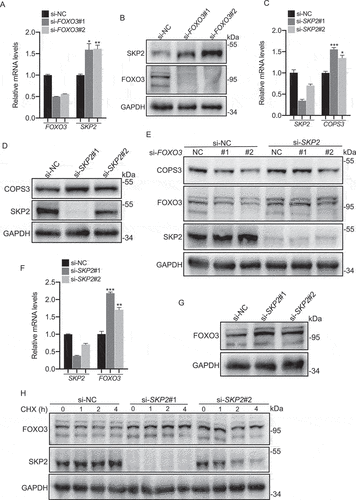
Subsequently, FOXO3-SKP2 axis involvement in the regulation of COPS3 was determined. SKP2 inhibition rescued the reduction of COPS3 protein caused by FOXO3 knockdown ( and Fig. S6C). Thus, we identified the FOXO3-SKP2 axis as a new mechanism crucial for the transcriptional regulation of COPS3. As shown in , we incidentally found that a reduction of SKP2 increased FOXO3 protein levels. Then we further validated that the inhibition of SKP2 enhanced both FOXO3 mRNA and protein levels in 143B cells ( and Fig. S6D). In addition, CHX treatment of si-SKP2-transfected 143B cells increased the half-life of FOXO3 (). These findings indicate that SKP2 is a repressor of FOXO3 expression, which is regulated not only in protein stability, but also at the transcriptional level in OS.
Discussion
The role of the COP9 signalosome in tumors has been widely studied. For example, COPS5 is associated with disease progression in prostate and pancreatic cancers [Citation41,Citation42], while overexpression of COPS3 correlates with tumor proliferation and metastasis [Citation30,Citation43]. In this study, we identified a new role of COPS3 in the promotion of cisplatin resistance and further clarified that COPS3-induced autophagy mediates this resistance. Our results showed that COPS3 was significantly upregulated in chemoresistant OS tissues and that COPS3 inhibition sensitized OS cells to cisplatin treatment. This may explain our previous finding that COPS3 promotes the metastasis of OS cells to the lung [Citation30]. In clinical application, OS is normally treated with a combination of cisplatin, doxorubicin, and methotrexate in order to improve the therapeutic effect and reduce toxicity. In the present study, we confirmed that OS cells with high expression of COPS3 showed resistance to cisplatin. However, whether COPS3 is related to the resistance to other chemotherapeutic agents needs further study.
Autophagy usually occurs at a basal level in most cells and can be rapidly induced by diverse stimuli and cellular stresses. In this study, we found a low basal level of autophagy in OS 143B cells but not in Saos-2 cells under normal conditions, and we also noted that the maintenance of the basal level of autophagy was associated with COPS3 expression. Moreover, we observed that COPS3-induced basic autophagy was amplified in response to cisplatin. The role of autophagy in tumors is highly context-dependent and can be either a pro-survival or a pro-death mechanism [Citation36]. In COPS3-expressing cell models, the combination treatment of cisplatin with autophagy inhibitors resulted in positive outcomes, indicating that COPS3-mediated autophagy is a survival mechanism and can therefore be a target with promising anti-tumor effect in combination therapy with cisplatin in OS. In our in vivo model, although the tumor volume and tumor weight were lower in nude mice grafted with 143B cells than in controls after treatment with CQ alone, the difference was not significant. However, the combination treatment of cisplatin with CQ obviously delayed OS xenograft growth compared with cisplatin alone, indicating that the positive outcome was mainly due to overcoming cisplatin resistance rather than gaining new sensitivity to an autophagy inhibitor. The positive benefits associated with combining an autophagy inhibitor with a kinase inhibitor or chemotherapy provide obvious evidence that combination therapy containing targeting autophagy is promising in specific patient populations [Citation44,Citation45]. However, whether autophagy inhibitors alone can be used as an anti-tumor treatment is still controversial. In our previous study, CQ has an inhibitory effect on OS growth [Citation46]. The discrepancy may be due to the time of the first injection after tumor inoculation and the interval of drug administration. In the previous study, CQ administration was initiated 3 days after tumor inoculation and dosed daily, whereas, in the present study, medications were administered every 3–4 days starting from day 11 of tumor inoculation. The difference may raise the hypothesis that a high frequency of CQ treatment in the early stage of OS is more effective, but more research is needed.
We have previously established the role of COPS3 in autophagy regulation [Citation30]. In the present study, we further provide mechanistic understanding. We noted that the mRNA and protein levels of RAB7 were downregulated upon siRNA-mediated depletion of COPS3. RAB7 plays an indispensable role in the fusion of the autophagosome with the lysosome [Citation35], and we subsequently confirmed the promoting role of COPS3 on autophagosome maturation. The result of RNA sequencing revealed that FOXO3 transcript abundance was downregulated by the inhibition of COPS3. FOXO3 activity can be regulated by different post-translational modifications. For example, FOXO3 is normally phosphorylated by AKT and localizes in the cytoplasm. When FOXO3 is dephosphorylated, it translocates from the cytoplasm to the nucleus and induces the transcription of autophagy genes [Citation47]. Interestingly, our findings showed that FOXO3 was mainly enriched in the nucleus after cisplatin treatment, and inhibition of COPS3 decreased the nuclear abundance of FOXO3. Subsequently, we demonstrated that the decrease in the nuclear expression level of FOXO3 was correlated with a decrease in the transcriptional activity, indicating that the regulation of FOXO3 activity by COPS3 is independent of post-translational modification. FOXO3 is a key molecule that controls the transcription of autophagy-related genes, such as LC3 [Citation38]. We also confirmed that COPS3 regulated LC3 expression by modulating FOXO3 transcriptional activity. Although one study has confirmed that FOXO3 can induce the expression of RAB7 [Citation39], our results further proved that RAB7 was the target gene of FOXO3 and provided the targeted promoter sequence. Thus, our data indicate that COPS3 plays an important role in the autophagic process by enhancing the expression of FOXO3-responsive genes, including LC3 and RAB7. In addition, immunohistochemistry results revealed that LC3 and RAB7 were highly expressed in chemoresistant OS samples. Correlation analyses showed significant positive correlations between the expression of COPS3 and both LC3 and RAB7. These findings indicated that autophagy was induced in COPS3-amplified chemoresistant OS and further confirmed that COPS3-induced autophagy was responsible for OS chemoresistance.
Last, we elucidated a regulatory mechanism that governed COPS3 expression. There were inconsistent changes in the mRNA and protein levels of COPS3 caused by the knockdown of FOXO3. More importantly, FOXO3 knockdown markedly shortened the half-life of COPS3 and increased COPS3 ubiquitination. The results indicated that FOXO3 maintained the protein stability of COPS3, but the ubiquitin ligase that targets COPS3 needs to be further identified. The COP9 signalosome is established to play a role in ubiquitin-mediated proteolysis [Citation48,Citation49]. In the present study, we have proposed for the first time the role of COPS3 as a substrate for degradation by the ubiquitin-proteasome pathway. SKP2, an F-box protein subunit of the SCF E3 ubiquitin ligase complex, is involved in ubiquitin-mediated degradation [Citation40]. However, our results showed that SKP2 inhibited both COPS3 mRNA and protein levels, indicating that SKP2 regulated COPS3 expression at the transcriptional level, not the post-translational modification level. Due to the transcriptional inhibition of SKP2 by FOXO3 [Citation40], we found that the high expression of FOXO3 attenuated the SKP2-mediated inhibition of COPS3, thereby identifying a new signaling cascade of the FOXO3-SKP2-COPS3 axis. In addition to the ubiquitin-mediated degradation of FOXO3 by SKP2 [Citation50], we also demonstrated that SKP2 inhibited FOXO3 expression at the transcriptional level. Overall, our data indicate that COPS3 regulates its own expression through feedback loop to activate autophagy in response to cisplatin (). The role of the COP9 signalosome in the regulation of autophagy is first established by finding that COPS8 gene deletion impairs autophagosome removal in mouse hearts. COPS8 is then proven to play an important role in autophagosome maturation [Citation51]. The current study showed that COPS3 not only promoted autophagosome maturation but was also required for autophagosome formation. We demonstrated that COPS3 increased the conversion of LC3B-I to LC3B-II and inhibited the MTOR pathway in autophagosome formation. This discrepancy is probably due to differences in species or diseases, or because different subunits of the COP9 signalosome do not have the same role in the regulation of autophagy. The current study shows the emerging role of COPS3 in autophagy regulation, but the specific function of other subunits in autophagy regulation remains unclear.
Figure 8. Schematic model of the role of the COPS3-induced autophagy in cisplatin resistance in osteosarcoma. Cisplatin treatment amplifies COPS3-mediated autophagy. Mechanistically, the model mainly depicts that the COPS3-FOXO3 positive feedback loop is involved in autophagy induction in response to cisplatin. COPS3 regulates the nuclear abundance of FOXO3 and enhances FOXO3-dependent transcription of LC3B and RAB7. In turn, FOXO3 inhibits COPS3 degradation mediated by the UPS and suppresses SKP2-mediated COPS3 inhibition to cooperatively maintain high COPS3 level. In addition, SKP2 exerts a negative feedback effect on FOXO3 through inhibition at the transcriptional level. In coordination with the feedback loop, inhibition of MTOR pathway by high COPS3 level also ensures rapid initiation of autophagy. High COPS3 level can activate cytoprotective autophagy, eventually leading to cisplatin resistance in osteosarcoma.
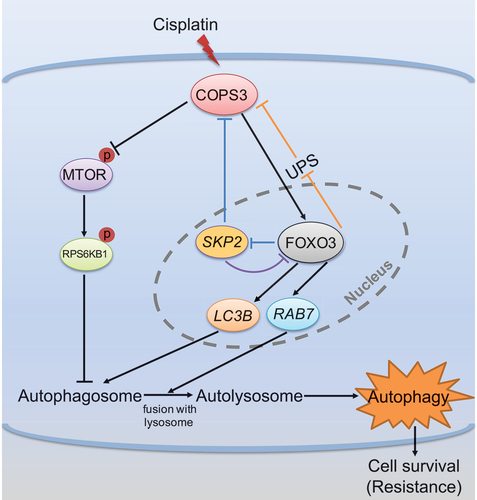
In summary, our study revealed a new connection between COPS3 expression, autophagy induction, and cisplatin resistance, and identified a positive feedback loop between COPS3 and FOXO3, which stimulated autophagy to promote cisplatin resistance. Our results also showed that inhibition of COPS3-mediated autophagy was sufficient to overcome cisplatin resistance in OS. These findings elucidate the molecular mechanisms of cisplatin resistance and further provide a novel therapeutic strategy to enhance cisplatin treatment efficacy in OS.
Materials and methods
Cell lines and cell culture
143B and HEK-293T cells were acquired from the American Type Culture Collection (CRL-8303 and CRL-11268). Saos-2 cells were purchased from the National Collection of Authenticated Cell Cultures (SCSP-5057). All cells were cultured in Dulbecco’s modified Eagle’s medium (Thermo Fisher Scientific, C11995500BT) supplemented with 10% fetal bovine serum (Thermo Fisher Scientific, 10099–141) and antibiotics (Thermo Fisher Scientific, 15140–122) in a 5% CO2 atmosphere at 37°C.
Construction of stable cell lines and RNA interference
Cells stably expressing COPS3 and FOXO3 genes were generated by lentivirus transfection followed by puromycin selection. COPS3-knockdown cells were generated by stable transfection of COPS3 shRNA into 143B cells. Cells expressing GFP-mRFP-LC3 adenoviral vectors were constructed. For the dual-luciferase reporter assays, cells were co-transfected with a FOXO luciferase reporter plasmid (1.5 μg; Yeasen, 11527ES03) and a Renilla luciferase control plasmid (0.5 μg; Promega, E2241). 143B cells were transiently transfected with HA-tagged ubiquitin using Lipofectamine 2000 (Invitrogen, 11668027). For RNA interference, siRNAs (20 nM) were transfected using GP-transfect-Mate (GenePharma, G04008), following the manufacturer’s instructions. At 48–72 h after transfection, cells were used for further experiments. The reagents and plasmids used are listed in . The siRNA oligonucleotides targeting COPS3, FOXO3, and SKP2 are listed in Table S3.
Table 1. Reagents used in this study.
Human tissues
Chemosensitive and chemoresistant OS samples were obtained from the Musculoskeletal Tumor Center, Peking University People’s Hospital (Beijing, China). All selected patients underwent preoperative chemotherapy and surgery. This study was approved by the Ethics Committee of Peking University People’s Hospital, and informed consent was obtained from all patients. The clinical information of the OS tissues used in this study is listed in Table S4.
Immunohistochemistry
Paraffin-embedded human OS sections were stained with the indicated antibodies, as described in our previous study [Citation30]. Staining intensity was scored as: 0, negative staining; 1, weak staining intensity; 2, moderate staining intensity; and 3, strong staining intensity. The ratio of positively stained cells was categorized according to the following proportion scores: 0, <5%; 1, 5–25%; 2, 26–50%; 3, 51–75%; and 4, >75%. The histoscore (H-score) was calculated by multiplying the staining intensity by the percentage of positive cells, and the final H-score for each case was calculated using the mean of the scores of 5 random areas. The antibodies used are listed in .
Cell viability assays
The cells were seeded into a 96-well plate at 5000 cells per well. On the following day, the medium was replaced with medium containing different concentrations of cisplatin (MedChemExpress, HY-17394). For the autophagy inhibition experiment, medium containing different concentrations of cisplatin with or without an autophagy inhibitor (CQ or 3-MA; MedChemExpress, HY-17589A and HY-19312) was added. CQ and 3-MA were used at a concentration of 10 μM and 5 mM, respectively. After 24 h of incubation, absorbance at 450 nm was measured using a water-soluble tetrazolium salt assay with the Cell Counting Kit-8 (Dojindo, CK04). We then established the cell viability curve and calculated the IC50 value using GraphPad Prism software (Prism 8). The reagents used are listed in .
Murine xenograft assays
Four-week-old female BALB/c nude mice were purchased from Vital River (Beijing, China) and housed under specific pathogen-free conditions. In experiment 1, 6 × 106 GFP-Control shRNA or GFP-COPS3 shRNA 143B cells were inoculated subcutaneously into the right flank of the mice (n = 4 mice per group). Cisplatin (3 mg/kg) was administered intraperitoneally from day 6 after injection, and administration was continued thereafter every 3 days. The long diameter (I) and short diameter (W) of tumors were measured five times every 3 days using Vernier calipers. Tumor volume was calculated as 0.52×(I× W2). On day 18, the mice were sacrificed, and the tumors were excised and weighed.
In experiment 2, 6 × 106 143B cells were injected subcutaneously into the right flank of BALB/c nude mice (n = 4 mice per group). Saline water (0.9% sodium chloride), CQ (60 mg/kg), cisplatin (3 mg/kg), or cisplatin + CQ were intraperitoneally injected once every 3–4 days starting from day 11 of tumor inoculation. Tumor size was measured using Vernier calipers, and tumor volume was calculated using the formula 0.52×(I× W2). Finally, the mice were euthanized, and the tumor tissues were harvested and weighed on day 26. All animal experiments were approved by the Animal Experiment Committee of Peking University People’s Hospital and were performed in accordance with all institutional and international animal protocols.
Western blotting
Cells were washed with phosphate-buffered saline (PBS; Meilunbio, MA0015) and lysed with cell lysis buffer (Cell Signaling Technology, 9803) containing protease and phosphatase inhibitors (Thermo Fisher Scientific, 78440). Total protein concentration was measured by the BCA protein assay kit (Solarbio, PC0020). Clarified cell extracts were mixed with 5X loading buffer (EpiZyme, LT101) and boiled at 99°C for 5 min. Equal amounts of protein were loaded into each lane, separated on sodium dodecyl sulfate–polyacrylamide gel electrophoresis gel, and transferred to a polyvinylidene fluoride membrane. Membranes were blocked in tris-buffered saline (Solarbio, T1080) with 5% milk and 0.1% Tween 20 (Solarbio, T8220), followed by overnight incubation with primary antibodies. Blots were detected after incubation with secondary antibodies for 1 h at 37°C using chemiluminescent assay. The antibodies used are listed in .
Confocal microscopy and transmission electron microscopy
For autophagy studies, cells were plated onto confocal dishes and transfected with GFP-mRFP-LC3 adenovirus 24 h prior to treatment. Cells were then treated with or without cisplatin for another 24 h. LC3 puncta were observed and photographed using a confocal microscope. For TEM, cells were seeded on 10-cm plates and treated with cisplatin for 24 h when cell confluence reached 60–70%. The cells were then washed with PBS, centrifuged, and collected in microcentrifuge tubes. Then, 400 μL of 3% glutaraldehyde was added to the microcentrifuge tubes and incubated overnight at 4°C. The samples were then postfixed with 1% OsO4 for 1 h at room temperature and dehydrated with 30, 50, 70, 90, and 100% ethanol concentrations in series. The dehydrated samples were infiltrated with LR white resin (Santa Cruz Biotechnology, sc-215266) and embedded. Resin-embedded blocks were cut into 60-nm-thick sections and stained with uranyl acetate and lead citrate. The sections were then observed under TEM. Five fields were randomly selected, and the number of autophagic vacuoles was quantified.
RNA sequencing
Total RNA was extracted from 143B cells transfected with either si-COPS3 or si-NC using TRIzol reagent (Invitrogen, 15596018). Cells were treated with 3 μM cisplatin for 24 h prior to RNA isolation. RNA concentration was detected using NanoDrop, and RNA integrity was assessed using an Agilent Bioanalyzer 2100 (Agilent Technologies, CA, USA). Libraries for transcriptome sequencing were generated using the VAHTS Stranded mRNA-seq Library Prep Kit (Illumina). After the library was established, the library concentration was quantified using a Qubit® 2.0 Fluorometer (Life Technologies, CA, USA) and quantitative PCR. The insert size was assayed on an Agilent Bioanalyzer 2100 system. The library was sequenced using the Illumina Novaseq PE150 after quality inspection. To acquire high-quality clean reads for the subsequent analysis, reads containing poly-N, adaptors, and low-quality reads were filtered using a FastQ screen (https://www.bioinformatics.babraham.ac.uk/projects/fastq_screen/). Gene expression levels were quantified using high-throughput sequencing analysis, and DEGs were identified using edgeR (|log2FoldChange|>0.4 and adjusted p-value <0.05). After identifying the DEGs, the transcription factors were screened using hmmsearch in the Hmmer software based on the AnimalTFDB (http://bioinfo.life.hust.edu.cn/AnimalTFDB/#!/) database. Among these, we selected several transcription factors related to autophagy regulation, and compared the significant differences according to the -log10 (p-value).
RNA extraction and qRT-PCR analysis
Total cellular RNA was extracted using TRIzol reagent (Invitrogen, 15596018), and RNA concentrations were quantified using NanoDrop. cDNA was then synthesized using the extracted RNA and a PrimeScript RT reagent Kit (TaKaRa, DRR037A). qRT-PCR was performed using a SYBR Green Premix Ex Taq kit (Bio-Rad, 1725121), and relative gene expression levels were determined using a CFX96 Real-Time PCR Detection System (Bio-Rad, CA, USA), with GAPDH as an internal control. All primers used for qPCR are listed in Table S5.
Immunofluorescence
Cells were seeded on coverslips and treated with cisplatin for 24 h, and then rinsed three times with PBS, fixed for 15 min in 4% paraformaldehyde, and again washed three times with PBS for 3 min per wash. The fixed cells were permeabilized with 0.1% Triton X-100 (Solarbio, T8200) in PBS for 20 min at room temperature, then blocked with normal goat serum for 30 min at room temperature. The cells were then incubated with the indicated primary antibodies overnight at 4°C, followed by incubation with fluorescent-tagged secondary antibodies for 1 h at 37°C in the dark. All nuclei were stained with DAPI for 15 min at room temperature in the dark. Cells were then visualized under a confocal microscope (Leica TCS-SP8, Germany), and the number of LC3 and LAMP1 double-positive dots was counted. The antibodies used are listed in .
Nuclear-cytosolic fractionation
Approximately 2 × 106 143B cells were harvested with trypsin-EDTA solution (Thermo Fisher Scientific, 25200072) and washed with PBS. Next, 200 μL ice-cold cytoplasmic extraction reagent I (Thermo Fisher Scientific, 78833) with protease inhibitor (Thermo Fisher Scientific, 78430) was added to the dried cell pellets. The tube was vortexed at the highest setting for 15 s and incubated on ice for 10 min. After adding 11 μL ice-cold cytoplasmic extraction reagent II (Thermo Fisher Scientific, 78833), the tube was vortexed at the highest setting for a further 5 s and incubated on ice for 1 min. After centrifugation at 13000 x g for 5 min, the supernatant (cytoplasmic extract) was transferred to a clean pre-chilled tube. The insoluble fraction was then suspended in 100 μL nuclear extraction reagent (Thermo Fisher Scientific, 78833) and protease inhibitor. The cell pellets were then left on ice and vortexed for 15 s every 10 min for a total of 40 min. After centrifugation at 13000 x g, for 10 min, the supernatant (nuclear extract) was transferred to a new tube. Cytoplasmic and nuclear proteins were analyzed using western blotting.
Dual-luciferase reporter assay
143B and HEK-293T cells were seeded in 12-well plates and co-transfected with a mixture of 1.5 μg FOXO luciferase reporter plasmid and 0.5 μg Renilla luciferase vector plasmid (pRL-TK) according to the manufacturer’s instructions. At 48 h after transfection, luciferase signals were measured using the dual luciferase reporter gene assay kit (Yeasen, 11402ES60*). The relative luciferase activity was determined by normalizing firefly luciferase activity to Renilla luciferase activity. The plasmids used are listed in .
ChIP-PCR assay
ChIP-PCR assay was performed using the ChIP-IT Express kit (Active Motif, 53008) following the manufacturer’s protocol. 143B cells were fixed with 37% formaldehyde (Sigma Aldrich, 252549) to crosslink endogenous proteins and DNA and sonicated to yield 200–1200 bp fragments. The protein-DNA complexes were immunoprecipitated overnight at 4°C using an anti-FOXO3 antibody (NOVUS, NBP2-16521). Immunoprecipitated DNA was reverse cross-linked, purified using DNA purification columns, and analyzed using PCR. The primers used for ChIP-PCR are listed in Table S5.
Statistical analysis
Data are presented as the mean ± standard error of the mean (SEM). Significant differences were determined using Student’s t test and one-way ANOVA. All statistical analyses were performed using Prism 8. Differences were considered statistically significant at p < 0.05 (significance levels: *p < 0.05; **p < 0.01; *** p < 0.001; **** p < 0.0001).
Supplemental Material
Download MS Word (9.3 MB)Acknowledgments
We are grateful to the Central Laboratory of Peking University Health Science Center for assistance with confocal microscopy and transmission electron microscopy.
Disclosure statement
No potential conflict of interest was disclosed.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/15548627.2022.2150003
Additional information
Funding
References
- Gorlick R, Khanna C. Osteosarcoma. J Bone Miner Res. 2010;25(4):683–691. PMID: 20205169.
- Bernthal NM, Federman N, Eilber FR, et al. Long-term results (> 25 years) of a randomized, prospective clinical trial evaluating chemotherapy in patients with high-grade, operable osteosarcoma. Cancer. 2012;118(23):5888–5893. PMID: 22648705.
- Shen TS, Hsu YK, Huang YF, et al. Licochalcone A suppresses the proliferation of osteosarcoma cells through autophagy and ATM-Chk2 activation. Molecules. 2019;24(13):12. PMID: 31269698.
- Davis AM, Bell RS, Goodwin PJ. Prognostic factors in osteosarcoma: a critical review. J Clin Oncol. 1994;12(2):423–431. PMID: 8113851.
- Rosen G, Marcove RC, Huvos AG, et al. Primary osteogenic sarcoma: eight years experience with adjuvant chemotherapy. J Cancer Res Clin Oncol. 1983;106:55–67. PMID: 6604058.
- Kansara M, Teng MW, Smyth MJ, et al. Translational biology of osteosarcoma. Nat Rev Cancer. 2014;14(11):722–735. PMID: 25319867.
- Link MP, Goorin AM, Miser AW, et al. The effect of adjuvant chemotherapy on relapse-free survival in patients with osteosarcoma of the extremity. N Engl J Med. 1986;314(25):1600–1606. PMID: 3520317.
- Meyers PA, Healey JH, Chou AJ, et al. Addition of pamidronate to chemotherapy for the treatment of osteosarcoma. Cancer. 2011;117(8):1736–1744. PMID: 21472721.
- Marina NM, Smeland S, Bielack SS, et al. Comparison of MAPIE versus MAP in patients with a poor response to preoperative chemotherapy for newly diagnosed high-grade osteosarcoma (EURAMOS-1): an open-label, international, randomised controlled trial. Lancet Oncol. 2016;17(10):1396–1408. PMID: 27569442.
- Xu J, Guo W, Xie L. Combination of gemcitabine and docetaxel: a regimen overestimated in refractory metastatic osteosarcoma? BMC Cancer. 2018;18(1):987. PMID: 30326879.
- Maiuri MC, Tasdemir E, Criollo A, et al. Control of autophagy by oncogenes and tumor suppressor genes. Cell Death Differ. 2009;16(1):87–93. PMID: 18806760.
- Chen JH, Zhang P, Chen WD, et al. ATM-mediated PTEN phosphorylation promotes PTEN nuclear translocation and autophagy in response to DNA-damaging agents in cancer cells. Autophagy. 2015;11(2):239–252. PMID: 25701194.
- Duan Y, Fang H. RecQL4 regulates autophagy and apoptosis in U2OS cells. Biochem Cell Biol. 2016;94(6):551–559. PMID: 27813658.
- Zhao GS, Gao ZR, Zhang Q, et al. TSSC3 promotes autophagy via inactivating the Src-mediated PI3K/Akt/mTOR pathway to suppress tumorigenesis and metastasis in osteosarcoma, and predicts a favorable prognosis. J Exp Clin Cancer Res. 2018;37(1):188. PMID: 30092789.
- Towers CG, Thorburn A. Therapeutic targeting of autophagy. EBiomedicine. 2016;14:15–23. PMID: 28029600.
- Xiao X, Wang W, Li Y, et al. HSP90AA1-mediated autophagy promotes drug resistance in osteosarcoma. J Exp Clin Cancer Res. 2018;37(1):201. PMID: 30153855.
- Kim M, Jung JY, Choi S, et al. GFRA1 promotes cisplatin-induced chemoresistance in osteosarcoma by inducing autophagy. Autophagy. 2017;13(1):149–168. PMID: 27754745.
- Meschini S, Condello M, Calcabrini A, et al. The plant alkaloid voacamine induces apoptosis-independent autophagic cell death on both sensitive and multidrug resistant human osteosarcoma cells. Autophagy. 2008;4(8):1020–1033. PMID: 18838862.
- Wei N, Deng XW. The COP9 signalosome. Annu Rev Cell Dev Biol. 2003;19(1):261–286. PMID: 14570571.
- Chamovitz DA, Wei N, Osterlund MT, et al. The COP9 complex, a novel multisubunit nuclear regulator involved in light control of a plant developmental switch. Cell. 1996;86(1):115–121. PMID: 8689678.
- Wei N, Deng XW. Making sense of the COP9 signalosome. A regulatory protein complex conserved from Arabidopsis to human. Trends Genet. 1999;15(3):98–103. PMID: 10203806.
- Petroski MD, Deshaies RJ. Mechanism of lysine 48-linked ubiquitin-chain synthesis by the cullin-RING ubiquitin-ligase complex SCF-Cdc34. Cell. 2005;123(6):1107–1120. PMID: 16360039.
- Bennett EJ, Rush J, Gygi SP, et al. Dynamics of cullin-RING ubiquitin ligase network revealed by systematic quantitative proteomics. Cell. 2010;143(6):951–965. PMID: 21145461.
- Lee MH, Zhao R, Phan L, et al. Roles of COP9 signalosome in cancer. Cell Cycle. 2011;10(18):3057–3066. PMID: 21876386.
- Pollmann C, Huang X, Mall J, et al. The constitutive photomorphogenesis 9 signalosome directs vascular endothelial growth factor production in tumor cells. Cancer Res. 2001;61(23):8416–8421. PMID:11731421.
- Luo J, Emanuele MJ, Li D, et al. A genome-wide RNAi screen identifies multiple synthetic lethal interactions with the Ras oncogene. Cell. 2009;137(5):835–848. PMID:19490893.
- Henriksen J, Aagesen TH, Maelandsmo GM, et al. Amplification and overexpression of COPS3 in osteosarcomas potentially target TP53 for proteasome-mediated degradation. Oncogene. 2003;22(34):5358–5361. PMID: 12917637.
- van Dartel M, Redeker S, Bras J, et al. Overexpression through amplification of genes in chromosome region 17p11.2 approximately p12 in high-grade osteosarcoma. Cancer Genet Cytogenet. 2004;152(1):8–14. PMID: 15193436.
- Yan TQ, Wunder JS, Gokgoz N, et al. COPS3 amplification and clinical outcome in osteosarcoma. Cancer. 2007;109(9):1870–1876. PMID: 17366602.
- Zhang F, Yan TQ, Guo W, et al. Novel oncogene COPS3 interacts with Beclin1 and Raf-1 to regulate metastasis of osteosarcoma through autophagy. J Exp Clin Cancer Res. 2018;37(1):135. PMID: 29970115.
- Kumar A, Singh UK, Chaudhary A. Targeting autophagy to overcome drug resistance in cancer therapy. Future Med Chem. 2015;7(12):1535–1542. PMID: 26334206.
- Sui X, Chen R, Wang Z, et al. Autophagy and chemotherapy resistance: a promising therapeutic target for cancer treatment. Cell Death Dis. 2013;4(10):e838. PMID: 24113172.
- Klionsky DJ, Abdelmohsen K, Abe A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy. 2016;12(1):1–222. PMID: 26799652.
- Nazio F, Strappazzon F, Antonioli M, et al. mTOR inhibits autophagy by controlling ULK1 ubiquitylation, self-association and function through AMBRA1 and TRAF6. Nat Cell Biol. 2013;15(4):406–416. PMID: 23524951.
- Hyttinen JMT, Niittykoski M, Salminen A, et al. Maturation of autophagosomes and endosomes: a key role for Rab7. Biochim Biophys Acta. 2013;1833(3):503–510. PMID: 23220125.
- Camuzard O, Santucci-Darmanin S, Carle GF, et al. Role of autophagy in osteosarcoma. J Bone Oncol. 2019;16:100235. PMID: 31011524.
- Kimura T, Takabatake Y, Takahashi A, et al. Chloroquine in cancer therapy: a double-edged sword of autophagy. Cancer Res. 2013;73(1):3–7. PMID: 23288916.
- Mammucari C, Milan G, Romanello V, et al. FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab. 2007;6(6):458–471. PMID: 18054315.
- Lu Z, Yang H, Sutton MN, et al. ARHI (DIRAS3) induces autophagy in ovarian cancer cells by downregulating the epidermal growth factor receptor, inhibiting PI3K and Ras/MAP signaling and activating the FOXo3a-mediated induction of Rab7. Cell Death Differ. 2014;21(8):1275–1289. PMID: 24769729.
- Shin HJR, Kim H, Oh S, et al. AMPK-SKP2-CARM1 signalling cascade in transcriptional regulation of autophagy. Nature. 2016;534(7608):553–557. PMID: 27309807.
- Mazzu YZ, Liao YR, Nandakumar S, et al. Prognostic and therapeutic significance of COP9 signalosome subunit CSN5 in prostate cancer. Oncogene. 2022;41(5):671–682. PMID: 34802033.
- Mao L, Le S, Jin X, et al. CSN5 promotes the invasion and metastasis of pancreatic cancer by stabilization of FOXM1. Exp Cell Res. 2019;374(2):274–281. PMID: 30352219.
- Yu YS, Tang ZH, Pan QC, et al. Inhibition of Csn3 expression induces growth arrest and apoptosis of hepatocellular carcinoma cells. Cancer Chemother Pharmacol. 2012;69(5):1173–1180. PMID: 22237956.
- Levy JM, Thompson JC, Griesinger AM, et al. Autophagy inhibition improves chemosensitivity in BRAF(V600E) brain tumors. Cancer Discov. 2014;4(7):773–780. PMID: 24823863.
- Wang J, Wu GS. Role of autophagy in cisplatin resistance in ovarian cancer cells. J Biol Chem. 2014;289(24):17163–17173. PMID: 24794870.
- Chen CL, Zhang HL, Yu YY, et al. Chloroquine suppresses proliferation and invasion and induces apoptosis of osteosarcoma cells associated with inhibition of phosphorylation of STAT3. Aging (Albany NY). 2021;13(13):17901–17913. PMID: 34170850.
- SenGupta A, Molkentin JD, Yutzey KE. FoxO transcription factors promote autophagy in cardiomyocytes. J Biol Chem. 2009;284(41):28319–28331. PMID: 19696026.
- Zhang XC, Chen J, Su CH, et al. Roles for CSN5 in control of p53/MDM2 activities. J Cell Biochem. 2008;103(4):1219–1230. PMID: 17879958.
- Berse M, Bounpheng M, Huang XH, et al. Ubiquitin-dependent degradation of Id1 and Id3 is mediated by the COP9 signalosome. J Mol Biol. 2004;343(2):361–370. PMID: 15451666.
- Wang F, Chan CH, Chen K, et al. Deacetylation of FOXO3 by SIRT1 or SIRT2 leads to Skp2-mediated FOXO3 ubiquitination and degradation. Oncogene. 2012;31(12):1546–1557. PMID: 21841822.
- Su HB, Li FQ, Ranek MJ, et al. COP9 Signalosome regulates autophagosome maturation. Circulation. 2011;124(19): 2117–28-U199. PMID: 21986281. DOI:10.1161/CIRCULATIONAHA.111.048934.