
Abstract
Context: Methanol poisoning induces acute optic neuropathy with possible long-term visual damage.
Objective: To study the dynamics and key determinants of visual pathway functional changes during 4 years after acute methanol poisoning.
Methods: A total of 42 patients with confirmed methanol poisoning (mean age 45.7 ± 4.4 years) were examined 4.9 ± 0.6, 25.0 ± 0.6, and 49.9 ± 0.5 months after discharge. The following tests were performed: visual evoked potential (VEP), retinal nerve fiber layer (RNFL) measurement, brain magnetic resonance imaging (MRI), complete ocular examination, biochemical tests, and apolipoprotein E (ApoE) genotyping.
Results: Abnormal VEP P1 latency was registered in 18/42 right eyes (OD) and 21/42 left eyes (OS), abnormal N1P1 amplitude in 10/42 OD and OS. Mean P1 latency shortening during the follow-up was 15.0 ± 2.0 ms for 36/42 (86%) OD and 14.9 ± 2.4 ms for 35/42 (83%) OS, with maximum shortening up to 35.0 ms. No significant change of mean N1P1 amplitude was registered during follow-up.
A further decrease in N1P1 amplitude ≥1.0 mcV in at least one eye was observed in 17 of 36 patients (47%) with measurable amplitude (mean decrease −1.11 ± 0.83 (OD)/−2.37 ± 0.66 (OS) mcV versus −0.06 ± 0.56 (OD)/−0.83 ± 0.64 (OS) mcV in the study population; both p < .001).
ApoE4 allele carriers had lower global and temporal RNFL thickness and longer initial P1 latency compared to the non-carriers (all p < .05). The odds ratio for abnormal visual function was 8.92 (3.00–36.50; 95%CI) for ApoE4 allele carriers (p < .001). The presence of ApoE4 allele was further associated with brain necrotic lesions (r = 0.384; p = .013) and brain hemorrhages (r = 0.395; p = .011).
Conclusions: Improvement of optic nerve conductivity occurred in more than 80% of patients, but evoked potential amplitude tended to decrease during the 4 years of observation. ApoE4 allele carriers demonstrated lower RNFL thickness, longer P1 latency, and more frequent methanol-induced brain damage compared to non-carriers.
Introduction
Methanol is one of the most widely applied toxic alcohols in industry, agriculture, and households. It is used as an organic solvent, biofuel, antifreeze fluid component, for car chemistry, and in the production of other chemical substances and compounds. Acute or subacute exposure to methanol leads to systemic intoxication and development of toxic optic neuropathy. Methanol poisoning outbreaks due to consumption of illicit alcoholic drinks present a challenge for healthcare systems throughout the world due to a high lethality rate and serious visual and central nervous system (CNS) damage in survivors [Citation1–5]. Treatment involves inhibition of methanol transformation to the highly toxic formic acid by alcohol dehydrogenase (ADH), with ethyl alcohol or 4-methylpyrazole [Citation6–9]. Formic acid accumulation results in mitochondrial cytochrome c oxidase inhibition, impairment of oxygen utilization, membrane lipid peroxidation, depletion of ATP in cells, and severe metabolic acidosis [Citation10–13].
The ocular retina is one of the tissues with the highest oxygen consumption, and the axons of retinal ganglion cells, which form the optic nerve, are selectively vulnerable to histotoxic hypoxia caused by formic acid because of their high energy dependence [Citation14–16]. The symptoms of methanol-induced optic neuropathy manifest after a latency period of 6 to 48 h, depending upon the amount of methanol ingested, possible ethanol co-ingestion, and body mass. Symptoms range from blurred or “snowfield” vision, reduced visual acuity (VA), photophobia, peripheral constriction of visual fields, central/centrocecal scotomata, loss of color vision, and complete blindness. In many cases, restoration of visual functions with the resolution of pathologic changes to the fundus and improvement of VA occurs 1–2 months after methanol exposure. However, long-term visual impairment may be present in 25–40% of patients [Citation17–19]. It is difficult to predict the character and degree of long-term visual damage in patients with acute or subacute methanol poisoning and new or initially unrecognized visual disturbances may manifest several years after poisoning [Citation20].
Acute demyelination of the optic nerve due to the toxic effects of formic acid may result in axonal degeneration due to the lack of trophic support from myelin and disruption of normal axon–myelin interactions. Optic neuritis studies demonstrate that spontaneous remyelination takes place after the acute episode of demyelination with a visual evoked potential (VEP) latency shortening of 6–7 ms during the first 6 months and a further reduction of 4 ms between 6 months and 2 years after the first attack [Citation21,Citation22]. VEP measurements provide important data on the dynamics of chronic optic nerve conductivity changes associated with myelin repair and restoration of visual pathway integrity. The prolongation of the wave P1 latency of full-field VEP reflects the degree of demyelination of the optic nerve fibers, and a decrease in N1P1 amplitude reflects the number of impaired axons after acute damage [Citation23].
Knowing the character and severity of visual pathway damage, recognizing long-term visual sequelae, and understanding the key determinants of chronic visual impairment during the years following discharge from hospital is important to evaluate the effectiveness of therapeutic interventions, prognosis, and timely indication of special devices to enhance the quality of life of methanol poisoning survivors. Nevertheless, there are no prospective longitudinal cohort studies on the prevalence, character, and dynamics of chronic functional changes of the visual pathway after methanol-induced optic neuropathy with the series of complete ophthalmological examinations, VEP, optical coherence tomography (OCT) of peripapillary retinal nerve fiber layer (RNFL) thickness, and magnetic resonance imaging (MRI) of the brain performed several times during the years following acute poisoning.
We present the data based on a methanol mass poisoning outbreak with more than 130 cases of poisoning and more than 50 deaths [Citation24]. In 2013, we performed a cross-sectional study of the prevalence and character of visual damage in the survivors of poisoning after discharge from hospital [Citation18]. During the 4 years after the methanol poisoning “epidemic”, we carried out three consecutive clinical examinations of the survivors according to the standardized clinical protocol in one medical facility to determine the dynamics of chronic changes of the visual pathway and its association with key clinical, genetic, and laboratory parameters measured during hospitalization and the follow-up observation period.
Materials and methods
Study design and setting
The prospective longitudinal cohort study included patients with confirmed methanol poisoning treated in 30 hospitals in the country during a mass methanol poisoning outbreak [Citation18,Citation24]. The initial clinical, toxicological, and biochemical data were obtained from treatment providers by applying a standardized data collection form and sent to the Toxicological Information Center (TIC) on the day following hospital admission. Information on pre-hospital and hospital therapeutic interventions and the outcomes was obtained from hospital discharge reports. The follow-up clinical examinations were performed three times in the same hospital approximately 6, 24, and 48 months after discharge from hospital. The study was approved by the General University Hospital Ethics Committee in Prague, Czech Republic.
Selection of participants and treatment
Mandatory reporting to the Ministry of Health and TIC on all cases of hospital admission with acute methanol poisoning and nationwide daily monitoring of the situation in all hospitals was established. All patients hospitalized with confirmed methanol poisoning were eligible for this study.
For controls, healthy subjects of the same age, ethnicity, and history of chronic alcohol abuse were recruited. Exclusion criteria for the controls were intraocular pressure ≥22 mmHg or glaucoma in either eye; evidence of a reproducible visual field (VF) defect (pattern standard deviation significant at the <5% level or abnormal glaucoma hemifield test result) in either eye as measured with Medmont automated perimeter M700 (Medmont International Pty Ltd, Vermont, Australia); unreliable VFs (false-positive or false-negative rate >15% or fixation losses >20%); any pattern of loss consistent with ocular disease; intraocular surgery in the study eye (except cataract or refractive surgery if performed more than one year before testing); best-corrected VA worse than 20/32 on the Early Treatment Diabetic Retinopathy Study scale; evidence of diabetic retinopathy, diabetic macular edema, or other vitreoretinal disease in either eye; evidence of optic nerve or RNFL abnormality in either eye; and use of a photosensitizing agent within 14 days.
Patients with methanol poisoning were treated in accordance with the American Academy of Clinical Toxicology (AACT) and European Association of Poisons Centres and Clinical Toxicologists (EAPCCT) practice guidelines for the treatment of methanol poisoning [Citation25]. Bicarbonate was administered as a buffer to patients with severe metabolic acidosis on admission. Ethyl alcohol or 4-methylpyrazole was used as an antidote to inhibit methanol oxidation by ADH [Citation26,Citation27]. Intermittent or continuous modalities of renal replacement therapy were applied in severely poisoned patients to eliminate formate and methanol and to correct acidemia [Citation28,Citation29]. Folic or folinic acid was administered to substitute the internal folate pool [Citation30].
Clinical examinations and laboratory analyses during hospital treatment
Detailed histories of poisoning and of ocular and systemic toxicity were received from the hospitalized patients or from relatives of critically ill patients upon admission to hospital. Diagnosis was established when (i) a history of recent ingestion of illicit spirits was available and serum methanol was higher than 200 mg/L, or (ii) there was a history or clinical suspicion of methanol poisoning, and serum methanol was above the lower limit of detection with at least two of the following: pH <7.3, serum bicarbonate <20 mmol/L, or anion gap (AG) ≥ 20 mmol/L [Citation25].
The standardized protocol of clinical examinations included ocular examination with standard ophthalmologic tests (VA, color vision, VFs, contrast sensitivity, and fundus examination), cerebral computed tomography (CT) or MRI of the brain, and standard neurological examination. Patients were considered to have visual sequelae of acute methanol poisoning if the symptoms of toxic optic neuropathy were documented on admission and/or during hospitalization, with pathologic findings on VA, perimeter, color vision, contrast sensitivity, and persisting lesions on fundoscopy on discharge from hospital. Further, patients were considered to have CNS sequelae of poisoning if symmetrical necrosis and hemorrhages of basal ganglia and subcortical white matter compatible with the diagnosis of acute methanol poisoning were present on CT or MRI of the brain.
Apolipoprotein E (ApoE) genotyping
DNA was isolated from frozen ethylenediaminetetraacetic acid (EDTA) blood using a modified method according to Miller et al. [Citation31]. ApoE genotyping was performed using polymerase chain reaction–restriction fragment length polymorphism (PCR-RFLP) analysis. The oligonucleotides 5′-ACA GAA TTC GCC CCG GCC TGG TAC AC-3′ and 5′-TAA GCT TGG CAC GGC TGC CAA GGA-3′ were used to amplify a 244 base pair (bp) fragment of the ApoE gene. CfoI restriction enzyme (Fermentas) was used to cut the PCR product. Restriction fragments (separated on a 15% polyacrylamide gel) of 91 bp and 83 bp are typical for the ApoE2 allele, fragments of 91 bp and 48 bp for the ApoE3 allele, and 72 bp and 48 bp fragments for the ApoE4 allele. Known ApoE genotype frequencies of a large group of an adult elderly population (HAPIEE study, n = 6230) were used for comparison [Citation32].
Follow-up clinical examination protocol
The follow-up clinical examination protocol was previously described in detail [Citation33–35]. The study protocol included complete ocular examination and standard ophthalmic tests, including VA measurement, slit-lamp examination, intraocular pressure measurement, fundus examination, color vision, VFs, OCT with RNFL, VEP, MRI of the brain, neurological and neuropsychological examinations, biochemical tests (glucose, glycohemoglobin, hepatic tests, cholesterol, lipids, thyroid-stimulating hormone (TSH), vitamins B12 and B1, carbohydrate-deficient transferrin, ethyl glucuronide (in urine), and standardized questionnaire forms.
The VEP examination was performed on a two-channel TruTrace 4 Alien Technik CZ device. Monocular full-field checkerboard pattern-reversal stimulation was used, with the reversal frequency of 1.5 c/s, angular size of the monitor 6°×5° from the fixation point, and angular size of checkerboard squares was 40´. Luminance of the white and black squares was 84 cd/m2 and 57 cd/m2, respectively, corresponding to the contrast between black and white squares of about 20%. Bandwidth of the amplifier was 1 Hz–1 kHz, and the evoked response was registered from the Oz-Fz derivation. For each eye, examination was performed twice in order to check reproducibility of the evoked complex. We evaluated the latency of the P1 wave and the N1P1 amplitude. Four criteria of abnormality were chosen: (1) missing evoked response, (2) P1 wave latency >117 ms, (3) interocular difference of P1 wave latencies >6 ms, and (4) amplitude of evoked complex <3 µV. The result was categorized as abnormal if at least one of the above-mentioned criteria was fulfilled.
The medical examiners which performed the follow-up clinical examinations were masked to the results of toxicological and biochemical measurements, severity of poisoning, clinical course, treatment measures, and clinical outcomes upon discharge from hospital.
Calculations and data analysis
Basic descriptive statistics (mean, median, confidence interval (CI), SD, skewness, and kurtosis) were computed for all variables, which were subsequently tested for normality using the Kolmogorov–Smirnov test. The Chi-squared (χ2) test was used to compare frequency counts of demographic and clinical categorical variables. The bivariate relationship was assessed using Pearson’s correlation coefficient. A linear mixed effects model was applied to study the longitudinal relationship between demographic, clinical, and laboratory parameters, and the results of VEP P1 latency measurements during the study period. The dependent variable in this model was P1 latency measured on both eyes (OD, OS – two measurements) over time (three measurements during four years), resulting in six measurements per individual. The results of repeated measurements were nested for each individual and an autoregressive covariance structure (AR-1) was used to model the relationships between observed variables. The independent variables included in the model were: age, sex, time, severity of metabolic acidosis (arterial blood pH), acute serum concentrations of methanol and ethanol on admission, and follow-up serum glucose, vitamins B1 and B12, and TSH measured during the study period. Due to the low number of ApoE2/E2 and ApoE4/E4 homozygotes, these individuals were pooled with adequate heterozygotes. Thus, three groups (ApoE2/E2 + ApoE3/E2 versus ApoE3/E3 versus ApoE4/E3 + ApoE4/E4) were compared.
Statistical significance was set at p < .05. Statistical analysis was performed in Excel (Microsoft, Redmond, WA), and formal calculations were produced in QC Expert software 3.1 (Trilobyte, Pardubice, Czech Republic) and IBM SPSS version 24.0 (Chicago, IL).
Results
Demographic and clinical data of the study population and the control group
Altogether, 108 patients were admitted to the hospital during a methanol mass poisoning outbreak. Twenty-four patients died during hospitalization and 84 patients survived and were approached and invited to join the group of volunteers in a prospective study of long-term health sequelae at discharge from the hospital. Fifty-five subjects with a mean age at discharge of 46.7 ± 3.6 years (46 men and 9 women) expressed their consent and were examined at least once during the observation period. In 49 patients, ApoE genotyping was performed. The follow-up clinical examinations were performed 4.9 ± 0.6 months, 25.0 ± 0.6 months, and 49.9 ± 0.5 months after discharge from hospital (means with standard deviation (SD)).
Of the 55 patients included in the study, eight subjects died before the third round of clinical examination, and in 5 patients less than three examinations were carried out because of a delay in joining the study program. These 13 patients were excluded from analysis of chronic visual pathway functional changes. Forty-two subjects, 34 men and eight women, underwent three consecutive clinical examinations during 4 years of observation. The control group of 41 individuals was examined according to the same clinical investigation protocol. Thus, 84 eyes from survivors of acute methanol poisoning and 82 eyes from controls were analyzed.
Demographic and clinical laboratory parameters of the studied population and controls are shown in . Patients exposed to methanol had higher follow-up glycemia because of the five subjects in this group with diabetes mellitus type 2. Blood cholesterol, but not triglycerides, was higher in the study population. The serum level of the vitamin B1 was relatively higher in the study population, but no patients with hypovitaminosis of B1 or B12 were present in either group.
Table 1. Demographic and laboratory data of the study population and the control group (means with SD).
The median ingested volume of toxic alcoholic drinks was 0.3 L (range 0.1–1.5 L) in the study population with approximately 50% methanol and 50% ethyl alcohol in various strong beverages with total alcohol content of approximately 40% by volume. Pre-hospital ethyl alcohol was administered as a “first aid antidote” by the ambulance medical or paramedic staff in 15 (36%) patients. Twenty-six per cent of the patients were admitted to hospital within 12 hours of methanol ingestion, 60% were admitted within 48 hours, and 10% were admitted more than 48 hours after ingestion. Twenty-two patients had severe acidemia on admission, 10 patients were hospitalized in a comatose state (Glasgow coma scale (GCS) of eight or lower), and 20 patients reported at admission subjective signs of visual impairment ranging from blurred and “snowfield” vision to complete blindness (three cases).
Dynamics of the P1 latency of VEPs during the observation period
The results of the measurements of the wave P1 latency of VEP during 4 years of observation are shown in . On first examination approximately 6 months after discharge, abnormal P1 latency was measured in 18/42 (43%) right eyes (oculus dexter, OD) and 21/42 (50%) left eyes (oculus sinister, OS), including five OD and four OS without elicitable evoked potential. Significant mean P1 latency shortening, with the most remarkable changes between the first and second examinations, or 2 years after methanol exposure, was observed in the study population with a return of the initially prolonged mean P1 latency to values similar to those measured in the control group ().
Figure 1. P1 latency of visual evoked potentials in the study population (n = 42) versus controls (n = 41). OD: oculus dexter; OS: oculus sinister; ms: : milliseconds; “6 months”, “24 months”, “48 months”: clinical examinations performed after discharge from hospital; ***p < .001; **p < .01; *p < .05.
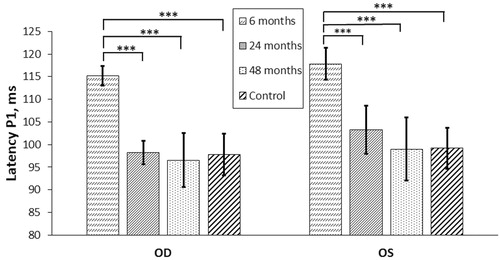
Mean P1 latency shortening in the study population was 15.0 ± 2.0 ms for 36/42 (86%) OD and 14.9 ± 2.4 ms for 35/42 (83%) OS, with maximum individual changes up to 29.0–35.0 ms. However, in five OD and four OS, visual potential could not be evoked, and in one OD and three OS, the evoked potential became non-elicitable at the second or the third examination. Worsening dynamics and non-elicitable VEP were registered in the most severely poisoned patients with pronounced acidemia on admission to hospital.
Arterial blood pH measured on admission (severity of acidemia) was significantly associated with P1 latency measured at the first and second examinations, 6 and 24 months after discharge (r1= −0.349; p = .04, and r2 = −0.379; p = .02), but not at the third one, four years after discharge, when optic nerve remyelination was completed. There was a similar association between serum methanol concentration and P1 latency: r1 = 0.324; p = .04, r2 = 0.526; p < .001 and r3 = 0.141; p = .41. Finally, there was a positive association between P1 latency measured at the first and second examinations and patient age (r1 = 0.509; p = .002, r2 = 0.494; p = .003). There were no associations for either P1 latency or the rate of latency shortening with sex or the follow-up laboratory parameters (serum glucose, cholesterol, lipids, vitamins B1, B12, TSH, and others). However, the rate of P1 latency shortening was slower in patients with high gamma glutamyl transferase (GGT) liver enzyme (r = 0.359; p = .03).
Regarding treatment modalities, the type of antidote administered in the hospital (ethanol or 4-methylpyrazol) was not significant for P1 latency, but patients treated with continuous modalities of hemodialysis had a longer P1 latency measured at the first and second examinations (r1 = 0.343; p = .03 and r2 = 0.34; p = .04). The effect of folate substitution on the dynamics of P1 latency was not significant.
For mixed model regression analysis of the dynamics of P1 latency during the observation period, the following parameters were included as predictors: age, sex, time of the follow-up examination, time between methanol exposure and hospital admission, side (reflecting interocular OD/OS P1 latency difference), acute laboratory parameters on admission (arterial blood pH, serum methanol, ethanol, and glucose), vitamins B1 and B12, and TSH measured during the follow-up period. The results of regression analysis of the variables significant to P1 latency dynamics are presented in . The observed data of P1 latency dynamics (summarized as means and SD) for arterial blood pH groupings (three groups: severe acidosis, pH ≤7.0; moderate acidosis, pH >7.0 ≤ 7.2; minor acidosis, pH >7.2 ≤ 7.4) over four years of observation are presented in .
Figure 2. Dynamics of P1 latency changes during the observation period depending on arterial blood pH at admission. OD: oculus dexter; OS: oculus sinister; Latency: : mean P1 latency, ms; pH: arterial blood pH measured on admission to hospital.
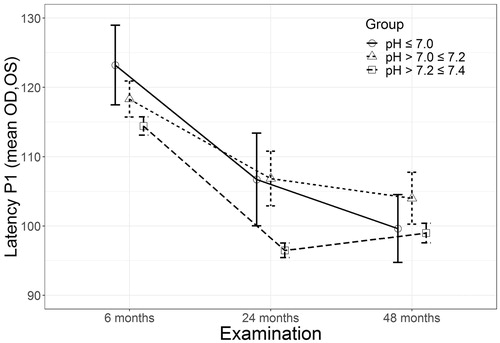
Table 2. Mixed effect model predicting changes of P1 latency during four years of observation.
Dynamics of N1P1 amplitude of VEPs during the observation period
The results of three consecutive measurements of N1P1 amplitude of VEPs in the study population are presented in . During the first examination, 6 months after discharge, abnormal N1P1 amplitude was measured in 10/42 (24%) OD and 10/42 (24%) OS, including five OD and four OS without elicitable evoked potential. At the third examination, four years after discharge, abnormal N1P1 amplitude was measured in 10/42 (24%) OD and in 14/42 (33%) OS, including six OD and seven OS without elicitable evoked potential. In one OD and three OS with abnormal N1P1 amplitude measured at the first examination, the evoked potential became non-elicitable during the second or the third examination.
Figure 3. (A) N1P1 amplitude of VEPs in the study population (n = 42) versus control group (n = 41). (B) N1P1 amplitude of VEPs in the patients with the amplitude of at least 1.0 mcV in at least one eye during the observation period (n = 17) versus control group (n = 41). OD: oculus dexter; OS: oculus sinister; ms: milliseconds; “6 months”, “24 months”, “48 months”: clinical examinations performed after discharge from hospital; ***p < .001; **p < .01; *p < .05.
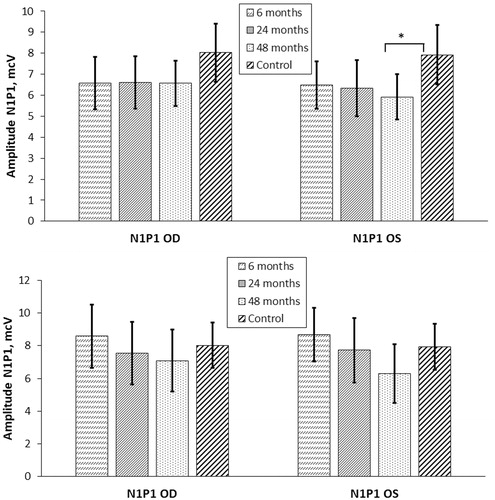
Mean N1P1 amplitude in the study population was lower than in the controls; however, the difference did not reach statistical significance (first examination: 6.6 ± 1.2 versus 8.0 ± 1.4 (OD); 6.5 ± 1.1 versus 7.9 ± 1.4 mcV (OS); second examination: 6.6 ± 1.2 versus 8.0 ± 1.4 (OD); 6.3 ± 1.3 versus 7.9 ± 1.4 mcV (OS); third examination: 6.6 ± 1.1 versus 8.0 ± 1.4 (OD); 5.9 ± 1.1 versus 7.9 ± 1.4 mcV (OS); all p > .05). No significant changes in mean N1P1 amplitude were registered during the observation period in the study population (). The mean decrease of amplitude was −0.06 ± 0.56 mcV for OD and −0.83 ± 0.64 mcV for OS.
In 17 patients from the study population, a decrease in amplitude of at least 1.0 mcV in at least one eye was observed during the observation period. In these 17 patients, the results of three consecutive measurements of N1P1 amplitude of VEPs are presented in . The mean amplitude decrease in this group (n = 17) was −1.11 ± 0.83 mcV for OD and −2.37 ± 0.66 mcV for OS, both of which were significantly higher than for the whole study population (p < .001). In eight of 17 patients, an amplitude decrease of at least 1.0 mcV was observed in both eyes, and in nine of them, the decrease occurred only in one eye, predominantly the left one. The highest rate of decrease was 4.6 mcV over 4 years for the right eye and 6.6 mcV for the left eye. None of the patients with a significant amplitude decrease exhibited abnormal N1P1 amplitude at the first examination, and only 3/17 had abnormal amplitude measured at the third examination. The remaining patients (14 of 17) had N1P1 amplitude values within the normal range during the entire observation period.
The negative association between N1P1 amplitude and P1 latency of VEP was strongest at the first examination, six months after discharge, less strong, but still significant, at the second examination, two years after discharge, and was not significant during the third examination, four years after discharge (for OD/OS: r1 = −0.398/−0.628; p = .02/.000; r2 = −0.354/−0.510; p = .037/.001; and r3 = 0.123/−0.036; p = .47/.84). Patient age negatively correlated with N1P1 amplitude, and this correlation was nearly unchanged during 4 years of observation (for OD/OS: r1 = −0.469/−0.474; p = .004/.003; r2= −0.436/−0.448; p = .009/.006; r3= −0.454/−0.422; p = .005/.01).
Patients with lower N1P1 amplitude were more severely poisoned and exhibited a lower GCS (r = 0.418; p = .009) and higher serum methanol concentration at admission to hospital (r=−0.414; p = .01). The effect of the type of antidote administered in the hospital on N1P1 amplitude was not significant, but patients treated with continuous modalities of hemodialysis had lower amplitude measured during the first and second examinations (r1= −0.495; p = .002, r2= −0.382; p = .02) compared to intermittent hemodialysis. The effect of folate substitution during treatment on N1P1 amplitude was not significant.
Chronic visual pathway changes and the effect of ApoE gene polymorphism
For the analysis of possible effects of ApoE gene polymorphism on the chronic visual pathway functional changes, 49 patients (98 eyes) with confirmed acute methanol poisoning, including 41 patients (82 eyes) with three follow-up examinations, from the study population were genotyped. Frequencies of individual ApoE genotypes in the study population did not differ significantly from the frequencies in the general Czech population ().
Table 3. Apolipoprotein E genotype and allele distribution between the study population and general czech populationa.
The association of ApoE genotype with chronic structural and functional changes of the visual pathway of the patients is presented in the . The baseline genotype group was ApoE2/E2(E3/E2): as long as only one patient had E2/E2 genotype (see ), he was added to the group of patients with E3/E2 genotype (n = 8). We compared the thickness of retinal nerve fiber layer (global one and in the temporal segments of retina) in three groups coded as “2” (genotypes E2/E2 + E3/E2), “3” (genotype E3/E3), and “4” (genotypes E4/E3 + E4/E4). Patients carrying the ApoE4 allele had lower global and temporal RNFL thickness at all three examinations compared to patients without the ApoE4 allele. The association between the genotype and the thickness of retina became even stronger with time (4 years after discharge compared to 6 months after discharge) what meant higher rate of RNFL decrease in the patients with E4/E3(E4/E4) genotype. The degree of acute demyelination of optic nerve presented by the latency P1 prolongation measured 6 months after discharge demonstrated significant association with ApoE genotype, but the process of remyelination led to the conductivity restoration and the loss of significance of this association with time.
Table 4. The association of ApoE genotype group (ApoE2/E2(E3/E2) versus ApoE3/E3 versus ApoE4/E3(E4/E4)) with chronic structural and functional changes of the visual pathway during the observation period (n = 41).
To demonstrate that both for retinal nerve fiber layer thickness and for the latency P1 prolongation, as well as for the dynamics of further RNFL decrease the presence of E4 allele was the key factor, we divided the population on two groups, ApoE4 carriers (genotypes E4/E3 and E4/E4) versus non-carriers. demonstrates the results of t-test between these two groups. The results of t-test confirmed the data on the effect of E4 allele on both morphological state of retina after methanol poisoning and on the function of visual pathway. Of five patients with non-elicitable VEP in at least one eye, four patients carried the ApoE4 allele. In the study population of methanol poisoning survivors, the odds ratio (95% CI) for abnormal VEP finding at the first examination (prolonged P1 latency and/or abnormal N1P1 amplitude or non-elicitable evoked potential) was 8.92 (3.00–36.50) for ApoE4 allele carriers compared to the non-carriers (p < .001).
Table 5. Apolipoprotein E genotype group (ApoE4 carriers versus non-carriers) and chronic structural and functional changes of the visual pathway during the observation period (n = 41).
The presence of the ApoE4 allele was further associated with brain necrotic lesions on MRI (r = 0.384; p = .01) and brain hemorrhages due to methanol poisoning (r = 0.395; p = .01). No association of ApoE alleles with sex, age, acute methanol, formic acid or ethanol serum concentration, as well as the treatment modalities (antidote, mode of hemodialysis, folate substitution) was observed.
Discussion
The prevalence of visual sequelae of acute methanol poisoning is high but typically underestimated. In our previous studies, we demonstrated that the population of patients with long-term visual damage after methanol-induced toxic optic neuropathy comprised up to 40% of the survivors, and acute retinal ganglion cells injury was followed by chronic retinal neurodegeneration and progressive axonal loss occurred in up to 25% of the patients. [Citation18,Citation35]. In the present study, we found that total or partial functional restoration of optic nerve conductivity after acute myelin sheath damage occurred in more than 80% of patients during 4 years, with the highest rate of remyelination within 2 years of discharge. The time between methanol exposure and hospital admission, severity of acidemia, and patient age were the most significant variables for the dynamics of optic nerve conductivity restoration. The VEP amplitude, abnormal in one fourth of the study population at discharge from hospital, demonstrated the tendency of this variable to progressively decrease during the following years. In half of the patients with measurable evoked potential, a decrease of N1P1 amplitude of more than 1.0 mcV was observed; none of these patients had abnormal amplitude at discharge from the hospital. In 2–7% of examined eyes, the progression of visual pathway changes led to worsening function, from abnormal, but still measurable N1P1 amplitude, to a non-elicitable VEP. The effect of the type of antidote administered in hospital and of folate substitution therapy was not significant in our study; however, patients treated with intermittent hemodialysis compared to the continuous modalities demonstrated better functional outcome: shorter P1 latency and higher N1P1 amplitude of the VEP. Finally, we demonstrated the association between the ApoE genotype polymorphism and chronic structural and functional changes of the visual pathway. Patients who carried the ApoE4 allele had reduced RNFL thickness and longer P1 latency compared to non-carriers. ApoE4 presence was associated with necrotic lesions and hemorrhages on MRI of the brain.
Acute methanol poisoning is a life-threatening condition with a lethality rate of up to 30–40%, and necrotic basal ganglia damage occurs in up to 50% of survivors [Citation36–39]. However, long-term visual sequelae due to optical nerve and retina damage by formic acid should not be underestimated and underdiagnosed due to their impact on the quality of life of the patients and their relatives, as well as on the medical facilities providing health care after discharge from the hospital [Citation17–19,Citation40]. Optic nerve axons are the most vulnerable structures for acute neurotoxic effects of formic acid; nevertheless, chronic functional changes of the visual pathway in patients after acute exposure to methanol have not yet been well studied. One reason is that typically after discharge from hospital contact with the patients is lost [Citation19]. In our previous studies, we demonstrated that structural changes of the ocular retina, namely chronic decrease of RNFL, may progress in the years following discharge from hospital [Citation35,Citation41]. These changes may affect visual pathway functions and may be registered by the series of VEP measurements within the longitudinal follow-up study.
In the case reports and small retrospective case series studies, both partial recovery and progression of visual loss were registered typically 6–9 months after discharge [Citation17,Citation19,Citation42]. We reported that the process of chronic optic nerve remyelination in patients with mild to moderate damage of myelin sheaths may lead to improvement of conductivity in survivors without a significant increase of VEP amplitude [Citation43,Citation44]. Our present study demonstrates that the process of chronic remyelination leads to the restoration of optic nerve conductivity in most survivors, with the highest rate of remyelination during the first 2 years after discharge. Patients without positive dynamics of optic nerve conductivity or with worsening visual pathway function exhibited the most severe initial neuronal damage with non-elicitable evoked potential at discharge or initial P1 latency longer than 120 ms. These patients typically had severe acidemia, high serum formate, lactate, and no protective ethanol in blood serum on admission to the hospital.
Severe acidemia is one of the prognostic factors of mortality in acute methanol poisoning [Citation45,Citation46]. In our study, the degree of acidemia on admission was associated with the dynamics of P1 latency and provided the basis for the model prediction of optic nerve conductivity restoration during the observation period. This model accurately predicted the actual data on remyelination dynamics for most of the patients in our study population. Undissociated formic acid, but not the formate anion, easily crosses the blood-brain barrier and neuronal cell membranes [Citation47]. The dissociation constant for formic acid is 3.8; therefore, for this weak acid, a pH drop of 0.3 would mean a doubling of undissociated formic acid levels, and hence a significant increase in ocular neurotoxicity [Citation10–12]. This fact may explain the association between the modality of dialysis and both P1 latency and N1P1 amplitude of VEP. Higher rates of formate elimination and acidemia correction during intermittent hemodialysis compared to the continuous modalities may have an impact on the toxic effects of formate on optical neurons [Citation28,Citation29,Citation48].
Our findings suggest that no restoration of the VEP amplitude occurs during the years following acute neuronal damage. During the observation period, the mean amplitude did not significantly change in the study population. The character of association between the VEP N1P1 amplitude and P1 latency, with an observed gradual loss of a significant correlation between these parameters during 4 years after discharge, reflected the process of conductivity restoration of the optic nerve due to remyelination without corresponding amplitude restoration after methanol-induced toxic neuropathy. Nevertheless, a further decrease of the amplitude, probably due to chronic axonal degeneration, was observed in almost half of the patients with measurable N1P1 amplitude during the observation period. The mean amplitude decrease in this subgroup was significantly higher than in the total study population, with the highest rate of up to 5–7 mcV over the 4 years. An abnormal N1P1 amplitude was measured in 24–33% of eyes at the third examination, a finding that reflected the proportion of patients with diagnosed chronic retinal neurodegeneration reported in our previous study [Citation35]. Therefore, progressive chronic axonal loss leads to a decrease in N1P1 amplitude over the years following acute methanol poisoning. It should be emphasized that the decrease in VEP amplitude was registered in patients with initially normal N1P1 values measured at the first examination.
Other conditions unrelated to methanol exposure may affect optic nerve conductivity and functions of the visual pathway in the patients from our study population. Vitamin B12 or B1 deficiency caused by chronic alcohol abuse or nutritional deficits, thyroid gland hypofunction, or insufficiently controlled glycemia in diabetes mellitus may negatively impact remyelination after methanol-induced optic neuropathy and may cause axonal damage due to inadequate restoration of myelin sheaths trophic and protective functions [Citation49–53]. However, in our study, no association was found between repeatedly measured concentrations of serum glucose, TSH, vitamins B12 or B1, and the dynamics of chronic visual pathway functional changes. Nevertheless, conductivity restoration was relatively slower in patients with high GGT liver enzyme, which is one of the clinical signs of chronic alcohol abuse.
In our previous studies, we demonstrated that genetic polymorphisms of the enzymes involved in methanol oxidation affects the outcome of poisoning [Citation54,Citation55]. In the present study, we focused on the ApoE polymorphism that is not directly involved in methanol metabolism but plays an important role in eye development and metabolism. ApoE, as a pleiotropic protein that influences the risk of Alzheimer’s and cardiovascular diseases, attracts attention as a potential determinant of the visual functions. This glycoprotein is synthetized in the retina by Muller glial cells and transported into the optic nerve by retinal ganglion cells, where it plays a key role in the local transport and distribution of cholesteryl ester-rich lipoproteins to supply the optic axons’ need for particular lipids, since apolipoprotein B, the other molecule with the same function, is not synthetized in the CNS [Citation56,Citation57]. ApoE produced by Muller glial cells donates lipids and cholesterol to the neurons engaged in synaptic remodeling following acute lesioning [Citation56].
The lipid transport function of E4 form of ApoE is worse compared to the other two isoforms of ApoE, as experimental studies indicate [Citation58]. Further, ApoE4 allele has been linked to impaired blood-brain barrier function, what may reflect a breakdown in the blood-retina barrier and higher exposure of retinal ganglion cells and optic nerve axons to the toxic metabolites of methanol [Citation59,Citation60]. Finally, in optic neurons, ApoE4 reduces expression of the protein subunits of mitochondrial respiratory complexes, such as subunit 1 of complex IV (mtCOX1) and subunit of complex V, resulting in a reduction in mitochondrial respiratory function [Citation61]. Similarly, ApoE4 perturbs neuronal function by reducing mitochondrial motility, decreasing neurite outgrowth, and inhibiting synaptogenesis [Citation62–64]. Therefore, patients with the E4 isoform may be more susceptible to the deleterious effects of formic acid on retinal ganglion cells and their axons.
ApoE genotype distribution was similar in our patients to the general Czech population [Citation32]. We found an association between ApoE polymorphism and chronic visual pathway changes in the survivors of methanol poisoning. ApoE4 is associated with both neuronal and vascular impairment of the retina. In experimental studies, the ApoE4 genotype was associated with retinal vascular pathology, reduced levels of vascular endothelial growth factor (VEGF), and a significant decrease in retinal synaptic density in developing mice retina [Citation65]. The synaptic density of the retinal neuronal synaptic layers was significantly lower in ApoE4 compared to ApoE3 mice, with reduced levels of the presynaptic vesicular glutamatergic transporter that suggested glutamatergic nerve terminals are preferentially affected in ApoE4 allele carriers [Citation66].
Our observation suggests that ApoE4 might be associated with chronic retinal neurodegeneration and progressive visual pathway dysfunction. Further, the association among ApoE polymorphism, chronic retinal neuronal degeneration, and MRI findings of brain hemorrhages and necrotic damage in patients exposed to methanol indicates possible pathophysiological relationships between long-term visual and brain sequelae of poisoning. Our study demonstrates that the signs of progressive chronic visual pathway dysfunction may indicate association with a genetic risk factor for chronic neurodegenerative diseases in the population of survivors of acute methanol poisoning.
Limitations of the study
There were several limitations of this study. The limited number of the patients with methanol-induced toxic optic neuropathy could have an effect on the power of the study. The study was not controlled for the time to hospital admission, treatment, and comorbidities of the patients hospitalized with acute methanol poisoning who were included in the study. However, the patients in the study population were relatively young; the number of patients with comorbidities was low, and the clinical and laboratory parameters monitored during the observation period provided us with sufficient evidence to exclude these comorbidities as possible causes of chronic visual pathway functional changes.
The aim of the study was to register the dynamics of visual pathway function after acute methanol-induced toxic optic neuropathy in poisoning survivors during 4 years after discharge, to compare this data with data from controls of the same age and alcohol abuse profile, and to find any association between the dynamics of optical functions and the clinical and laboratory parameters followed during hospitalization and after discharge. Approximately one third of methanol poisoning survivors did not join the follow-up study; therefore, selection bias is possible, with fewer severely poisoned subjects participating in follow-up. However, the admission laboratory data demonstrated that at least half of the patients were severely poisoned and reported at admission subjective signs of visual impairment.
Despite these limitations, this study represents the first prospective longitudinal cohort study of the dynamics and determinants of chronic visual pathway functional changes after methanol-induced optic neuropathy performed during 4 years after discharge by applying a standardized clinical examination protocol and advanced technological measurements in the same medical facility.
Disclosure statement
The authors report no conflict of interest. The authors alone are responsible for the content and writing of this paper. The manuscript has been read and approved by all authors.
The authors certify that the submission (aside from an abstract) is not under review at any other publication.
The authors certify that the authors have no other submissions and previous reports that might be regarded as overlapping with the current work.
Additional information
Funding
References
- Paasma R, Hovda KE, Tikkerberi A, et al. Methanol mass poisoning in Estonia: outbreak in 154 patients. Clin Toxicol. 2007;45:152–157.
- Hovda KE, Hunderi OH, Tafjord AB, et al. Methanol outbreak in Norway 2002–2004: epidemiology, clinical features and prognostic signs. J Intern Med. 2005;258:181–190.
- Rostrup M, Edwards JK, Abukalish M, et al. The methanol poisoning outbreaks in Libya 2013 and Kenya 2014. PLoS One. 2016;11:e0152676.
- Bezdicek O, Michalec J, Vaneckova M, et al. Cognitive sequelae of methanol poisoning involve executive dysfunction and memory impairment in cross-sectional and long-term perspective. Alcohol. 2017;59:27–35.
- Peterova K, Brozova H, Klempir J, et al. Gait and balance impairment after acute methanol poisoning. Basic Clin Pharmacol Toxicol. 2018;122:176–182.
- McMartin K, Jacobsen D, Hovda KE. Antidotes for poisoning by alcohols that form toxic metabolites. Br J Clin Pharmacol. 2016;81:505–515.
- Zakharov S, Pelclova D, Navratil T, et al. Fomepizole versus ethanol in the treatment of acute methanol poisoning: comparison of clinical effectiveness in a mass poisoning outbreak. Clin Toxicol. 2015;53:797–806.
- Zakharov S, Pelclova D, Urban P, et al. Use of out-of-hospital ethanol administration to improve outcome in mass methanol outbreaks. Ann Emerg Med. 2016;68:52–61.
- Zakharov S, Nurieva O, Kotikova K, et al. Positive serum ethanol concentration on admission to hospital as the factor predictive of treatment outcome in acute methanol poisoning. Monatsh Chem. 2017;148:409–419.
- Liesivuori J, Savolainen H. Methanol and formic acid toxicity: biochemical mechanisms. Pharmacol Toxicol. 1991;69:157–163.
- Eells JT, Henry MM, Lewandowski MF, et al. Development and characterization of a rodent model of methanol-induced retinal and optic nerve toxicity. Neurotoxicology. 2000;21:321–330.
- Zakharov S, Kurcova I, Navratil T, et al. Is the measurement of serum formate concentration useful in the diagnostics of acute methanol poisoning? A prospective study of 38 patients. Basic Clin Pharmacol Toxicol. 2015;116:445–451.
- Hlusicka J, Loster T, Lischkova L, et al. Role of activation of lipid peroxidation in the mechanisms of acute methanol poisoning. Clin Toxicol (Phila). 2018;56(10):893–903.
- Yu DY, Cringle SJ. Oxygen distribution and consumption within the retina in vascularized and avascular retinas and in animal models of retinal disease. Prog Retin Eye Res. 2001;20:175–208.
- Carelli V, Ross-Cisneros FN, Sadun AA. Mitochondrial dysfunction as a cause of optic neuropathies. Prog Retin Eye Res. 2004;23:53–89.
- Seme MT, Summerfelt P, Neitz J, et al. Differential recovery of retinal function after mitochondrial inhibition by methanol intoxication. Invest Ophthalmol Vis Sci. 2001;42:834–841.
- Sanaei-Zadeh H, Zamani N, Shadnia S. Outcomes of visual disturbances after methanol poisoning. Clin Toxicol. 2011;49:102–107.
- Zakharov S, Pelclova D, Diblik P, et al. Long-term visual damage after acute methanol poisonings: longitudinal cross-sectional study in 50 patients. Clin Toxicol. 2015;53:884–892.
- Desai T, Sudhalkar A, Vyas U, et al. Methanol poisoning: predictors of visual outcomes. JAMA Ophthalmol. 2013;131:358–364.
- Paasma R, Hovda KE, Jacobsen D. Methanol poisoning and long term sequelae - a six years follow-up after a large methanol outbreak. BMC Clin Pharmacol. 2009;9:5.
- Brusa A, Jones SJ, Kapoor R, et al. Long-term recovery and fellow eye deterioration after optic neuritis, determined by serial visual evoked potentials. J Neurol. 1999;246:776–782.
- Brusa A, Jones SJ, Plant GT. Long-term remyelination after optic neuritis: a 2-year visual evoked potential and psychophysical serial study. Brain. 2001;124:468–479.
- Jones SJ, Brusa A. Neurophysiological evidence for long-term repair of MS lesions: implications for axon protection. J Neurol Sci. 2003;206:193–198.
- Zakharov S, Pelclova D, Urban P, et al. Czech mass methanol outbreak 2012: epidemiology, challenges and clinical features. Clin Toxicol. 2014;52:1013–1024.
- Barceloux DG, Bond GR, Krenzelok EP, et al. American Academy of Clinical Toxicology practice guidelines on the treatment of methanol poisoning. J Toxicol Clin Toxicol. 2002;40:415–446.
- Zakharov S, Navratil T, Pelclova D. Fomepizole in the treatment of acute methanol poisonings: experience from the Czech mass methanol outbreak 2012–2013. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub. 2014;158:641–649.
- Zakharov S, Navratil T, Salek T, et al. Fluctuations in serum ethanol concentration in the treatment of acute methanol poisoning: a prospective study of 21 patients. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub. 2015;159:666–676.
- Zakharov S, Pelclova D, Navratil T, et al. Efficiency of acidemia correction on intermittent versus continuous hemodialysis in acute methanol poisoning. Clin Toxicol. 2017;55:123–132.
- Zakharov S, Rulisek J, Nurieva O, et al. Intermittent versus continuous renal replacement therapy in acute methanol poisoning: comparison of clinical effectiveness in mass poisoning outbreaks. Ann Intensive Care. 2017;7:77.
- Zakharov S, Nurieva O, Navratil T, et al. Acute methanol poisonings: folates administration and visual sequelae. J Appl Biomed. 2014;12:309–316.
- Miller SA, Dykes DD, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988;16:1215.
- Hubacek JA, Piper BJ, Pikhart H, et al. Lack of an association between left-handedness and APOE polymorphism in a large sample of adults: results of the Czech HAPIEE study. Laterality. 2013;18:513–519.
- Zakharov S, Kotikova K, Nurieva O, et al. Leukotriene-mediated neuroinflammation, toxic brain damage, and neurodegeneration in acute methanol poisoning. Clin Toxicol. 2017;55:249–259.
- Zakharov S, Kotikova K, Vaneckova M, et al. Acute methanol poisoning: prevalence and predisposing factors of haemorrhagic and non-haemorrhagic brain lesions. Basic Clin Pharmacol Toxicol. 2016;119:228–238.
- Nurieva O, Diblik P, Kuthan P, et al. Progressive chronic retinal axonal loss following acute methanol-induced optic neuropathy: four-year prospective cohort study. Am J Ophthalmol. 2018;191:100–115.
- Vaneckova M, Zakharov S, Klempir J, et al. Imaging findings after methanol intoxication (cohort of 46 patients). Neuro Endocrinol Letters. 2015;36:737–744.
- Bezdicek O, Klempir J, Liskova I, et al. Sequelae of methanol poisoning for cognition. Cesk Slov Neurol N. 2014;77/110:320–325.
- Zakharov S, Hlusicka J, Nurieva O, et al. Neuroinflammation markers and methyl alcohol induced toxic brain damage. Toxicol Lett. 2018;pii:S0378–4274(18)30175–9.
- Vaneckova M, Zakharov S, Klempir J, et al. Methanol intoxication on magnetic resonance imaging – case reports. Cesk Slov Neurol Neurochir. 2014;77:235–239.
- Rulisek J, Balik M, Polak F, et al. Cost-effectiveness of hospital treatment and outcomes of acute methanol poisoning during the Czech Republic mass poisoning outbreak. J Crit Care. 2017;39:190–198.
- Zakharov S, Nurieva O, Kotikova K, et al. Factors predicting optic nerve axonal degeneration after methanol-induced acute optic neuropathy: a two-year prospective study in 54 patients. Monatsh Chem. 2016;147:251–261.
- Zhao XJ, Lu L, Li M, et al. Ophthalmic findings in two cases of methanol optic neuropathy with relapsed vision disturbance. Int J Ophthalmol. 2015;8:427–429.
- Nurieva O, Kotikova K, Urban P, et al. Prevalence, dynamics, and biochemical predictors of optic nerve remyelination after methanol-induced acute optic neuropathy: a two-year prospective study in 54 patients. Monatsh Chem. 2016;147:239–249.
- Urban P, Zakharov S, Diblík P, et al. Visual evoked potentials in patients after methanol poisoning. Int J Occup Med Environ Health. 2015;29:471–478.
- Paasma R, Hovda KE, Hassanian-Moghaddam H, et al. Risk factors related to poor outcome after methanol poisoning and the relation between outcome and antidotes - a multicenter study. Clin Toxicol. 2012;50:823–831.
- Zakharov S, Navratil T, Pelclova D. Analysis of serum anion gap and osmolal gap in diagnosis and prognosis of acute methanol poisoning: clinical study in 86 patients. Monatsh Chem. 2015;146:787–794.
- Drangsholt E, Vangstad M, Zakharov S, et al. The hypothesis of circulus hypoxicus and its clinical relevance in patients with methanol poisoning - an observational study of 35 patients. Basic Clin Pharmacol Toxicol. 2018. doi:10.1111/bcpt.13074
- Zakharov S, Pelclova D, Navratil T, et al. Intermittent hemodialysis is superior to continuous veno-venous hemodialysis/hemodiafiltration to eliminate methanol and formate during treatment for methanol poisoning. Kidney Int. 2014;86:199–207.
- Misra UK, Kalita J, Das A. Vitamin B12 deficiency neurological syndromes: a clinical MRI and electrodiagnostic study. Electromyogr Clin Neurophysiol. 2003;43:57–64.
- Fei G-q, Zhong C, Jin L, et al. Clinical characteristics and MR imaging features of nonalcoholic Wernicke encephalopathy. AJNR Am J Neuroradiol. 2008;29:164–169.
- Yeh W-Y, Lian L-M, Chang A, et al. Thiamine-deficient optic neuropathy associated with Wernicke’s encephalopathy in patients with chronic diarrhea. J Formos Med Assoc. 2013;112:165–170.
- Fernandez M, Giuliani A, Pirondi S, et al. Thyroid hormone administration enhances remyelination in chronic demyelinating inflammatory disease. Proc Natl Acad Sci USA. 2004;101:16363–16368. 16
- Veselinovic D, Jovanovic M. Diabetes mellitus and optic nerve diseases. Acta Fac Med NAISS. 2005;22:145–148.
- Hubacek JA, Jirsa M, Bobak M, et al. Aldehyde dehydrogenase 2 polymorphism affects the outcome of methanol poisoning in exposed humans. Clin Genet. 2018;94(5):445–449.
- Hubacek JA, Pelclova D, Seidl Z, et al. Rare alleles within the CYP2E1 (MEOS system) could be associated with better short-term health outcome after acute methanol poisoning. Basic Clin Pharmacol Toxicol. 2015;116:168–172.
- Poirier J. Apolipoprotein E in animal models of CNS injury and in Alzheimer's disease. Trends Neurosci. 1994;17:525–530.
- Amaratunga A, Abraham CR, Edwards RB, et al. Apolipoprotein E is synthesized in the retina by Müller glial cells, secreted into the vitreous, and rapidly transported into the optic nerve by retinal ganglion cells. J Biol Chem. 1996;271:5628–5632.
- Nathan BP, Bellosta S, Sanan DA, et al. Differential effects of apolipoproteins E3 and E4 on neuronal growth in vitro. Science. 1994;264:850–852.
- Fullerton SM, Shirman GA, Strittmatter WJ, et al. Impairment of the blood-nerve and blood-brain barriers in apolipoprotein E knockout mice. Exp Neurol. 2001;169:13–22.
- Wong TY, Mitchell P. Hypertensive retinopathy. N Engl J Med. 2004;351:2310–2317.
- Chen HK, Ji ZS, Dodson SE, et al. Apolipoprotein E4 domain interaction mediates detrimental effects on mitochondria and is a potential therapeutic target for Alzheimer disease. J Biol Chem. 2011;286:5215–5221.
- Bellosta S, Nathan BP, Orth M, et al. Stable expression and secretion of apolipoproteins E3 and E4 in mouse neuroblastoma cells produces differential effects on neurite outgrowth. J Biol Chem. 1995;270:27063–27071.
- Brodbeck J, Balestra ME, Saunders AM, et al. Rosiglitazone increases dendritic spine density and rescues spine loss caused by apolipoprotein E4 in primary cortical neurons. Proc Natl Acad Sci USA. 2008;105:1343–1346.
- Chen HK, Liu Z, Meyer-Franke A, et al. Small molecule structure correctors abolish detrimental effects of apolipoprotein E4 in cultured neurons. J Biol Chem. 2012;287:5253–5266.
- Maharshak I, Salomon-Zimri S, Antes R, et al. The effects of the apoE4 genotype on the developing mouse retina. Exp Eye Res. 2016;145:17–25.
- Antes R, Ezra-Elia R, Weinberger D, et al. ApoE4 induces synaptic and ERG impairments in the retina of young targeted replacement apoE4 mice. PLoS One. 2013;8:e64949.