
ABSTRACT
Tuning and assimilation of endoplasmic reticulum (ER) content in each cell of the human body is an essential part of organismal homeostasis and adaptation to stress. As such, the lysosomal turnover of ER (reticulophagy) needs to be regulated in a spatio-temporal as well as cell-type specific manner. We recently identified CSNK2/CK2 (casein kinase 2) as the enzyme that phosphorylates the reticulophagy receptors RETREG1/FAM134B and RETREG3/FAM134C and regulates their activity. Phosphorylation of the receptors is a prerequisite for their subsequent functional ubiquitination and the formation of high-density clusters, presumably representing active macroautophagy/autophagy sites at the ER membrane. Consistently, treatment with kinase inhibitor SGC-CK2-1, knockdown of endogenous CSNK2, or mutation of respective phospho-sites prevents ubiquitination, the formation of high-density clusters as well as reticulophagy flux. We hypothesize that CSNK2 has a broader impact on ER and Golgi content in a cell-type and context-specific manner by orchestrating the activity of several autophagy receptors and potentially also factors of the ER-associated protein degradation pathway.
The ER is a continuum of membrane structures that spans the cell, forms contacts with other organelles, and is involved in protein synthesis and folding, lipid and steroid synthesis, and calcium storage. The sequestration of discrete ER fragments into phagophores, precursors to autophagosomes, with the help of specific autophagy receptors and subsequent lysosomal degradation (selective macroautophagy of the ER; reticulophagy) has emerged as a major quality control and stress response pathway to achieve and maintain optimal function of the ER.
We recently reported that the function of the reticulophagy receptors RETREG1/FAM134B and RETREG3/FAM134C is regulated through phosphorylation-dependent ubiquitination [Citation1]. The regulation of RETREG/FAM134-driven reticulophagy was analyzed via a cell-based screen for kinase inhibitors that prevent reticulophagy flux induction upon torin1 treatment (MTOR inhibition). We biased our screen toward RETREG/FAM134-driven reticulophagy by overexpressing RETREG1/FAM134B and RETREG3/FAM134C in a U2OS reporter cell line for reticulophagy flux expressing a KDEL-based fluorescent reporter. SGC-CK2-1, an inhibitor of CSNK2, was a common hit of the screens and phosphorylation sites on RETREG/FAM134 proteins identified by mass spectrometry (MS) match well with the target sequence of CSNK2.
Phosphorylation events identified in our mass spectrometry data set are in close proximity to transmembrane domains 3/4 of the reticulon homology domain (RHD) of RETREG1/FAM134B (S149, S151 and S153) and RETREG3/FAM134C (S258 and S260). Mutation of phosphorylation sites (RETREG1S149,151,153A; RETREG3S258A) leads to a strong reduction in torin1-induced, RETREG/FAM134-mediated reticulophagy flux. Of note, overexpression of mutant RETREG1S149,151,153A in the presence of endogenous RETREG1/FAM134B leads to an increase in basal reticulophagy flux, which hints toward a dominant negative effect of the RETREG1/FAM134B mutant and an activation of a stress response pathway to compensate for RETREG1/FAM134B deficiency. In mouse embryonic retreg knockout fibroblasts, the wild-type protein but not the phospho-mutant is able to facilitate the degradation of accumulated COL1A1, an endogenous substrate of RETREG/FAM134-driven reticulophagy.
We could further show that phosphorylation by CSNK2 is a prerequisite for subsequent ubiquitination of overexpressed as well as endogenous RETREG/FAM134. Mechanistically, the phosphorylation-dependent ubiquitination of RETREG1/FAM134B and RETREG3/FAM134C is essential for their activity and clustering. Consistently, ubiquitination of RETREG/FAM134 is induced upon activation of reticulophagy by torin1 and prevented by co-treatment with CSNK2 inhibitor, knockdown of endogenous CSNK2, or mutation of the respective phospho-sites on RETREG/FAM134. A recent study by the Dikic lab identified and analyzed multi-monoubiquitination of RETREG1/FAM134B as an essential part of its activation mechanism. In our study, we have used super-resolution microscopy to determine the size/diameter of RETREG1/FAM134B and RETREG3/FAM134C nanoclusters as well as the density of RETREG/FAM134 molecules within clusters of a given size. In these analyses we noticed that clusters formed by RETREG1/FAM134B but not RETREG3/FAM134C increased in size upon reticulophagy induction and that this phenotype can be suppressed by CSNK2 inhibition or mutation of RETREG1/FAM134B phosphorylation sites. In addition, we observed that the density of both RETREG1/FAM134B- and RETREG3/FAM134C-clusters increases upon reticulophagy induction by torin1 treatment. The formation of such high-density clusters can be suppressed by co-treatment with CSNK2 inhibitor or by mutation of respective phosphorylation sites. The relative abundance of high-density clusters is correlative to the ubiquitination state of the receptors, indicating that phosphorylation is a trigger for the recruitment and/or activation of a ubiquitin ligase (complex). In a previous manuscript, we described RETREG3/FAM134C as an enhancer of RETREG1/FAM134B activity. The fact that the size of RETREG1/FAM134B – but not RETREG3/FAM134C – clusters rapidly increases upon torin1 treatment, led us to hypothesize that inactive RETREG3/FAM134C clusters may serve as nucleation points for the formation of larger RETREG1/FAM134B clusters upon reticulophagy induction.
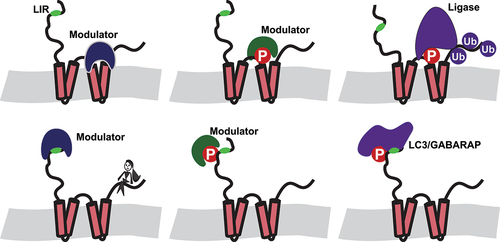
CSNK2 is a constitutive active kinase, which seems to contradict the idea of a regulatory kinase with the capability to facilitate cell-type specific responses. However, phosphorylation efficiency as well as the consequence of a phosphorylation can be easily altered by either cell-type specific/stress-inducible binders of the non-phosphorylated target restricting access to the phosphorylation site or by cell-type specific/stress-inducible modulators with very high affinity to the phosphorylated region. In this way, phosphorylation of, e.g., an LC3-interacting region (LIR) motif could have different outcomes: enhanced binding of Atg8-family proteins and subsequently full activation of the respective autophagy reporter in a cell type lacking the modulator as well as inhibition of even basal activity of the receptor in a cell type expressing the modulator ().
Figure 1. Schematic overview of putative, cell type specific regulatory pathways for reticulophagy involving CSNK2. Modulators, expressed in a cell-type specific manner, may bind to either non-phosphorylated or phosphorylated regions of RETREG/FAM134 proteins. This can facilitate or prevent the recruitment of ubiquitin ligases and Atg8-family proteins to the RHD and LIR domain, respectively. The possibility that two modulators, one targeting the LIR and the other targeting the RHD domain, can be expressed in the same cell further increases the number of cell-type specific responses.
Besides regulating RETREG/FAM134 proteins, CSNK2 has been reported by the Mizushima lab to phosphorylate TEX264 in close proximity to its LIR motif thereby enhancing binding affinity for LC3/GABARAPs. Next to CSNK2, a handful of other kinases including ATR were shared as hits between our RETREG1/FAM134B and RETREG3/FAM134C screens. Further comparative analysis on global proteome changes using TMT-based mass spectrometry can be found in an Addendum article published in Autophagy. The data are supportive of our hypothesis that CSNK2 is a specific regulator of ER (and Golgi) content that may facilitate adaptation in a cell-type specific manner.
Acknowledgements
I thank all the co-authors and collaboration partners of our study for their wonderful work. In particular Rayene Berkane, the leading author of our original publication. This work was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) grant number 259130777 (SFB1177).
Disclosure statement
No potential conflict of interest was reported by the author(s).