
ABSTRACT
L. monocytogenes is a widely used infection model for the research on pathogenesis and host defense against gram-positive intracellular bacteria. Emerging evidence indicates that posttranslational modifications play a critical role in the regulation of macroautophagy/autophagy. However, little is known about the posttranslational modifications of ATG7, the essential protein in the autophagy process. In this study, we demonstrated that the RING-type E3 ligase TRIM7/RNF90 positively regulated autophagosome accumulation by promoting the ubiquitination of ATG7 at K413, thereby affecting L. monocytogenes infection. TRIM7 expression was induced by a variety range of conditions, including starvation, rapamycin stimulation, and L. monocytogenes infection. TRIM7 deficiency in mice or cells resulted in elevated innate immune responses and increased L. monocytogenes infection. ATG7 was associated with TRIM7 and the positive regulatory role of TRIM7 in L. monocytogenes infection-, starvation- or rapamycin-induced autophagosome accumulation was suggested by TRIM7 deficiency, TRIM7 overexpression, and TRIM7 knockdown. Further mechanistic investigation indicated that TRIM7 promoted the K63-linked ubiquitination of ATG7 at K413 and ubiquitination at this site was required for the function of ATG7 in autophagy and L. monocytogenes infection. Thus, our findings suggested a new regulator in intracellular bacterial infection and autophagy, with a novel posttranslational modification targeting ATG7. This research may expand our understanding of host anti-bacterial defense and the role of autophagy in intracellular bacterial infection.
Abbreviations: ATG3: autophagy related 3; ATG5: autophagy related 5; ATG7: autophagy related 7; ATG10: autophagy related 10; ATG12: autophagy related 12; ATG16L1: autophagy related 16 like 1; Baf A1: bafilomycin A1; CQ: chloroquine; BMDC: bone marrow-derived dendritic cell; BMDM: bone marrow-derived macrophage; CFUs: colony-forming units; CXCL10/IP-10: C-X-C motif chemokine ligand 10; EBSS: Earle’s balanced salt solution; ELISA: enzyme-linked immunosorbent assay; IFIT1/ISG56: interferon induced protein with tetratricopeptide repeats 1; IFNB/IFN-β: interferon beta; IL6: interleukin 6; IRF3, interferon regulatory factor 3; Lm: L. monocytogenes; MAP1LC3/LC3: microtubule associated protein 1 light chain 3; MEF: mouse embryonic fibroblast; MOI: multiplicity of infection; PLA: proximity ligation assay; PMA: phorbol myristate acetate; PMA-THP1, PMA-differentiated THP1; PMs: peritoneal macrophages; PTMs: posttranslational modifications; STING1, stimulator of interferon response cGAMP interactor 1; TBK1, TANK binding kinase 1; TNF/TNF-α: tumor necrosis factor; TRIM7/RNF90: tripartite motif containing; Hainan Provincial Natural Science Foundation of China.
Introduction
Macroautophagy, hereafter called autophagy, is a fundamental biological process that contributes to cellular homeostasis by targeting cytoplasmic materials for a lysosome-dependent cellular degradation [Citation1,Citation2]. The initiation of autophagy can be induced by a variety of stress conditions, including nutrient starvation, energy deprivation, infection, and hypoxia [Citation1]. Intracellular macromolecules, organelles, and invading pathogens that are captured and targeted for autophagic destruction are sequestered into double-membrane vesicles called autophagosomes and then delivered to the lysosome for clearance [Citation3].
Listeria monocytogenes (L. monocytogenes), a gram-positive intracellular bacteria, is a food-borne pathogen capable of infecting humans and other animal species. It leads to gastroenteritis that might be complicated by bacteremia and central nervous system infection which is associated with up to 30% mortality [Citation4]. In research, L. monocytogenes is considered an infection model for gram-positive intracellular bacteria and is extensively used in the laboratory to study bacterial pathogenesis and host defense [Citation5]. It has been clear that L. monocytogenes induces an autophagic response, which targets L. monocytogenes in listeriolysin O (LLO)-damaged phagosomes and also in the cytosol under some experimental conditions [Citation6]. Although L. monocytogenes is thought to evolve mechanisms to evade autophagic cellular surveillance, it has been reported that autophagy limits its intracellular growth in the early phase of primary infection [Citation7]. In accordance with this, mice with macrophages- and granulocyte-specific deletion exhibited increased susceptibility to infection with L. monocytogenes, indicating the involvement of autophagy in bacterial infections [Citation8].
It is well established that autophagy is orchestrated by several Atg (autophagy related) proteins, including Atg1 to Atg10, Atg12 to Atg14, Atg16 to Atg18, Atg29, and Atg31, which are essential for the formation of canonical autophagosomes [Citation9]. Accumulating evidence well demonstrates that posttranslational modifications (PTMs) play a pivotal role in cellular processes [Citation10]. In autophagy, PTMs, such as ubiquitination, phosphorylation, and acetylation, are widely involved in the exquisite regulation of autophagy in a timely and efficient way [Citation11]. Recently, emerging evidence has identified ubiquitination as an important regulatory mechanism for the initiation, execution, and termination of autophagy [Citation12]. Ubiquitination is the covalent conjugation of ubiquitin to the substrates through a series of enzymes, including E1 ubiquitin-activating enzyme, E2 ubiquitin-conjugating enzymes, and E3 ubiquitin-protein ligases [Citation13]. Among these three types of enzymes, E3 ubiquitin-protein ligases determine the high substrate specificity for protein ubiquitination [Citation14]. Generally, E3 ubiquitin-protein ligases can be divided into three main groups, HECT-, RING-, and RBP-type [Citation15,Citation16]. Several RING-type E3 ubiquitin-protein ligases have been demonstrated in the regulation of autophagy. For example, TRIM27 ubiquitinates ULK1 directly for proteasomal turnover to maintain control over basal ULK1 levels or catalyzes the ubiquitination of STK38L to inhibit ULK1-mediated autophagy [Citation17]. Another RING-type E3 ligase, RNF216, interacts with BECN1 (beclin 1) and promotes its K48-linked ubiquitination, which contributes to BECN1 degradation and the inhibition of autophagy [Citation18]. However, most of the identified modifications by ubiquitination in autophagy are targeting ULK1 or BECN1, and little is known about the regulation of other ATG proteins by ubiquitination.
In this study, we identified for the first time a pivotal role for TRIM7 (tripartite motif containing 7)-mediated ubiquitination of ATG7 in autophagy and clearance of L. monocytogenes. TRIM7, is a recognized RING-type E3 ubiquitin-protein ligase, involved in the development, progression, and metastasis of cancers, enterovirus replication, and TLR4-mediated innate response [Citation19–24]. Our previous research work has demonstrated that TRIM7 plays an important role in innate immune responses against invading pathogens. TRIM7 inhibits DNA virus-triggered innate immune responses by promoting K48-linked ubiquitination and subsequent degradation of STING1, whereas TRIM7 targets MAVS for degradation and negatively regulates antiviral responses against RNA virus [Citation25,Citation26]. However, the role of TRIM7 in the clearance of intracellular bacteria remains unclear. We showed here that TRIM7 promoted the K63-linked ubiquitination of ATG7 at lysine residues 413 under a variety of conditions and positively regulates autophagy. TRIM7 was induced by diverse autophagy activation conditions, including starvation, rapamycin stimulation, and L. monocytogenes infection. Trim7-deficient mice and cells showed increased susceptibility to L. monocytogenes infection, with enhanced innate immune response. Further study indicated that TRIM7 promoted the accumulation of autophagosomes during starvation, rapamycin stimulation, or L. monocytogenes infection. Collectively, our findings suggest a new regulatory role of TRIM7 in autophagy and host defenses against intracellular bacteria.
Results
TRIM7 deficiency promotes IFNB production but reduces the survival rate in mice following L. monocytogenes infection in vivo
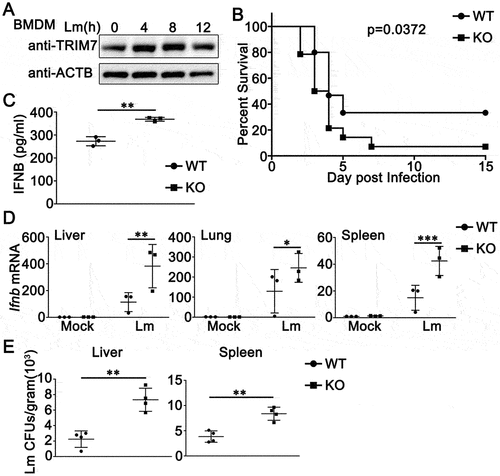
L. monocytogenes induces IFNB/IFN-β expression through a STING1-dependent pathway [Citation27]. Our previous research work indicated TRIM7 negatively regulates DNA virus- or cytosolic DNA-triggered signaling pathways by promoting the degradation of STING1 [Citation26]. Thus, we wondered whether TRIM7 played a role during the process of L. monocytogenes infection. Firstly, we examined the expression pattern of TRIM7 upon L. monocytogenes infection. As shown in , TRIM7 expression increased in protein levels 4 h after L. monocytogenes infection and reached the peak at 8 h in bone marrow-derived macrophages (BMDMs), suggesting that a relationship existed between TRIM7 expression and L. monocytogenes infection. Next, we used trim7-deficient mice to investigate the role of TRIM7 in L. monocytogenes infection. Compared to wild-type mice, trim7-deficient mice exhibited a lower survival rate following L. monocytogenes infection (). Serum and various organs were collected for the measurement of type I interferon production. As shown in , the induction of IFNB by L. monocytogenes infection was enhanced in the serum of trim7-deficient mice. Consistently, trim7 deficiency upregulated IFNB expression in the liver, lung, and spleen following L. monocytogenes infection (). In addition, in both the liver and spleen, increased L. monocytogenes infections were observed in trim7-deficient mice (). Taken together, these data indicate that whereas trim7 deficiency increases the type I IFN production against L. monocytogenes infection in vivo, the resistance of L. monocytogenes is reduced in trim7-deficient mice.
Figure 1. TRIM7 protects mice from L. monocytogenes infection. (A) BMDMs were stimulated with L. monocytogenes (Lm, MOI = 10) for indicated time periods. Afterward, the cells were lysed for immunoblot assays. ACTB/β-actin was used as a loading control. (B) Wild-type (WT) and trim7-deficient (KO) mice (n = 14) were intravenously (i.v.) infected with Lm (1.5 × 105 CFUs/mouse). Mouse survival rates were monitored twice a day during 15 days of infection. (C) ELISA of IFNB in serum of wild-type (WT) and trim7-deficient (KO) mice (n = 3) 6 h after intravenous infection with Lm (1.5 × 105 CFUs/mouse). (D) Wild-type (WT) and trim7-deficient (KO) mice (n = 3) were infected with Lm (1.5 × 105 CFUs/mouse) for 24 h, and then the livers, lungs, and spleens of mice were subjected to real-time PCR analysis of Ifnb mRNA. (E) Quantification of bacterial loads in target organs (liver and spleen) from wild-type (WT) and trim7-deficient (KO) mice (n = 4) on the 3rd day after injection with Lm (1.5 × 105 CFUs/mouse). Bacterial loads were measured in CFUs per gram of liver (left panel) and gram of spleen (right panel). The data are representative of three independent experiments and are presented as mean ± SD. *, p < 0.05, **, p < 0.01, ***, p < 0.001.
TRIM7-deficiency or TRIM7 knockdown increases both bacterial infection and innate immune responses following L. monocytogenes infection
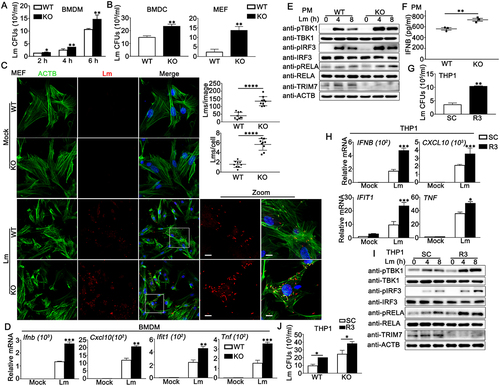
Next, we explored the role of TRIM7 at the cellular level. BMDMs, bone marrow-derived dendritic cells (BMDCs), and mouse embryonic fibroblasts (MEFs) were isolated and cultured from the trim7-deficient mice [Citation26]. TRIM7-deficient HaCaT cell line was generated by CRISPR/Cas9 strategy [Citation26]. Wild-type or trim7-deficient BMDMs, BMDCs, MEFs, and HaCaT cells were infected with L. monocytogenes or left untreated, with colony-forming units (CFUs) corresponding to intracellular live bacteria. Time-course analysis showed that differences in L. monocytogenes infection between wild-type and trim7-deficient BMDMs were observed in all the three time points detected, with the largest differences occurring at 6 h after infection (). Thus, 6 h was used in the following experiments and the increase in L. monocytogenes infection caused by trim7 deficiency was confirmed in BMDCs, MEFs, and HaCaT cells ( and Figure S1a). Next, wild-type and trim7-deficient MEFs or HaCaT cells were infected with L. monocytogenes or left uninfected and then imaged by confocal microscopy. The red dots indicated L. monocytogenes infection as suggested by an antibody against L. monocytogenes. Compared to that in wild-type cells, more red dots were detected in trim7-deficient MEFs and HaCaT cells, suggesting trim7 deficiency promoted L. monocytogenes infection (, S1b, and S1c). Similarly, flow cytometry using the same antibody against L. monocytogenes revealed the enhancement of L. monocytogenes infection by trim7 deficiency in MEFs (Figure S1D). Meanwhile, the host innate immune responses against L. monocytogenes infection were examined. During L. monocytogenes infection, increased production of IFNB, CXCL10/IP-10, IFIT1/ISG56, and TNF/TNF-α was observed in RNA90-deficient BMDMs (). Consistently, Trim7 deficiency in HaCaT cells enhanced the production of IFNB and CXCL10 upon L. monocytogenes infection (Figure S1E). In peritoneal macrophages (PMs), trim7 deficiency resulted in elevated phosphorylation levels of TBK1, IRF3, and RELA/p65 after L. monocytogenes infection (). Upon L. monocytogenes infection, compared to wild-type PMs, trim7-deficient PMs exhibited higher levels of IFNB in ELISA assays (). To further evaluate the role of TRIM7 in L. monocytogenes infection, bacterial entry and L. monocytogenes triggered innate immune responses were investigated in TRIM7-silenced phorbol-12-myristate-13-acetate (PMA)-differentiated THP1 (PMA-THP1, a human macrophage-like cell line) cells. TRIM7 was silenced by TRIM7-specific siRNA R3, the efficiency of which had been proved in our previous research [Citation26]. As shown in , an increased bacterial load of L. monocytogenes was observed in TRIM7-silenced cells. At the same time, TRIM7 knockdown enhanced L. monocytogenes-induced innate immune responses, including the production of IFNB, CXCL10, IFIT1, and TNF () and the phosphorylation of TBK1, IRF3, and RELA (). Finally, considering that TRIM7 regulated STING1-mediated innate immune responses [Citation26], we examined whether the inhibitory role of TRIM7 in L. monocytogenes infection was dependent on STING1. Wild-type or STING1-deficient PMA-THP1 cells were transfected with control siRNA (SC) or TRIM7-specific siRNA (R3) and CFUs were obtained after transfection. As shown in , although STING1 deficiency resulted in elevated L. monocytogenes infection, it did not make a significant change in the enhancement in L. monocytogenes infection caused by TRIM7 knockdown, suggesting TRIM7 negatively regulated L. monocytogenes infection independent on STING1. Taken together, all these findings suggest that both L. monocytogenes infection and innate immune responses are enhanced by TRIM7 deficiency or TRIM7 knockdown.
Figure 2. TRIM7 negatively regulated Lm infection in cells. (A) CFUs obtained at indicated time points from wild-type (WT) and trim7-deficient (KO) BMDMs infected with L. monocytogenes (Lm) (MOI = 10). (B) CFUs obtained at 6 h from wild-type (WT) and trim7-deficient (KO) BMDCs (left panel) or MEFs (right panel) infected with L. monocytogenes (Lm) (MOI = 10). (C) Confocal microscopy images of Lm-infected wild-type (WT) and trim7-deficient (KO) MEFs at 6 h after infection. The left panel shows DAPI (blue), Lm (red), ACTB (green), and merged views of three channels. The bottom right panel shows magnified views of the boxed areas from the merged view. Yellow indicates the co-localization of Lm with ACTB. Scale bars: 10 μm (magnified views). The top right panel shows the statistical analysis of the total number of bacteria and the number of bacteria per cell in all pictures (9 pictures per genotype). (D) Wild-type (WT) and trim7-deficient (KO) BMDMs were infected with Lm for 8 h, and then subjected to real-time PCR analysis. (E) Wild-type (WT) and trim7-deficient (KO) PMs were infected with Lm (MOI = 10) for indicated time periods. Afterward, the cells were lysed for immunoblot assays. (F) Wild-type (WT) and trim7-deficient (KO) PMs were infected with Lm (MOI = 10) for 24 h. The secretion of IFNB was measured by ELISA. (G) PMA-THP1 cells were transfected with control siRNA (SC) or TRIM7-specific siRNA (R3). At 24 h after transfection, the cells were infected with Lm (MOI = 10) for 6 h, and then colony forming assay was performed. (H) PMA-THP1 cells were transfected with control siRNA (SC) or TRIM7-specific siRNA (R3). At 24 h after transfection, the cells were infected with Lm (MOI = 10) for 8 h and then subjected to real-time PCR analysis. (I) PMA-THP1 cells were transfected with control siRNA (SC) or TRIM7-specific siRNA (R3). At 24 h after transfection, the cells were infected with Lm (MOI = 10) for indicated time periods. Afterward, the cells were lysed for immunoblot assays. (J) Wild-type (WT) or STING1-deficient (KO) PMA-THP1 cells were transfected with control siRNA (SC) or TRIM7-specific siRNA (R3). At 24 h after transfection, the cells were infected with Lm (MOI = 10) for 6 h, and then colony forming assay was performed. The data are representative of three independent experiments and are presented as mean ± SD. *, p < 0.05, **, p < 0.01, ***, p < 0.001, ****, p < 0.0001.
TRIM7 interacts with ATG7
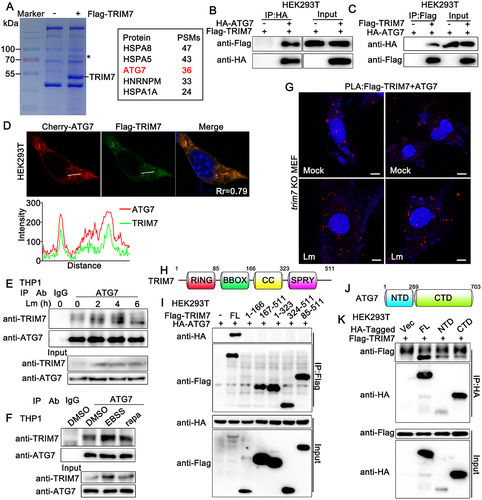
TRIM7 deficiency promoted L. monocytogenes infection with elevated innate immune responses and in a STING1-independent way, suggesting that other mechanisms existed for the regulation of L. monocytogenes infection. We wondered whether TRIM7 colocalized with or modified the bacterial surface directly. Flag-TRIM7 was transfected into trim7-deficient MEFs and then infected with L. monocytogenes. However, no significant colocalization between TRIM7 and L. monocytogenes was observed, indicating it was unlikely that TRIM7 modified the bacterial surface directly (Figure S1F). Then, to explore the underlying mechanism by which TRIM7 regulated L. monocytogenes infection, we tried to identify the potential substrate of TRIM7 by co-immunoprecipitation. HEK293T cells were transfected with or without Flag-TRIM7 and then subjected to immunoprecipitation with an anti-Flag antibody. The immunoprecipitates were sent for mass spectroscopy analysis and a few proteins were identified as potential TRIM7-associated proteins (). ATG7, which has been reported to be a core autophagy-related protein that is indispensable to classical autophagy [Citation28], was identified to be associated with TRIM7 (). To investigate the association between TRIM7 and ATG7, Flag-TRIM7 and HA-ATG7 were transfected into HEK293T cells. Subsequent co-immunoprecipitation experiments were performed and the results suggested exogenous expressed TRIM7 bound to ATG7 (). Consistently, confocal microscopy assays indicated that Flag-TRIM7 was colocalized with Cherry-ATG7 in HEK293T cells, as much overlap between the two signals at the pixel level was observed (). To confirm the endogenous interaction between TRIM7 and ATG7, PMA-THP1 cells were treated with L. monocytogenes infection, starvation with EBSS, rapamycin or left untreated and then subjected to co-immunoprecipitation analysis. As shown in , endogenous TRIM7 interacted with ATG7 in untreated PMA-THP1 cells and more TRIM7-ATG7 interaction was observed after L. monocytogenes infection, starvation, or rapamycin stimulation, which might be caused at least partly by the increased expression of TRIM7. In addition, we performed in situ-proximity ligation assay (PLA) to validate the TRIM7-ATG7 interaction. Trim7-deficient MEFs were transfected with Flag-TRIM7 and then left uninfected or infected with L. monocytogenes. PLA was performed using anti-Flag and anti-ATG7 antibodies. The detected red dots depicted the co-localization of TRIM7 and ATG7. As shown in , the red dots indicating TRIM7-ATG7 interaction were detected in untreated cells and more red dots were observed after L. monocytogenes infection. Next, we explored the region of TRIM7 responsible for its interaction with ATG7. Surprisingly, co-immunoprecipitation assays indicated only the full-length TRIM7 interacted with ATG7 (). We also mapped the binding regions on ATG7 for the TRIM7 association. As shown in , only very weak interaction was detected between the C-terminal domain (CTD) or N-terminal domain (NTD) of ATG7 and TRIM7. In all, our findings suggest that TRIM7 interacts with ATG7.
Figure 3. TRIM7 interacted with ATG7. (A) HEK293T cells were transfected with Flag-TRIM7 (+) or Vector (-) for 24 h, and then the cell lysates were subjected to immunoprecipitation with an anti-Flag antibody. The immunoprecipitates were resolved in SDS-PAGE followed by coomassie blue staining. The specific band highlighted by an asterisk was excised for mass spectroscopy identification. PSMs, peptide spectrum matches. (B, C) HEK293T cells were transfected with Flag-TRIM7 and HA-ATG7. At 24 h after transfection, the cell lysates were subjected to immunoprecipitation (IP) and immunoblot (IB) analysis as indicated. (D) HEK293T cells were transfected with Flag-TRIM7 and mCherry-ATG7. At 24 h after transfection, immunofluorescence was performed using anti-Flag (green). Nuclei were stained with DAPI. Plots of pixel intensity along the white line were shown in the right panel. Pearson’s correlation coefficient was calculated using ImageJ software. Rr, Pearson’s correlation coefficient. (E) PMA-THP1 cells were infected with Lm for indicated time periods, and then the cell lysates were subjected to immunoprecipitation (IP) and immunoblot (IB) analysis as indicated. (F) PMA-THP1 cells were treated with rapamycin for 12 h or EBSS for 2 h, and then the cell lysates were subjected to immunoprecipitation (IP) and immunoblot (IB) analysis as indicated. (G) trim7-deficient (KO) MEFs were transfected with Flag-TRIM7. At 24 h after transfection, the cells were infected with Lm for 4 h or left uninfected, and then in situ PLA assay was performed to examine the colocalization of TRIM7 and ATG7. TRIM7-ATG7 complex, red; nuclei, blue. (H) A schematic presentation of full-length TRIM7. RING, ring-finger domain; BB, B-box domain; CC, coiled-coil domain; SPRY, SPRY domain. (I) HEK293T cells were transfected with indicated plasmids. At 24 h after transfection, the cell lysates were subjected to immunoprecipitation (IP) and immunoblot (IB) analysis as indicated. (J) A schematic presentation of full-length ATG7. NTD, N-terminal domain, 1–289aa; CTD, C-terminal domain, 290–703aa. (K) HEK293T cells were transfected with indicated plasmids. At 24 h after transfection, the cell lysates were subjected to immunoprecipitation (IP) and immunoblot (IB) analysis as indicated. The data are representative of three independent experiments.
TRIM7 promotes L. monocytogenes-triggered autophagy
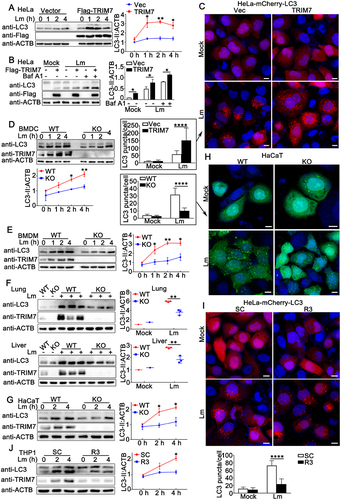
Given that TRIM7 interacted with ATG7, the protein essential to autophagy, and autophagy plays an important role in host defense against intracellular bacteria, we proposed the hypothesis that TRIM7 regulates autophagy targeting ATG7. To test our hypothesis, the effect of TRIM7 overexpression on L. monocytogenes-induced autophagy was evaluated by LC3-II levels in immunoblot assays. As shown in , TRIM7 overexpression in HeLa cells resulted in enhanced LC3-II accumulation in a dose-dependent pattern during L. monocytogenes infection. In addition, to distinguish between effects on autophagosome accumulation and degradation, we pretreated the cells with bafilomycin A1 (Baf A1), which impaired lysosome activity and allowed us to assess LC3-II/autophagosome formation. TRIM7 promoted L. monocytogenes infection-triggered LC3-II accumulation to a similar extent with or without Baf A1, suggesting TRIM7 regulated the autophagosome formation, but not the degradation by lysosome (). Further, the effect of TRIM7 overexpression on L. monocytogenes-induced LC3-II puncta formation was examined by confocal microscopy. HeLa cells expressing mCherry-LC3 (HeLa-mCherry-LC3) were transfected with TRIM7 or a control vector, and then infected with L. monocytogenes or left uninfected. As shown by confocal microscopy, LC3-II puncta formation was augmented by TRIM7 overexpression ().
Figure 4. TRIM7 positively regulated Lm infection-induced autophagy. (A) HeLa cells were transfected with 0.5 μg Vector or TRIM7 plasmid. At 24 h after transfection, the cells infected with Lm (MOI = 10) or PBS for indicated time periods, and then subjected to immunoblot analysis. LC3-II density was shown in the right panel. (B) HeLa cells were transfected with 0.5 μg Vector (-) or TRIM7 (+) plasmid. At 24 h after transfection, the cells were treated with PBS, Lm (MOI = 10), or Lm with bafilomycin A1 (400 nM) for 2 h, and then subjected to immunoblot analysis. LC3-II density was shown in the right panel. (C) HeLa-mCherry-LC3 cells were transfected with Vector (Vec) or TRIM7. At 24 h after transfection, the cells were infected with Lm (MOI = 10) or left uninfected for 4 h, and then subjected to confocal microscopy analysis. Scale bars: 10 μm. LC3 puncta quantification was shown in the left panel. (D, E) Wild-type (WT) and trim7-deficient (KO) BMDCs (D) and BMDMs (E) were stimulated with Lm (MOI = 10) for the indicated time periods. Afterward, the cells were lysed for immunoblot assays (left panels). LC3-II density was shown at the bottom (D) or right (E) panels. (F) Wild-type (WT) and trim7-deficient (KO) mice (n = 3) were intravenously (i.v.) infected with Lm (1.5 × 105 CFUs/mouse) or left uninfected for 2 days, then the livers and lungs of the mice were subjected to immunoblot assays. LC3-II density was shown in the right panels. (g) Wild-type (WT) and TRIM7-deficient (KO) HaCaT cells were stimulated with Lm (MOI = 10) for indicated time periods. Afterward, the cells were lysed for immunoblot assays. LC3-II density was shown in the right panels. (h) Wild-type (WT) and TRIM7-deficient (KO) HaCaT cells were transfected with GFP-LC3 plasmid. At 24 h after transfection, the cells were infected with Lm (MOI = 10) or left uninfected for 4 h, and then subjected to confocal microscopy analysis. Scale bars: 10 μm. LC3 puncta quantification was shown in the left panel. (i) HeLa-mCherry-LC3 cells were transfected with control siRNA (SC) or TRIM7-specific siRNA (R3). At 24 h after transfection, the cells were infected with Lm (MOI = 10) or left uninfected for 4 h, and then subjected to confocal microscopy analysis. Scale bars: 10 μm. LC3 puncta quantification was shown in the bottom panel. (j) PMA-THP1 cells were transfected with control siRNA (SC) or TRIM7-specific siRNA (R3). At 24 h after transfection, the cells were infected with Lm (MOI = 10) for indicated time periods. Afterward, the cells were lysed for immunoblot assays. LC3-II density was shown in the right panel. The data are representative of three independent experiments and are presented as mean ± SD. *, p < 0.05, **, p < 0.01, ****, p < 0.0001.
To confirm the positive regulatory role of endogenous TRIM7 in L. monocytogenes-induced autophagosome accumulation, trim7-deficient BMDCs or BMDMs were infected with L. monocytogenes and then the autophagosome accumulation was evaluated by LC3-II levels. Induction of LC3-II accumulation by L. monocytogenes infection was observed in both BMDCs and BMDMs and this induction was significantly decreased by trim7 deficiency, suggesting TRIM7 promoted L. monocytogenes-triggered autophagosome accumulation (). Then, the effect of TRIM7 on LC3-II accumulation was examined in vivo. Wild-type or trim7-deficient mice were infected with L. monocytogenes and LC3-II accumulation in lungs and lives was detected by immunoblots. In both lungs and livers, compared to wild-type mice, trim7-deficient mice exhibited lower levels of LC3-II accumulation in these organs (). Further, we investigated the role of TRIM7 in L. monocytogenes-triggered autophagosome accumulation in TRIM7-deficient HaCaT cells, the generation of which had been demonstrated in our previous research [Citation26]. As shown in , TRIM7 deficiency significantly decreased the LC3-II accumulation in HaCaT cells upon L. monocytogenes infection. Consistently, confocal microscopy analysis indicated that decreased LC3-II puncta formation was observed in TRIM7-deficient HaCaT cells than that in wild-type cells after L. monocytogenes infection ().
Finally, the effect of TRIM7 on L. monocytogenes-induced autophagosome accumulation was examined in TRIM7-silenced cells. TRIM7 knockdown by siRNA R3 in Hela cells expressing mCherry-LC3 inhibited LC3-II puncta formation as suggested by confocal microscopy (). Consistent with these findings, LC3-II accumulation was decreased by TRIM7 knockdown via the transfection of R3 (). In all, the positive regulatory role of TRIM7 in L. monocytogenes-induced autophagosome accumulation is suggested by TRIM7 deficiency, TRIM7 overexpression, and TRIM7 knockdown.
TRIM7 promotes autophagy in the basal state and response to a variety of stress conditions
Given that TRIM7 manipulated L. monocytogenes-triggered autophagy, we wondered whether TRIM7 regulated the autophagy that was induced by other conditions, including nutrient starvation and rapamycin stimulation. Considering that the expression levels of TRIM7 are usually very low in rest cells and can be induced by virus or bacterial infection, we examined the expression patterns of TRIM7 protein in these conditions. Immunoblot assays indicated that TRIM7 protein expression was induced by nutrient starvation using EBSS (Figure S2A) or rapamycin stimulation (Figure S2B) in PMA-THP1 cells. Next, HeLa cells were transfected with TRIM7 and then treated with EBSS (starvation) or rapamycin. Immunoblot assays demonstrated that TRIM7 overexpression promoted LC3-II accumulation upon starvation or rapamycin stimulation (). Using Baf A1 to inhibit lysosome activity, we found that TRIM7 enhanced the LC-II accumulation to a similar extent with or without Baf A1, an indication that TRIM7 increased autophagosome accumulation, but not lysosomal degradation upon starvation or rapamycin stimulation (). Further, in basal conditions, TRIM7 overexpression caused an increase in LC3-II levels with or without Baf A1, suggesting positive regulation of TRIM7 in autophagosome formation (). In addition, TRIM7 overexpression in HeLa-mCherry-LC3 significantly increased LC3-II puncta accumulation after nutrient starvation or rapamycin treatment ().
Figure 5. TRIM7 positively regulated autophagy. (A) HeLa cells were transfected with Vector (Vec) or Flag-TRIM7 and then treated with EBSS for indicated time periods. Afterward, the cell lysates were subjected to immunoblot analysis as indicated. LC3-II density was shown in the right panel. (B) HeLa cells were transfected with vector (Vec) or Flag-TRIM7. At 24 h after transfection, the cells were treated with rapamycin (200 nM) for indicated time periods. Afterward, the cells were lysed for immunoblot assays. LC3-II density was shown in the right panel. (C) HeLa cells were transfected with Vector (-) or Flag-TRIM7 (+) plasmids. At 24 h after transfection, the cells were treated with DMEM (with 10% FBS), EBSS, or EBSS with bafilomycin A1 (Baf A1, 400 nM) for 2 h, and then subjected to immunoblot analysis. LC3-II density was shown in the right panel. (D) HeLa cells were transfected with Vector (-) or Flag-TRIM7 (+). At 24 h after transfection, the cells were treated with DMSO (Mock) or rapamycin (200 nM) for 12 h. Cells with rapamycin stimulation were treated with or without bafilomycin A1 (Baf A1, 400 nM) for 2 h before collection. Then, the cells were subjected to immunoblot analysis. LC3-II density was shown in the right panel. (E) HeLa cells were transfected with different doses of Flag-TRIM7. At 24 h after transfection, the cells were treated with bafilomycin A1 (Baf A1, 400 nM) or DMSO for 4 h, and then the cells were subjected to immunoblot analysis. LC3-II density was shown in the right panel. (F) HeLa-mCherry-LC3 cells were transfected with Vector (Vec) or TRIM7 plasmids. At 24 h after transfection, the cells were treated with rapamycin (200 nM) for 12 h, EBSS for 2 h, or DMEM with 10% FBS for 2 h, and then subjected to confocal microscopy analysis. Scale bars: 10 μm. LC3 puncta quantification was shown in the right panel. (G) Wild-type (WT) and trim7-deficient (KO) BMDMs were treated with EBSS for indicated time periods, and then the cells were lysed for immunoblot assays. LC3-II density was shown in the right panel. (H) Wild-type (WT) and trim7-deficient (KO) BMDMs were treated with different doses of rapamycin (0, 100, 200 nM) for 12 h. Afterward, the cells were lysed for immunoblot assays. LC3-II density was shown in the right panel. (I) Wild-type (WT) and TRIM7-deficient (KO) HaCaT cells, were transfected with GFP-LC3 plasmid, and then treated with rapamycin (200 nM) or DMSO as a control for 12 h, and then subjected to confocal microscopy analysis. Scale bars: 10 μm. LC3 puncta quantification was shown in the bottom panel. (J) Wild-type (WT) and TRIM7-deficient (KO) HaCaT cells were treated with CQ (100 μM), bafilomycin A1 (Baf A1, 400 nM) or DMSO as a control for 4 h, and then the cells were subjected to immunoblot analysis. LC3-II density was shown in the bottom panel. The data are representative of three independent experiments and are presented as mean ± SD. *, p < 0.05, **, p < 0.01, ***, p < 0.001.
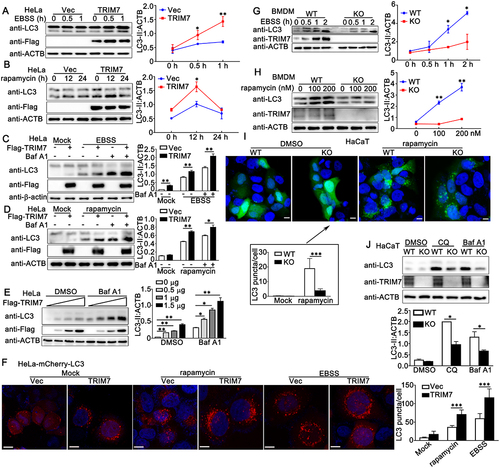
Then the role of TRIM7 in starvation- or rapamycin-triggered autophagosome accumulation was explored in TRIM7-deficient cells. As suggested by immunoblots, trim7 deficiency in BMDMs, MEFs or HaCaT cells resulted in decreased LC3-II accumulation upon starvation (, Figure S2c, and Figure S2e) or rapamycin stimulation (, Figure S2d and Figure S2f), indicating that TRIM7 played a positive role in the regulation of autophagosome formation under these conditions. In addition, compared to wild-type cells, TRIM7-deficient HaCaT cells displayed lower levels of LC3-II puncta formation after stimulation with rapamycin (). Further, in basal condition, although the effect of TRIM7 deficiency on LC3-II levels was not significant because of the low levels of LC3-II in basal condition, TRIM7 deficiency caused a decrease in LC3-II levels in the presence of lysosomal inhibitor chloroquine (CQ) or Baf A1, suggesting positive regulation of TRIM7 in autophagosome formation (). Finally, TRIM7 knockdown prevented the LC3-II puncta accumulation that was triggered by nutrient starvation or rapamycin stimulation (Figure S2G). Consistently, TRIM7 knockdown impaired autophagosome formation in basal conditions (Figure S2H). Taken together, TRIM7 promotes basal autophagy and autophagy induced by a variety of stress conditions.
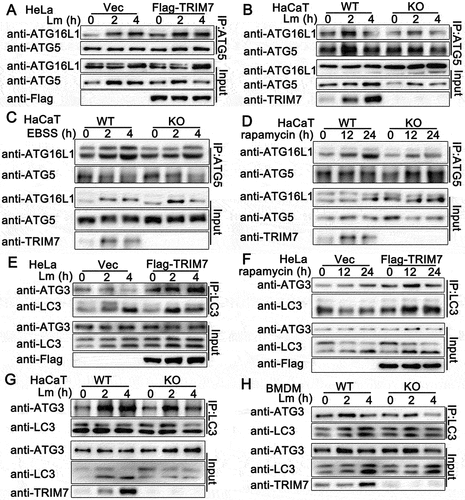
TRIM7 regulates ATG7-related ubiquitin-like conjugation systems in autophagy
As an E1-like enzyme, ATG7 is involved in two ubiquitin-like conjugation systems in classical autophagy, ATG12–ATG5-ATG16L1 conjugation system with ATG10 as an E2-like enzyme, and LC3-PE conjugation system with ATG3 as E2-like enzyme [Citation29]. Given that TRIM7 interacted with ATG7 and promoted autophagosome formation, we examined whether TRIM7 regulated ATG7-related ubiquitin-like conjugation systems in autophagy by co-immunoprecipitation. As shown in , TRIM7 overexpression promoted but TRIM7 deficiency impaired the association of ATG5 with ATG16L1 in autophagy that was triggered by L. monocytogenes infection. Consistently, after the treatment of starvation or rapamycin, compared to wild-type cells, TRIM7-deficient HaCaT cells exhibited decreased ATG5-ATG16L1 association (). Next, we investigated the effects of TRIM7 on the second conjuration system and found that TRIM7 overexpression resulted in enhanced LC3-ATG3 association after L. monocytogenes infection or rapamycin stimulation (), whereas TRIM7 deficiency impaired the LC3-ATG3 association after L. monocytogenes infection in both HaCaT cells and BMDMs (). In all, TRIM7 positively regulates ATG7-related ubiquitin-like conjugation systems in autophagy.
Figure 6. TRIM7 regulated ATG7-related ubiquitin-like conjugation systems in autophagy. (A) HeLa cells were transfected with Vector (Vec) or Flag-TRIM7, and then infected with Lm (MOI = 10) for indicated time periods. Afterward, the cell lysates were subjected to immunoprecipitation (IP) and immunoblot (IB) analysis as indicated. (B) Wild-type (WT) and TRIM7-deficient (KO) HaCaT cells were infected with Lm (MOI = 10) for indicated time periods. Afterward, the cell lysates were subjected to immunoprecipitation (IP) and immunoblot (IB) analysis as indicated. (C, D) Wild-type (WT) and TRIM7-deficient (KO) HaCaT cells were treated with EBSS (C) or rapamycin (D) for indicated time periods. Afterward, the cell lysates were subjected to immunoprecipitation (IP) and immunoblot (IB) analysis as indicated. (E) HeLa cells were transfected with Vector (Vec) or TRIM7-encoding plasmids, and were infected with Lm (MOI = 10) for indicated time periods. Afterward, the cell lysates were subjected to immunoprecipitation (IP) and immunoblot (IB) analysis as indicated. (F) HeLa cells were transfected with Vector (Vec) or Flag-TRIM7, and then treated with rapamycin for indicated time periods. Afterward, the cell lysates were subjected to immunoprecipitation (IP) and immunoblot (IB) analysis as indicated. (G, H) Wild-type (WT) and TRIM7-deficient (KO) HaCaT cells (G) or BMDMs (H) were infected with Lm (MOI = 10) for indicated time periods. Afterward, the cell lysates were subjected to immunoprecipitation (IP) and immunoblot (IB) analysis as indicated. The data are representative of three independent experiments.
TRIM7 promoted the K63-linked ubiquitination of ATG7
Considering TRIM7 serves as an E3 ligase in several signaling pathways [Citation19,Citation25,Citation26], we thought TRIM7 might have regulated the ubiquitination of ATG7. To examine this hypothesis, TRIM7 was co-transfected with ATG7 and HA-tagged ubiquitin, and the ubiquitination of ATG7 was detected by immunoprecipitation. As shown in , TRIM7 overexpression increased the ubiquitination of ATG7 in a dose-dependent manner. To exclude the possibility that the observed ubiquitination was on ATG7-interacting proteins, and not ATG7 itself, we performed the reverse IP and detected the modification of ATG7. As shown in Figure S3A, increased high molecular weight smears of ATG7 were observed with TRIM7 overexpression, indicating TRIM7 promoted the ubiquitination of ATG7. The activity of RING-type ubiquitin-protein ligases is dependent on their RING domains which contain conserved cysteine and histidine residues essential for recruiting the E2 ubiquitin-conjugating enzymes [Citation30]. Our previous research indicated that the C29, 32A mutant (the integrity of the RING domain was destroyed) and the ∆R mutant (lacking RING domain) lost the ability of ubiquitination targeting STING1 [Citation26]. Therefore, we investigated the effects of these mutants on the ubiquitination and the results suggested both C29,32A and ∆R mutants barely increased the ubiquitination of ATG7, suggesting the RING domain of TRIM7 was critical to the ubiquitination of ATG7 (). Ubiquitin contains seven Lys residues (K6, K11, K27, K29, K33, K48, and K63), so seven different polyubiquitin chains can be generated, among which K48-and K63-linked ubiquitination is the most studied types of linkage [Citation31]. K48-linked ubiquitination usually mediates the degradation of protein in a proteasome-dependent way, whereas K63-linked modification mainly regulates signal transduction in a non-proteolytic manner [Citation32]. To identify the type of linkage that was promoted by TRIM7, we used expression plasmids for ubiquitin mutants retaining only a single lysine residue. Immunoprecipitation and immunoblot analysis indicated that TRIM7 promoted only K63 mutants (only the Lys residue 63 was retained) mediated ubiquitination of ATG7, suggesting TRIM7 enhanced the K63-linked ubiquitination of ATG7 (). This phenomenon was further confirmed by the usage of K48R (only the Lys residue 48 was mutated to Arg) and K63R (only the Lys residue 63 was mutated to Arg). As shown in , TRIM7 increased K48R mediated ubiquitination of ATG7, but not K63R, indicating the Lys residue 63 was essential to the TRIM7-triggered linkage of ATG7 with ubiquitin (). Further, in vitro ubiquitination assays suggested that TRIM7 promoted the ubiquitination of ATG7 directly using UBC5 as a ubiquitin-conjugating E2 enzyme, but the C29,32A and ∆R mutants of TRIM7 did not ( and S3b).
Figure 7. TRIM7 promoted the ubiquitination of ATG7. (A) HEK293T cells were transfected with Myc-ATG7, HA-Ubi, and increasing doses of Flag-TRIM7 (0, 0.5, 1, and 2 μg). At 24 h after transfection, the cells were lysed and subjected to immunoprecipitation (IP) and immunoblot (IB) analysis. (B) HEK293T cells were transfected with the indicated plasmids. At 24 h after transfection, immunoprecipitation (IP) and immunoblot (IB) analysis were performed as indicated. CA, TRIM7 C29, 32A mutant; ΔR, 85–511aa of TRIM7. (C, D) HEK293T cells were transfected with plasmids as indicated. 24 h later, immunoprecipitation (IP) and immunoblot (IB) analysis were performed as indicated. (E) Immunoblot analysis of ATG7 ubiquitination in vitro. ATG7 and wild-type TRIM7 were quickly translated in vitro, and then, the biotin-ubiquitin E1 and indicated E2s were added for in vitro ubiquitination assays. Ubiquitination of ATG7 was detected by anti-Ubi. (F) Wild-type (WT) and TRIM7-deficient (KO) HaCaT cells, were transfected with plasmids as indicated and then infected with Lm for 4 h. Afterward, the cells were lysed for immunoprecipitation (IP) and immunoblot (IB) assays. CA, TRIM7 C29, 32A mutant. (G, H) Wild-type (WT) and trim7-deficient (KO) BMDMs (G) or BMDCs (H) were infected with Lm for indicated time periods. Afterward, the cells were lysed for immunoprecipitation (IP) and immunoblot (IB) assays. (i) Wild-type (WT) and trim7-deficient (KO) MEFs were transfected with Vector (-) or TRIM7 (+) plasmids, and were stimulated with rapamycin for 12 h. Afterward, the cell lysates were subjected to immunoprecipitation (IP) and immunoblot (IB) analysis as indicated. The data are representative of three independent experiments.
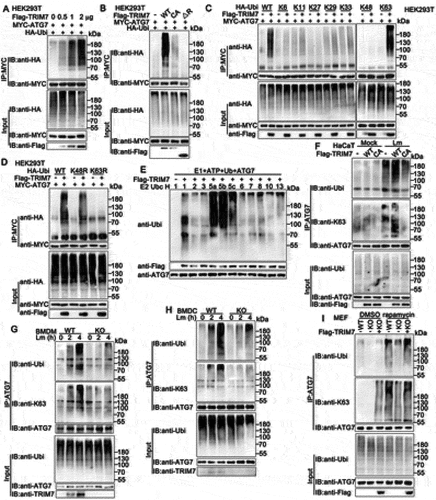
Next, we investigated the effects of TRIM7 on the ubiquitination of ATG7 during autophagy that was triggered by L. monocytogenes infection, starvation, or rapamycin. Firstly, we examined whether the ubiquitination of ATG7 was induced in these conditions. As shown in and Figure S3c, upon the treatment of L. monocytogenes infection, starvation, or rapamycin, the ubiquitination of ATG7 was significantly induced. Then, the role of TRIM7 in the ubiquitination of ATG7 in these conditions was evaluated. Wild-type or trim7-deficient BMDMs, BMDCs, or MEFs were infected with L. monocytogenes and then subjected to the examination of ubiquitination of ATG7. Immunoprecipitation and immunoblots assays indicated that L. monocytogenes infection induced the ubiquitination of ATG7 and the ubiquitination was impaired by trim7-deficiency ( and S3d). Further, in MEFs, trim7 deficiency decreased rapamycin-triggered ubiquitination of ATG7 and this decrease was rescued by TRIM7 transfection (). Meantime, upon the treatment of starvation or rapamycin, TRIM7 overexpression in HeLa cells increased the K63-linked ubiquitination of ATG7 (Figure S3E) whereas TRIM7 silence in PMA-THP1 cells inhibited it (Figure S3F). Taken together, these findings suggest TRIM7 promotes the K63-linked ubiquitination of ATG7 with the treatment of L. monocytogenes infection, starvation, or rapamycin.
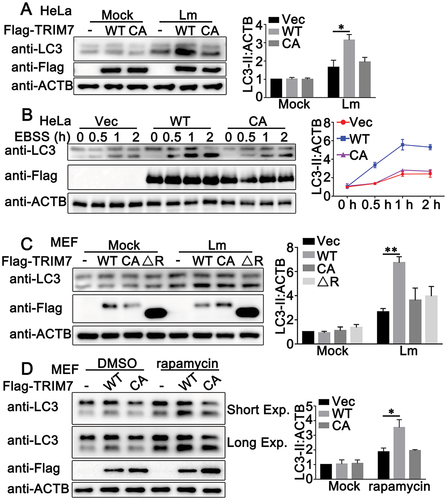
E3 ligase activity of TRIM7 is critical to its role in the regulation of autophagy
Considering that TRIM7 regulated the ubiquitination of ATG7 as an E3 ligase and TRIM7 manipulated autophagy, we examined whether the E3 ligase activity of TRIM7 was essential to its role in autophagy. TRIM7 or its C29, 32A mutant were transfected into HeLa cells. After L. monocytogenes infection, LC3-II was detected by immunoblots and the results demonstrated that C29, 32A mutant, which lost the ability to increase the ubiquitination with the destroyed RING domain (), made little difference in LC3-II accumulation whereas wild-type TRIM7 promoted the accumulation of LC3-II (). Similar results were observed in HeLa cells with the treatment of starvation (). Further, to exclude the possible effects of endogenous TRIM7, TRIM7 or its mutants were transfected into trim7-deficient MEFs, followed by L. monocytogenes infection or rapamycin stimulation. As shown in , in trim7-deficient MEFs with L. monocytogenes infection, whereas TRIM7 transfection increased LC3-II accumulation, the C32, 29A, and the ∆R mutant of the TRIM7 barely affected LC3-II accumulation. Consistently, in response to the stimulation of rapamycin, C29, 32A mutant of TRIM7 did not significantly affect the accumulation of LC3-II as wild-type TRIM7 did (). In all, these results indicate that the E3 ligase activity of TRIM7 is essential to its regulation of autophagy.
Figure 8. Ubiquitination is critical to the effect of TRIM7 on autophagy. (A) HeLa cells, were transfected with Vector (-), wild-type TRIM7 (WT) or C29, 32A mutant (CA), and then infected with Lm (MOI = 10) for 2 h. Afterward, the cells were lysed for immunoblot assays. LC3-II density was shown in the right panel. (B) HeLa cells were transfected with Vector (Vec), wild-type TRIM7 (WT) or C29, 32A mutant (CA), and then treated with EBSS for indicated time periods. Afterward, the cells were lysed for immunoblot assays. LC3-II density was shown in the right panel. (C) trim7-deficient (KO) MEFs, were transfected with Vector (-), wild-type TRIM7 (WT), C29, 32A mutant (CA) or 85–511aa of TRIM7 (ΔR), and then infected with Lm or left uninfected for 2 h. Afterward, the cells were lysed for immunoblot assays. LC3-II density was shown in the right panel. (d) trim7-deficient (KO) MEFs, were transfected with Vector (-), wild-type TRIM7 (WT) or C29, 32A mutant (CA), and then treated with DMSO or rapamycin (200 nM) for 12 h. LC3-II density was shown in the right panel. The data are representative of three independent experiments and are presented as mean ± SD. *, p < 0.05, **, p < 0.01.
TRIM7 promotes ubiquitination of ATG7 at K413
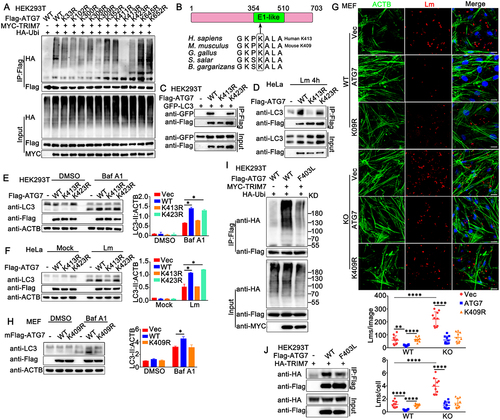
To investigate whether the ubiquitination of ATG7 by TRIM7 was essential to its function in autophagy, we explored the ubiquitination site at ATG7 that was targeted by TRIM7. Potential ubiquitination sites at ATG7 were predicted by CPLM4.0, a database for protein lysine modifications [Citation33]. Then ATG7 mutants were constructed with these potential lysine sites replaced by arginine separately. As shown in , A series of ATG7 mutants were transfected into HEK293T cells, together with Myc-TRIM7 and HA-Ubi, and the ubiquitination of ATG7 was examined. Immunoprecipitation and immunoblot analysis indicated that the K413R mutant of ATG7 could not be ubiquitinated by TRIM7, suggesting TRIM7 promoted the ubiquitination of ATG7 at K413 (). Protein alignments revealed that the K413 was located in the E1-like domain of ATG7 and was highly conserved among species (). Next, we investigated whether the mutation at K413 affected the function of ATG7. Wild-type, K413R or K423R (the mutant with the same level of ubiquitination by TRIM7 as wild-type ATG7) mutants of ATG7 were co-transfected with LC3 and immunoprecipitation and immunoblot analysis were performed. The results indicated that the ATG7-LC3 interaction was impaired by the mutation at K413, suggesting K413 was critical to the association of ATG7 with LC3 (). This phenomenon was confirmed by the impairment in the interaction of K413R with endogenous LC3 in L. monocytogenes-infected HeLa cells (). Next, we examined the effect of the K413 mutation on autophagy and L. monocytogenes infection. In HEK293T cells, with inhibition of lysosome activity by Baf A1, ATG7 overexpression resulted in elevated autophagosome accumulation, whereas the K413R mutant ATG7 barely affected LC-II protein accumulation (). Similar results were observed in L. monocytogenes infection-induced LC-II protein accumulation in HeLa cells (). In addition, ATG7 transfection in MEFs inhibited L. monocytogenes infection and the K413R mutant could not inhibit L. monocytogenes infection as much as the wild-type ATG7 did (Figure S4). Finally, given that the K413 of ATG7 is conserved among species and is located at K409 in mice (), we investigated the effect of the K409R mutant of mouse ATG7 on L. monocytogenes infection and autophagy in MEFs. As expected, the K409R mutant of mouse ATG7 could not inhibit L. monocytogenes infection to the same extent as the wild-type mouse ATG7 did (). However, in trim7-deficient MEFs, wild-type and K409R mutant of mouse ATG7 did not show a significant difference in their effect on L. monocytogenes infection, suggesting the modification on K409 by TRIM7 played an important role in the function of ATG7 (). Consistent with the results from K413R, the ATG7-promoted autophagosome accumulation was impaired by the mutation at K409 in MEFs (). It has been reported that the Phe403Leu (F403L) variant of ATG7 impairs autophagy and is detected in patients with primary ovarian insufficiency/POI, but the underlying mechanism by which the F403L mutant impaired the role of ATG7 in autophagy is unclear [Citation34,Citation35]. Considering that the site of F403 is very close to K413, both of which are in the E1-like domain of ATG7, we examined whether the mutation at the site of 403 affected the association of ATG7 with TRIM7 and its ubiquitination. Our findings demonstrated that compared to wild-type ATG7, the F403L mutant exhibited decreased ubiquitination and weaker interaction with TRIM7, suggesting TRIM7 might contribute to the impairment of autophagy by the F403L mutant of ATG7 (). In all, our findings demonstrate that TRIM7 promotes the ubiquitination of ATG7 at K413, and ubiquitination at this site is critical to the function of ATG7 in autophagy and L. monocytogenes infection.
Figure 9. TRIM7 targeted ATG7 for K63-mediated ubiquitination at K413. (A) HEK293T cells were transfected with the indicated plasmids. At 24 h after transfection, immunoprecipitation (IP) and immunoblot (IB) analysis were performed as indicated. (B) Schematic representation of ATG7 protein and interspecific sequence alignments of genomic regions carrying the interchanged amino acids. E1-like domain, E1 activating enzyme-like. (C) HEK293T cells were transfected with the indicated plasmids. At 24 h after transfection, immunoprecipitation (IP) and immunoblot (IB) analysis were performed as indicated. (D) HeLa cells were transfected with indicated plasmids, and then stimulated with Lm (MOI = 10) for 4 h. Afterward, the cells were lysed and subjected to immunoprecipitation (IP) and immunoblot analysis as indicated. (E) HEK293T cells, were transfected with Flag-tagged ATG7 or its mutants as indicated, and then treated with Baf A1 (400 nM) or DMSO as a control for 4 h. The cells were subjected to immunoblot analysis. LC3-II density was shown in the right panel. (F) HeLa cells were transfected with indicated plasmids, and then stimulated with Lm, (MOI = 10) for 4 h. Afterward, the cells were lysed for immunoblot analysis. LC3-II density was shown in the right panel. (G) Wild-type (WT) or trim7-deficient (KO) MEFs were transfected with Flag-tagged mouse ATG7 or its mutant mK409R. 24 h later, the cells were infected with Lm or left uninfected for another 6 h, and then subjected to confocal analysis. The top panel shows DAPI (blue), Lm (red), ACTB (green), and merged views of three channels. Scale bars: 20 μm. The bottom panel shows the statistical analysis of the total number of bacteria and the number of bacteria per cell in all pictures (10 pictures per genotype). (H) Wild-type MEFs were transfected with Flag-tagged mouse ATG7 or its mutant and then treated with Baf A1 (400 nM) or DMSO as a control for 4 h. The cells were subjected to immunoblot analysis. LC3-II density was shown in the right panel. (I) HEK293T cells were transfected with indicated plasmids, and then the cell lysates were subjected to immunoprecipitation (IP) and immunoblot. (IB) analysis as indicated. (J) HEK293T cells were transfected with indicated plasmids, and then the cell lysates were subjected to immunoprecipitation (IP) and immunoblot. The data are representative of three independent experiments and are presented as mean ± SD. *, p < 0.05, **, p < 0.01, ****, p < 0.0001.
Discussion:
Based on our previous work, TRIM7 plays an important role in the anti-viral innate immune responses via two different mechanisms [Citation25,Citation26]. TRIM7 negatively regulates DNA virus or RNA virus-triggered innate immune responses targeting STING1 or MAVS separately, by promoting the K48-linked ubiquitination and subsequent proteasome-dependent degradation of target proteins [Citation25,Citation26]. Given the similarities between the innate responses to viruses and intracellular bacteria, these findings about TRIM7 prompted us to explore the role of TRIM7 in intracellular bacterial infection, using L. monocytogenes as a model. Consistent with the results from virus infection, TRIM7 deficiency in mice resulted in elevated IFNB production upon L. monocytogenes infection. Surprisingly, with higher IFNB expression levels than wild-type mice, TRIM7-deficient mice exhibited a lower survival rate and an increase in L. monocytogenes infection following L. monocytogenes infection. Confused with these findings in mice, we further examined the role of TRIM7 in L. monocytogenes infection at the cellular level. Consistent with the results from trim7-deficient mice, TRIM7 knockdown in PMA-THP1 cells or trim7 deficiency in BMMs, BMDCs, MEFs, PMs, or HaCaT cells promoted both L. monocytogenes infection and L. monocytogenes triggered innate immune responses, including the production of IFNB, CXCL10, IFIT1 and TNF and the phosphorylation of TBK1, IRF3, and RELA. Given that STING1 is required for the type I IFN response to L. monocytogenes infection [Citation36], to exclude the regulatory effects of TRIM7 on STING1, we further tested the role of TRIM7 on L. monocytogenes infection in STING1-deficient THP1 cells and the results demonstrated that TRIM7 could negatively regulate L. monocytogenes infection in a STING1-independent way. These in vivo and in vitro observations indicated that TRIM7 might manipulate L. monocytogenes infection through some mechanism other than the regulation of innate signaling pathways.
Considering that TRIM7 is an E3-ligase, to explore the underlying mechanism by which TRIM7 regulated L. monocytogenes infection, we first tried to identify the potential substrate of TRIM7. Comparing the proteins that were immunoprecipitated with or without TRIM7, a few TRIM7-associated proteins were identified by mass spectrum. Among these proteins, ATG7, which is a well-known essential protein to autophagy, was found at the top of the protein “hit list”. Central to canonical autophagy are two ubiquitination-like conjugation systems, the ATG12 conjugation system, and the LC3/ATG8 lipidation system, both of which are activated by the same E1-like enzyme, ATG7 [Citation37]. With ATG7 as an E1-like enzyme and ATG10 as an E2-like enzyme, ATG12 is attached to ATG5 [Citation38]. Then the ATG12–ATG5-ATG16L1 complex assists the conjugation of LC3 to PE, catalyzed by ATG7 and ATG3 as E1-like and E2-like enzymes respectively [Citation39]. Therefore, the association of TRIM7 with ATG7 provided us hint that TRIM7 might be involved in the regulation of autophagy.
It has become clear that autophagy is a major mechanism by which the host cells eliminate invading pathogens, including viruses and intracellular bacteria [Citation39]. In addition to host cell recognition of L. monocytogenes PAMPs by DNA sensors or NLRs, L. monocytogenes and its secreted toxin, LLO, can be targeted by the Ub-SQSTM1/p62-LC3 pathway to autophagy [Citation4,Citation36]. The deficiency in autophagy genes, such as Atg5 and Atg7, increases susceptibility to intracellular bacteria, including Mycobacterium (M) tuberculosis and L. monocytogenes [Citation40–43]. Thus, after the confirmation of the TRIM7-ATG7 interaction, we examined the effects of TRIM7 on L. monocytogenes infection-triggered autophagy by the overexpression, knockdown, or deficiency of TRIM7 in a variety range of cells, including HeLa, BMDMs, BMDCs, PMs, and HaCaT cells. Both the level of LC3-II in immunoblot assays and the amounts of LC3-II puncta by confocal microscopy suggest a positive regulatory role of TRIM7 in L. monocytogenes-induced autophagy. Further, the treatment with bafilomycin A1, an inhibitor of vascular proton pump that indirectly blocks the fusion of autophagosomes to lysosomes, and with chloroquine, the lysosome acidification inhibitors, indicated that TRIM7 promoted L. monocytogenes-induced autophagosome accumulation, but not the phagosome-lysosome fusion or lysosomal degradation. In addition to the role in L. monocytogenes-induced autophagy, our findings demonstrated that TRIM7 also positively manipulated basal, and starvation- or rapamycin-activated autophagy.
Given that ATG7 is critical to canonical autophagy, modifications of ATG7 may be an efficient way to regulate autophagic response under a variety of conditions. However, till now, few posttranslational modifications have been identified to manipulate autophagy targeting ATG7. Our previous study has revealed that the beta chain of the non-classical MHC-II protein HLA-DM interacts with ATG7 and increases its acetylation, leading to reduced autophagosome accumulation and human T-cell leukemia virus type-1/HTLV-1 infection [Citation44]. Recently, NEDD4 was identified to promote the ubiquitination and subsequent degradation of ATG7 [Citation38]. In this study, we demonstrated that the K63-linked ubiquitination of ATG7 was induced by a variety range of conditions, including starvation, rapamycin stimulation, and L. monocytogenes infection. This ubiquitination could be significantly promoted by ATG7 and the RING domain of TRIM7 was required for the ubiquitination of ATG7. The examination of the association of the ATG5-ATG16L1 and ATG3-LC3 indicated that TRIM7 regulated ATG7-involved ubiquitination-like conjugation systems in autophagy. Further, the C29, 32A, and ∆R mutants of TRIM7 lacking the ability of ubiquitination lost their effects on the regulation of autophagosome accumulation. Finally, we identified the ubiquitination site at ATG7 that was catalyzed by TRIM7. K413R mutant of ATG7 lost not only the ubiquitinated modification by TRIM7 but also the capability to promote autophagy, leading to less efficient inhibition of L. monocytogenes infection compared to wild-type ATG7. These findings indicated that the ubiquitination of ATG7 at K413 by TRIM7 was required for the function of ATG7 in autophagy and L. monocytogenes infection. Further, the reported F403L variant in primary ovarian insufficiency patients as a loss of function mutant of ATG7 was found to have impairment in ubiquitination and interaction with TRIM7, suggesting an essential role of TRIM7-mediated ubiquitination in autophagy.
A few limitations should be noted. In this study, we screened TRIM7-associated proteins in unstimulated HEK293T cells, therefore some other proteins might be overlooked. In addition, future screening for immune or epithelial cells’ response upon L. monocytogenes infection, may further expand our understanding of the role of TRIM7 in L. monocytogenes infection in different tissues. Notably, the fact that the ubiquitination of ATG7 could be detected in TRIM7-deficient cells, suggests that other E3 ligases responsible for ATG7 ubiquitination may exist. Thus, screening for additional ATG7-associated E3 ligases, may shed more light on the modification of ATG7.
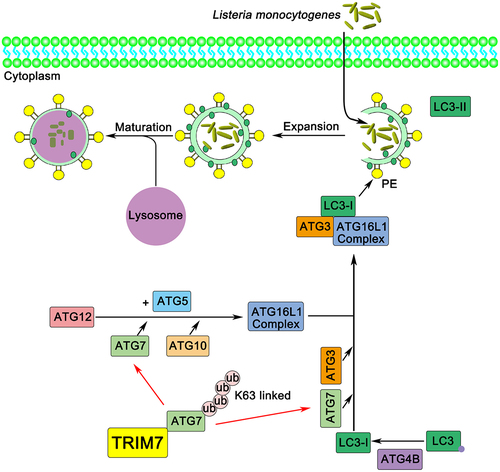
Together, our study demonstrated that TRIM7 positively regulated autophagosome accumulation by promoting the ubiquitination of ATG7 at K413, thereby affecting L. monocytogenes infection (). Our findings suggested a new regulator in intracellular bacterial infection and autophagy, with a novel posttranslational modification that targeted ATG7 at K413. This research may expand our understanding of host anti-bacterial defense and the role of autophagy in intracellular bacterial infection.
Figure 10. Working model of the mechanism by which TRIM7 regulates autophagy. In this model, TRIM7 enhances K63-linked poly-ubiquitination of ATG7, which in turn promotes autophagy, thereby increasing the clearance of L. monocytogenes.
Materials and methods
Mice
We generated trim7-deficient mice as described previously [Citation26]. Mice were housed in a facility with access to food and water and were maintained under a 12-h light/12-h dark cycle. All animal procedures were performed according to guidelines approved by the committee on animal care at Xinxiang Medical University, China. The age- and sex-matched wild-type and trim7-deficient mice were used in the experiments.
cDNA constructs and reagents
Human ATG7 or mouse Atg7 were amplified by PCR using cDNA from THP1 or MEF cells and were subsequently cloned into a pcDNA3 vector (Invitrogen, V79520). ATG7 mutants were constructed by PCR and also subcloned into a pcDNA3 vector. Human TRIM7 and its mutants were generated as described previously [Citation26]. GFP-LC3, HA-Ubi, HA-K6-Ubi, HA-K11-Ubi, HA-K27-Ubi, HA-K29-Ubi, HA-K33-Ubi, HA-K48-Ubi, HA-K63-Ubi, HA-K48R-Ubi, and HA-K63R-Ubi were obtained as described previously [Citation44–46]. The following antibodies were used for immunoblot analysis or immunoprecipitation: anti-Flag (Sigma-Aldrich, 3165,), anti-HA (Biolend, 901,515), anti-MYC (Proteintech, 66,004-1-Ig), anti-TRIM7/RNF90 (Santa Cruz Biotechnology, sc-109,107; Abcam, ab170538), anti-ATG7 (Cell Signaling Technology, 8588), anti-p-TBK1 (Cell Signaling Technology, 5483 T), anti-TBK1 (Flarbio, CSB-PA024154LA01HU), anti-p-IRF3 (Cell Signaling Technology, 4947), anti-IRF3 (Proteintech, 11,312-1-AP), anti-p-RELA/p65 (Cell Signaling Technology, 3033), anti-RELA/p65 (Proteintech, 10,745-1-AP), anti-Ubi (Santa Cruz Biotechnology, sc-8017), anti-Ubi-K63 (Millipore, 05–1313), anti-LC3 (Proteintech, 14,600-1-AP; MBL, PM036), anti-ATG3 (Cell Signaling Technology, 3415), anti-ATG5 (Cell Signaling Technology, 12,994), anti-ATG16L1 (8089, Cell Signaling Technology), anti-Lm (Abcam, ab35132), and anti-ACTB/β-actin (Proteintech, 60,008–1). The PMA (S1819) and rapamycin (S1842) were purchased from Beyotime Biotechnology. Bafilomycin A1 (11,038) was obtained from Cayman Chemical. Chloroquine (C6628) was purchased from Sigma-Aldrich.
Cell culture, transfection, and stimulation
Human embryonic kidney (HEK) 293 T and Tohoku Hospital Pediatrics (THP) 1 cells were kindly provided by Stem Cell Bank, Chinese Academy of Sciences. HaCaT keratinocytes were purchased from Procell Life Science & Technology Co., Ltd., (CL-0090). HeLa-mCherry-LC3 cells were kindly provided by Yuna Niu (Xinxiang Medical University). STING1-deficient THP1 cells were purchased from InvivoGen (thpd-kostg). HaCaT and HEK293T cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Sigma-Aldrich, D6429). THP1 cells were grown in RPMI 1640 (Gibco, C22400500BT). PMA-THP1 cells referred to THP1 cells that were pretreated with 100 ng/ml PMA for 24 h. All cells were supplemented with 10% FBS (Gibco, 10,099–141), 4 mM L-glutamine, 100 μg/ml penicillin, and 100 U/ml streptomycin under humidified conditions with 5% CO2 at 37°C. Transfection of HaCaT, HEK293T, THP1, MEF, and BMDM cells was performed with Lipofectamine 2000 (Invitrogen, 11,668–019) according to the manufacturer’s instructions.
Preparations of BMDMs, BMDCs, peritoneal macrophages and MEFs
The procedure for generating BMDMs, BMDCs, and MEFs has been described previously [Citation25,Citation47,Citation48]. For the generation of peritoneal macrophages (PMs), wild-type and TRIM7-deficient mice were injected intraperitoneally with 2.5 mL of a 4% Brewers TG solution (Sigma-Aldrich, 70,157). After 4 days, when most recruited cells were macrophages, mice were killed and peritoneal cells were collected by lavage with 5 mL complete RPMI 1640 media. The lavage fluid was centrifuged, then the supernatant was aspirated, and the cell pellet was resuspended in a complete medium. Macrophages were further purified by adherence for 3 to 4-h at 37°C, at which point all nonadherent (nonmacrophage) cells were eliminated. Cells were maintained in RPMI 1640 media.
Immunoprecipitation and immunoblot analysis
Immunoprecipitation and immunoblot analysis were performed as described previously [Citation49,Citation50]. In short, after the treatment, the cells were lysed in lysis buffer containing 1.0% (vol:vol) Nonidet P40 (Solarbio, N8030), 20 mM Tris-HCl, pH 8.0, 10% (vol:vol) glycerol, 150 mM NaCl, 0.2 mM Na3VO4, 1 mM NaF, 0.1 mM sodium pyrophosphate and a protease inhibitor “cocktail” (Roche, 04693132001). After centrifugation for 20 min at 14,000 g, supernatants were collected and incubated with the indicated antibodies together with protein A/G Plus-agarose immunoprecipitation reagent (Sc-2003, Santa Cruz Biotechnology) at 4°C for 3 h or overnight. After three washes, the immunoprecipitates were boiled in SDS sample buffer for 10 min and analyzed by immunoblot.
Real-time PCR
Total RNA was extracted from the cultured cells with TRIzol reagent (Invitrogen, 15,596,026) as described by the manufacturer. All gene transcripts were quantified by real-time PCR with SYBR Green qPCR Master Mix (Vazyme, Q711-02) using a 7500 Fast real-time PCR system (Applied Biosystems). The relative fold induction was calculated using the 2−ΔΔCt method. The primers used for real-time PCR were as follows:
Human IFNB,
Forward, 5’- CACGACAGCTCTTTCCATGA −3’;
Reverse, 5’ – AGCCAGTGCTCGATGAATCT −3’
Human CXCL10,
Forward, 5’- GGTGAGAAGAGATGTCTGAATCC −3’;
Reverse, 5’- GTCCATCCTTGGAAGCACTGCA −3’
Human TNF,
Forward, 5’- GGCGTGGAGCTGAGAGATAAC −3’;
Reverse, 5’- GGTGTGGGTGAGGAGCACAT −3’
Human IFIT1,
Forward, 5’- GCCATTTTCTTTGCTTCCCCTA −3’;
Reverse, 5’- TGCCCTTTTGTAGCCTCCTTG −3’
Human GAPDH,
Forward, 5’-TCAACGACCACTTTGTCAAGCTCA-3’;
Reverse, 5’-GCTGGTGGTCCAGGTCTTACT-3’
Mouse Ifnb,
Forward, 5’- TCCTGCTGTGCTTCTCCACCACA −3’;
Reverse, 5’- AAGTCCGCCCTGTAGGTGAGGTT −3’
Mouse Cxcl10,
Forward, 5’- ATCATCCCTGCGAGCCTATCCT −3’;
Reverse, 5’- GACCTTTTTTGGCTAAACGCTTTC −3’
Mouse Ifit1,
Forward, 5’-TACAGGCTGGAGTGTGCTGAGA −3’;
Reverse, 5’- CTCCACTTTCAGAGCCTTCGCA-3’
Mouse Tnf,
Forward, 5’- TCTTCTCATTCCTGCTTGTGG −3’;
Reverse, 5’- GGTCTGGGCCATAGAACTGA −3’
Mouse Gapdh,
Forward, 5’- ACGGCCGCATCTTCTTGTGCA-3’;
Reverse, 5’- ACGGCCAAATCCGTTCACACC-3’.
ELISA
HaCaT cells were infected with L. monocytogenes for 24 h. The culture media were collected for measurement of IFNB (Proteintech, KE00044). Eight-week-old wild-type and trim7-deficient mice were intravenously infected with L. monocytogenes for 6 h, and then the serum of mice was collected for measurement of IFNB by ELISA.
RNA interference
TRIM7 knockdown experiments were performed as described previously [Citation26]. TRIM7 Stealth-RNAi siRNA was designed by the Invitrogen BLOCKiT RNAi Designer. The small interfering RNA (siRNA) sequences used were as follows: R3,
Forward, 5’-CAGUCUCUUCUGAGAUGAAGAAUAA-3’;
Reverse, 5’-UUAUUCUUCAUCUCAGAAGAGACUG −3’
The Silencer Select negative control siRNA was purchased from Invitrogen (4,390,843). PMA-THP1, or HaCaT cells were transfected with siRNA using Lipofectamine 2000 according to the manufacturer’s instructions. At 24 h after transfection, the cells were used for further experiments.
Generation of TRIM7 knockout cell line
TRIM7-deficient HaCaT cell line was generated by the CRISPR-Cas9 system as described previously [Citation26]. The following oligonucleotides specifically targeting the TRIM7 gene were used:
Oligonucleotide 1, 5’-CACCGCTCACCTTGGATCCCGACA −3’;
Oligonucleotide 2, 5’-AAACTGTCGGGATCCAAGGTGAGC −3’.
L. monocytogenes infections and enumeration of bacterial load
L. monocytogenes (Lm, 10403s) were kindly provided by Professor Yunwei Lou, Xinxiang Medical University. Bacteria were prepared by shaking in brain heart infusion (BHI) broth (OXOID, CM1135) with 50 µg/ml streptomycin overnight at 37°C. For infections, L. monocytogenes were grown to the mid-log phase and then resuspended in PBS (Solarbio, P1020). Age and sex-matched mice (6–10 weeks of age) were intravenously infected with L. monocytogenes (1.5 × 105 CFUs/mouse) and killed at day 3. To assess bacterial number in organs, lungs, livers, and spleens were homogenized and bacterial load was determined by serial dilution on BHI agar plates. For survival experiments, wild-type (WT) and trim7-deficient (KO) mice were intravenously infected with L. monocytogenes (1.5 × 105 CFUs/mouse). Mouse survival rates were monitored twice a day during 15 days of infection. BMDC, BMDMs, and HaCaT Cells were infected with L. monocytogenes at a multiplicity of infection (MOI) of 10 for 30 min at 37°C. To determine the number of bacteria entering the cells, extracellular bacteria were killed by treatment with 100 μg/ml gentamicin (Sigma-Aldrich, 1045–41-0) for an additional 30 min at 37°C. Then, infected cells were washed with PBS three times and lysed with 0.05% Triton X-100 (Sigma-Aldrich, 9036–19-5) in distilled water. Serial dilutions were seeded on BHI agar plates and CFUs were counted after 36 h.
In vitro ubiquitination assay
ATG7, TRIM7, and its mutants were expressed with a TNT Quick-coupled Transcription/Translation Systems kit (Promega, L1171). In vitro ubiquitination assay was performed with a ubiquitination kit (Enzo Life Science, BML-UW9920) following the manufacturer’s instructions.
Confocal microscopy
After treatment, HEK293T, MEFs, HeLa, and HaCaT cells were fixed with 4% PFA in PBS, permeabilized with Triton X-100, and then blocked with 1% BSA (Solarbio, A8020) in PBS. Nuclei were stained with 4, 6-diamidino-2-phenylindole (DAPI; Solarbio, C0065). ACTB was labeled with a phalloidin conjugate (ThermoFisher, A22287). Images were taken using Nikon A1R microscopy.
In situ PLA
MEFs were seeded on Thermanox plastic coverslips (NUNC, 174,950) and cultured overnight. After L. monocytogenes infection for 4 h, cells were fixed in 4% PFA in PBS for 15 min and washed with PBS three times. After that, cells were permeabilized with 0.1% Triton X-100 on ice for 5 min, washed in PBS and blocked in 5% BSA in PBS for 1 h at 37°C, and incubated in primary antibodies overnight at 4°C. On the next day, cells were washed with the wash buffer (Sigma-Aldrich, DUO82049) three times and incubated with the secondary antibodies with PLA probes (Sigma-Aldrich, DUO92001, DUO92005) for 2 h at 37°C followed by three times wash with the wash buffer. Cells were incubated with the Duolink in situ detection reagents red (Sigma-Aldrich, DUO92008) for 2 h at 37°C. Finally, Nuclei were stained with DAPI and covered onto slides. Images were taken using Nikon A1R microscopy and the Image Pro Plus 6.0 was used for quantitative analyses.
Mass spectrometry
Ten million HEK293T cells were seeded in a T75 flask overnight, and then the cells were transfected with Flag-TRIM7 plasmids for 24 h. Cell lysates were subjected to immunoprecipitation with anti-Flag and agarose beads at 4°C overnight. Proteins were separated by SDS-PAGE gel. After Coomassie Brilliant Blue staining, the gel bands of interest were excised from the gel and analyzed by liquid chromatography-mass spectrometry, which was performed by Applied Protein Technology Company.
Statistics
The data are presented as the means ± SD from at least three independent experiments. The statistical comparisons between the different treatments were performed using the Student t-test, and p < 0.05 was considered statistically significant.
Supplemental Material
Download MS Word (14.1 MB)Acknowledgments
We want to thank Dr. Yinming Liang for providing trim7-deficient mice and all the members of Henan Key Laboratory of immunology and targeted drugs for sharing valuable material and research support.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/15548627.2022.2162706
Additional information
Funding
References
- Galluzzi L, Baehrecke EH, Ballabio A, et al. Molecular definitions of autophagy and related processes. EMBO J. 2017;36(13):1811–1836.
- Hansen M, Rubinsztein DC, Walker DW. Autophagy as a promoter of longevity: insights from model organisms. Nat Rev Mol Cell Biol. 2018;19(9):579–593.
- Boya P, Reggiori F, Codogno P. Emerging regulation and functions of autophagy. Nat Cell Biol. 2013;15(7):713–720.
- Ogawa M, Yoshikawa Y, Mimuro H, et al. Autophagy targeting of Listeria monocytogenes and the bacterial countermeasure. Autophagy. 2011;7(3):310–314.
- Hamon M, Bierne H, Cossart P. Listeria monocytogenes: a multifaceted model. Nat Rev Microbiol. 2006;4(6):423–434.
- Birmingham CL, Higgins DE, Brumell JH. Avoiding death by autophagy: interactions of Listeria monocytogenes with the macrophage autophagy system. Autophagy. 2008;4(3):368–371.
- Py BF, Lipinski MM, Yuan J. Autophagy limits Listeria monocytogenes intracellular growth in the early phase of primary infection. Autophagy. 2007;3(2):117–125.
- Zhao Z, Fux B, Goodwin M, et al. Autophagosome-independent essential function for the autophagy protein Atg5 in cellular immunity to intracellular pathogens. Cell Host Microbe. 2008;4(5):458–469.
- Mizushima N, Yoshimori T, Ohsumi Y. The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol. 2011;27(1):107–132.
- Chiang C, Gack MU. Post-translational control of intracellular pathogen sensing pathways. Trends Immunol. 2017;38(1):39–52.
- Xie Y, Kang R, Sun X, et al. Posttranslational modification of autophagy-related proteins in macroautophagy. Autophagy. 2015;11(1):28–45.
- Chen RH, Chen YH, Huang TY. Ubiquitin-mediated regulation of autophagy. J Biomed Sci. 2019;26(1):80.
- Berndsen CE, Wolberger C. New insights into ubiquitin E3 ligase mechanism. Nat Struct Mol Biol. 2014;21(4):301–307.
- Zheng N, Shabek N. Ubiquitin ligases: structure, function, and regulation. Annu Rev Biochem. 2017;86(1):129–157.
- Deshaies RJ, Joazeiro CA. RING domain E3 ubiquitin ligases. Annu Rev Biochem. 2009;78(1):399–434.
- Goto J, Otaki Y, Watanabe T, et al. The role of HECT-Type E3 ligase in the development of cardiac disease. Int J Mol Sci. 2021;23(1):22.
- Yang Y, Zhu Y, Zhou S, et al. TRIM27 cooperates with STK38L to inhibit ULK1-mediated autophagy and promote tumorigenesis. EMBO J. 2022;41(14):e109777.
- Xu C, Feng K, Zhao X, et al. Regulation of autophagy by E3 ubiquitin ligase RNF216 through BECN1 ubiquitination. Autophagy. 2014;10(12):2239–2250.
- Chakraborty A, Diefenbacher ME, Mylona A, et al. The E3 ubiquitin ligase Trim7 mediates c-Jun/AP-1 activation by Ras signalling. Nat Commun. 2015;6(1):6782.
- Lu M, Zhu X, Yang Z, et al. E3 ubiquitin ligase tripartite motif 7 positively regulates the TLR4-mediated immune response via its E3 ligase domain in macrophages. Mol Immunol. 2019;109:126–133.
- Jin J, Lu Z, Wang X, et al. E3 ubiquitin ligase TRIM7 negatively regulates NF-kappa B signaling pathway by degrading p65 in lung cancer. Cell Signal. 2020;69:109543.
- Zhu L, Qin C, Li T, et al. The E3 ubiquitin ligase TRIM7 suppressed hepatocellular carcinoma progression by directly targeting Src protein. Cell Death Differ. 2020;27(6):1819–1831.
- Zhou C, Zhang Z, Zhu X, et al. N6-Methyladenosine modification of the TRIM7 positively regulates tumorigenesis and chemoresistance in osteosarcoma through ubiquitination of BRMS1. EBioMedicine. 2020;59:102955.
- Fan W, Mar KB, Sari L, et al. TRIM7 inhibits enterovirus replication and promotes emergence of a viral variant with increased pathogenicity. Cell. 2021;184(13):3410–25 e17.
- Yang B, Zhang G, Qin X, et al. Negative regulation of RNF90 on RNA virus-triggered antiviral immune responses targeting MAVS. Front Immunol. 2021;12:730483.
- Yang B, Liu Y, Cui Y, et al. RNF90 negatively regulates cellular antiviral responses by targeting MITA for degradation. PLoS Pathog. 2020;16(3):e1008387.
- Hansen K, Prabakaran T, Laustsen A, et al. Listeria monocytogenes induces IFNβ expression through an IFI16-, cGAS- and STING-dependent pathway. EMBO J. 2014;33(15):1654–1666.
- Collier JJ, Suomi F, Olahova M, et al. Emerging roles of ATG7 in human health and disease. EMBO Mol Med. 2021;13(12):e14824.
- Kuang E, Qi J, Ronai Z. Emerging roles of E3 ubiquitin ligases in autophagy. Trends Biochem Sci. 2013;38(9):453–460.
- Vittal V, Stewart MD, Brzovic PS, et al. Regulating the regulators: recent revelations in the control of E3 ubiquitin ligases. J Biol Chem. 2015;290(35):21244–21251.
- Park Y, Jin HS, Aki D, et al. The ubiquitin system in immune regulation. Adv Immunol. 2014;124:17–66.
- Liu X, Wang Q, Chen W, et al. Dynamic regulation of innate immunity by ubiquitin and ubiquitin-like proteins. Cytokine Growth Factor Rev. 2013;24(6):559–570.
- Zhang W, Tan X, Lin S, et al. CPLM 4.0: an updated database with rich annotations for protein lysine modifications. Nucleic Acids Res. 2022;50(D1):D451–D9.
- Feng Y, Yao Z, Klionsky DJ. How to control self-digestion: transcriptional, post-transcriptional, and post-translational regulation of autophagy. Trends Cell Biol. 2015;25(6):354–363.
- Delcour C, Amazit L, Patino LC, et al. ATG7 and ATG9A loss-of-function variants trigger autophagy impairment and ovarian failure. Genet Med. 2019;21(4):930–938.
- Marinho FV, Benmerzoug S, Oliveira SC, et al. The emerging roles of STING in bacterial infections. Trends Microbiol. 2017;25(11):906–918.
- Ohsumi Y. Molecular dissection of autophagy: two ubiquitin-like systems. Nat Rev Mol Cell Biol. 2001;2(3):211–216.
- Zheng H, Lu X, Li K, et al. ATG ubiquitination is required for circumsporozoite protein to subvert host innate immunity against rodent malaria liver stage. Front Immunol. 2022;13:815936.
- Gomes LC, Dikic I. Autophagy in antimicrobial immunity. Mol Cell. 2014;54(2):224–233.
- Castillo EF, Dekonenko A, Arko-Mensah J, et al. Autophagy protects against active tuberculosis by suppressing bacterial burden and inflammation. Proc Natl Acad Sci U S A. 2012;109(46):E3168–76.
- Henault J, Martinez J, Riggs JM, et al. Noncanonical autophagy is required for type I interferon secretion in response to DNA-immune complexes. Immunity. 2012;37(6):986–997.
- Lee HK, Mattei LM, Steinberg BE, et al. In vivo requirement for Atg5 in antigen presentation by dendritic cells. Immunity. 2010;32(2):227–239.
- Mostowy S, Sancho-Shimizu V, Hamon MA, et al. p62 and NDP52 proteins target intracytosolic Shigella and Listeria to different autophagy pathways. J Biol Chem. 2011;286:26987–26995.
- Wang J, Song D, Liu Y, et al. HLA-DMB restricts human T-cell leukemia virus type-1 (HTLV-1) protein expression via regulation of ATG7 acetylation. Sci Rep. 2017;7(1):14416.
- Yang B, Wang J, Wang Y, et al. Novel function of Trim44 promotes an antiviral response by stabilizing VISA. J Immunol. 2013;190(7):3613–3619.
- Wang J, Yang B, Hu Y, et al. Negative regulation of Nmi on virus-triggered type I IFN production by targeting IRF7. J Immunol. 2013;191(6):3393–3399.
- Wang Y, Lian Q, Yang B, et al. TRIM30alpha is a negative-feedback regulator of the intracellular DNA and DNA virus-triggered response by targeting STING. PLoS Pathog. 2015;11(6):e1005012.
- Lou Y, Han M, Liu H, et al. Essential roles of S100A10 in Toll-like receptor signaling and immunity to infection. Cell Mol Immunol. 2020;17(10):1053–1062.
- Wang J, Kang L, Song D, et al. Ku70 senses HTLV-1 DNA and modulates HTLV-1 replication. J Immunol. 2017;199(7):2475–2482.
- Yang B, Song D, Liu Y, et al. IFI 16 regulates HTLV −1 replication through promoting HTLV-1 RTI -induced innate immune responses. FEBS Lett. 2018;592(10):1693–1704.