
ABSTRACT
Differentiation and fate decisions are critical for the epithelial cells lining the proximal tubule (PT) of the kidney, but the signals involved remain unknown. Defective cystine mobilization from lysosomes through CTNS (cystinosin, lysosomal cystine transporter), which is mutated in cystinosis, triggers the dedifferentiation and dysfunction of the PT cells, causing kidney disease and severe metabolic complications. Using preclinical models and physiologically relevant cellular systems, along with functional assays and a generative artificial intelligence (AI)-powered engine, we found that cystine storage imparted by CTNS deficiency stimulates Ragulator-RRAG GTPase-dependent recruitment of MTORC1 and its constitutive activation. In turn, this diverts the catabolic trajectories and differentiating states of PT cells toward growth and proliferation, disrupting homeostasis and their specialized functions. Therapeutic MTORC1 inhibition by using low doses of rapamycin corrects lysosome function and differentiation downstream of cystine storage and ameliorates PT dysfunction in preclinical models of cystinosis. These discoveries suggest that cystine may act as a lysosomal fasting signal that tailors MTORC1 signaling to direct fate decisions in the kidney PT epithelium, highlighting novel therapeutic paradigms for cystinosis and other lysosome-related disorders.
The epithelial cells lining the proximal tubule (PT) of the kidney play a critical role in homeostasis by reabsorbing ultrafiltered, low-molecular weight (LMW) plasma proteins through receptor-mediated endocytosis and lysosomal pathways, preventing the loss of vital nutrients in the urine. The lysosomal degradation of disulfide-rich proteins, such as ALB (albumin), accounts for the supply of cystine that is exported to the cytoplasm through the lysosomal cystine transporter called CTNS. Loss-of-function mutations in CTNS cause cystinosis, a lysosomal storage disease characterized by loss of differentiation and dysfunction of the tubular cells, causing life-threatening complications and chronic kidney disease/CKD. The precise mechanisms by which the elevated levels of cystine within lysosomes, resulting from defective export through CTNS, affect cell fate decisions remain unknown.
Studies using preclinical disease models (mouse, rat, and zebrafish) and physiologically relevant PT cellular systems, along with organelle-based function assays, have revealed that lysosomes accumulating cystine due to the loss of CTNS display profound defects in their proteolytic activities, with accumulation of intracellular constituents, including misfolded proteins and damaged mitochondria that are normally recycled through macroautophagy/autophagy. These catabolic abnormalities are mirrored by the upregulation of anabolic programs for growth and proliferation, altering the differentiation of the PT cells, hence disrupting their homeostatic functions (). These changes are reflected by LMW proteinuria – the earliest manifestation of the epithelial cell disease.
Figure 1. The CTNS-cystine-MTORC1 axis as a lysosomal signaling node for fate decisions in the kidney tubule epithelium and targetable pathway in cystinosis. Preclinical disease modeling in cystinosis and physiologically-relevant cellular systems, aligned with organelle-based function assays and artificial intelligence (AI)-powered target discovery pipeline can enable the identification of pathophysiological mechanisms contributing to disease pathology and highlight novel therapeutic strategies. In kidney tubular cells lacking CTNS, the storage of cystine triggers the Ragulator-RRAG-dependent translocation of MTORC1 and its constitutive activation at the surface of the lysosome. This diverts the catabolic trajectories and differentiating states of the cells towards anabolic programs for growth and proliferation, disrupting their reabsorptive activities and causing tubular dysfunction and kidney disease in cystinosis. Therapeutic inhibition of hyperactive MTORC1 by using low-doses of rapamycin rescues the lysosome proteolysis and rewires the proliferating trajectories of CTNS deficient/cystinosis-affected tubular epithelial cells towards differentiation and homeostasis. LC/MS-MS, LC-MS/MS, liquid chromatography with tandem mass spectrometry. TMEM192–3×HA/LysoTag marks lysosomes with a triple HA epitope, enabling their immuno-isolation with anti-HA antibodies.
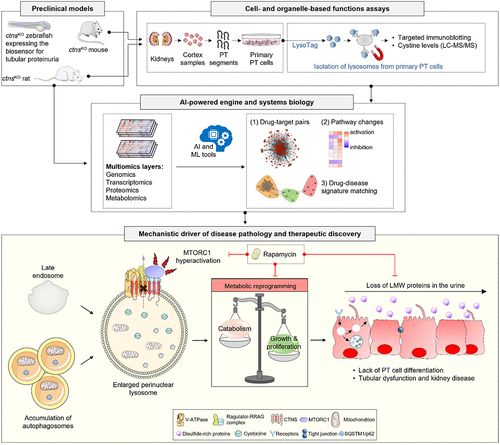
To identify the factors driving the metabolic switch induced by CTNS deficiency and cystine storage, we conducted proteomics- and metabolomics-based profiling of cystinosis-affected PT cells [Citation1]. Integrated biological network analysis crossing differentially produced proteins with metabolites have identified a significant enrichment of pathways involved in the regulation of the MTORC1 signalosome, such as glucose and amino acids, MTORC1 signaling itself, and the downstream anabolic (protein, fatty acid, and nucleotide biosynthesis) or catabolic (autophagy) processes in ctns KO versus WT Ctns tubular cells. These results were confirmed when applying an artificial intelligence (AI) engine (PandaOmics) that uses machine learning tools and statistical validation to rank disease-target associations and prioritize actionable drug targets. Accordingly, this computational platform identified MTOR on the top of the list of predicted drug targets potentially reversing dysregulated homeostasis and tubular dysfunction downstream of CTNS loss and cystine storage ().
Inspired by the biological evidence and in silico predictions, we hypothesized that the recycling of lysosomal cystine through CTNS regulates MTORC1 signaling in the tubular cells. Supporting this hypothesis, CTNS deficiency renders MTORC1 less sensitive to nutrient starvation and constitutively active, as indicated by persistent phosphorylation of RPS6 (ribosomal protein S6; p-RPS6 [Ser235/236]) and EIF4EBP1 (eukaryotic translation initiation factor 4E binding protein 1; p-EIF4EBP1 [Ser65]). Increased MTORC1activity is also observed in the PT segments of the ctns KO mouse and rat kidneys, as well as in zebrafish lacking the CTNS ortholog, demonstrating the evolutionary conservation of this signaling connection.
We next tested whether cystine storage per se forces MTORC1 to remain on the lysosomal surface. Treatment of CTNS-defective PT cells with cysteamine, which facilitates the export of cystine from lysosomes through SLC66A1/PQLC2 (solute carrier family 66 member 1) or the exposure of wild-type mPTCs to cystine methyl ester derivates, which accumulate within lysosomes independently of CTNS, reduce and stimulate, respectively, MTORC1 activation at the lysosomal surface. In line with this finding, the reconstitution with WT CTNS, but not the mutant CTNSG339R lacking cystine transport function, restores nutrient-dependent regulation of MTORC1 signaling in ctns knockout cells. These defects in lysosome dynamics and (MTORC1-driven) geroprotective signaling pathways, which occur early in the course of the disease, disrupt the autophagy-mediated turnover of dysfunctional and/or damaged mitochondria, in turn curbing PT metabolism and the actions of kidney epithelial cell fate-determining transcriptional programs ().
A key question remains: how does CTNS tailor the activation of MTORC1 signaling in response to lysosomal cystine levels? In the presence of nutrients, such as amino acids, a multiprotein complex composed of heterodimeric RRAG (Ras-related GTP binding) GTPases (consisting of RRAGA or RRAGB bound to RRAGC or RRAGD), Ragulator, and the vacuolar-type H+-translocating ATPase (V-ATPase) tethers MTORC1 to the lysosomal surface, enabling its activation by the (growth factor-induced) GTP-binding protein RHEB (Ras homolog enriched in brain). Remarkably, lysosomes from CTNS-deficient tubular cells contain elevated levels of V-ATPase subunits ATP6V0D1 and ATP6V1B2, Ragulator complex subunit LAMTOR2 and RRAGC GTPase proteins, which are rescued by re-expressing WT CTNS but not the CTNSG339R mutant. Co-immunoprecipitation studies in WT Ctns cells exposed to cystine methyl ester derivates or in ctns KO cells expressing CTNSG339 mutant revealed that, in both contexts, increased levels of cystine enable the interaction of CTNS with components of the V-ATPase and Ragulator-RRAG GTPase scaffold complex that recruits MTORC1 at the surface of the lysosome. The latter evidence indicates that, through interactions with Ragulator-RRAG GTPase lysosomal scaffold, CTNS may regulate MTORC1 signaling in response to variations in the levels of lysosomal cystine.
As children affected by cystinosis often evolve to kidney failure, a complication that cannot be prevented by the standard cysteamine therapy, there is an urgent need for more tailored, therapeutic interventions. We then tested whether the inhibition of hyperactive MTORC1 signaling rewires physiological homeostasis in cystinosis. In mouse cultured cells, PT segments of ctns KO rat kidneys, and mutant ctns KO zebrafish incorporating a bona fide biosensor for tubular proteinuria, the therapeutic inhibition of MTORC1 activity by using low doses of the licensed drug rapamycin recovers lysosome function and catabolic autophagy. These functional rescues steer the proliferative states of the PT cells toward differentiation and homeostasis, ameliorating PT dysfunction and LMW proteinuria downstream of CTNS loss and cystine storage (). Taken together, these preclinical proof-of-concept studies highlight the therapeutic value for modulators of MTORC1 signaling in the treatment of cystinosis.
In summary, our work describes a fundamental role of the CTNS-cystine-MTORC1 signaling node in homeostasis and identifies an evolutionarily conserved lysosome-based signal that instructs fate specification in the epithelial cells of the kidney tubule. Modulating this pathway may yield therapeutic strategies for cystinosis and other lysosomal disorders, while creating opportunities to regulate homeostasis in specialized cell types.
Disclosure statement
No potential conflict of interest was reported by the author(s).