
Abstract
Acute lymphoblastic leukemia (ALL) is the most common cancer diagnosed in children under the age of 15. In addition to genetic aberrations, epigenetic modifications such as DNA methylation are altered in cancer and impact gene expression. To identify epigenetic alterations in ALL, genome-wide methylation profiles were generated using the methylated CpG island recovery assay followed by next-generation sequencing. More than 25,000 differentially methylated regions (DMR) were observed in ALL patients with ∼90% present within intronic or intergenic regions. To determine the regulatory potential of the DMR, whole-transcriptome analysis was performed and integrated with methylation data. Aberrant promoter methylation was associated with the altered expression of genes involved in transcriptional regulation, apoptosis, and proliferation. Novel enhancer-like sequences were identified within intronic and intergenic DMR. Aberrant methylation in these regions was associated with the altered expression of neighboring genes involved in cell cycle processes, lymphocyte activation and apoptosis. These genes include potential epi-driver genes, such as SYNE1, PTPRS, PAWR, HDAC9, RGCC, MCOLN2, LYN, TRAF3, FLT1, and MELK, which may provide a selective advantage to leukemic cells. In addition, the differential expression of epigenetic modifier genes, pseudogenes, and non-coding RNAs was also observed accentuating the role of erroneous epigenetic gene regulation in ALL.
Abbreviations
| ALL | = | acute lymphoblastic leukemia |
| CD | = | cluster of differentiation |
| CpG | = | CG dinucleotide |
| CpGI | = | CpG island |
| DMRs | = | differentially methylated regions |
| FDR | = | false discovery rate |
| H3K4me1 | = | histone H3 lysine 4 monomethylation |
| H3K27ac | = | histone H3 lysine 27 acetylation |
| HCB | = | human umbilical cord blood |
| MBDs | = | methyl CpG binding domains |
| lincRNA | = | long intergenic non-coding RNA |
| MIRA-seq | = | methylated CpG island recovery assay (MIRA) followed by next generation sequencing |
| miRNA | = | microRNA |
| Pre-B | = | precursor B-cell |
| ROIs | = | regions of interest |
| TFs | = | transcription factors. |
Introduction
Acute lymphoblastic leukemia (ALL) is a hematological malignancy associated with precursor B-cells. ALL is the most common type of cancer in children with an annual occurrence rate of 35 to 40 cases per 1 million people in the United States.Citation1 The development and differentiation of B-cells comprises numerous stages and is a highly synchronized and controlled process governed by stage-specific gene expression.Citation2,3 Any deviation from normal stage-specific gene expression could lead to disease conditions including ALL. The general known mechanisms underlying the induction of ALL include chromosomal translocation, hyperdiploidy, and aberrant expression of proto-oncogenes. Advancement in deciphering additional mechanisms that may be responsible for the induction of all ALL is lacking. Therefore, the identification of key regulatory regions in the genome that may impact the development of ALL is critical to gaining a better understanding of ALL pathogenesis.
DNA methylation is responsible for tissue specific gene expression and plays a significant role in hematopoiesisCitation4,5 and malignant transformation.Citation6,7 A reduced level of CpG methylation was one of the first epigenetic alterations to be found in human cancer when compared with normal-tissue counterparts.Citation8 Although hypermethylation of CpG islands within gene promoters has been the main focus of studies on malignant cells, the role of differential DNA methylation in other regions is gaining favor.Citation9,10 One such region harbors transcriptional enhancers, which reside within non-coding regions of the genome and are known to work over long distances to promote cell/tissue type specific gene expression. Active enhancers are often accompanied by DNA demethylation,Citation11 and alterations in enhancer methylation are seen in malignant transformation. Recently, it has been shown that differential methylation of these regions exhibit a higher correlation with gene expression than differential promoter methylation.Citation12
As a step toward better understanding the consequence of altered DNA methylation on gene expression in pre-B ALL, MIRA-seq was used to identify altered DNA methylation throughout the genome and then correlated with transcriptome data. We show that differential comparisons of DNA methylation between normal and diseased tissue can identify potential regulatory regions of the genome and that when paired with gene expression data the functionality of the regulatory regions can be determined.
Results
Genome-wide DNA methylation profiles
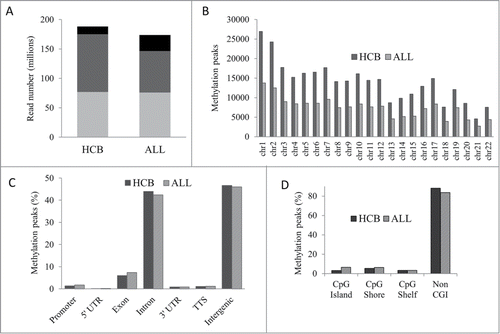
MIRA-seq was utilized to generate genome-wide DNA methylation profiles for 19 pre-B ALL patient samples from diagnostic bone marrow. Normal precursor B-cell populations (pre-BI and pre-BII) were isolated from 10 human umbilical cord blood (HCB) samples to generate methylation profiles for healthy tissue to be used as a comparator.Citation4 On average, 188 million reads were generated for HCB samples and 174 million reads were generated for ALL patient samples (, Table S1). Methylation peaks were more abundant in HCB samples (305,736) than in ALL samples (162,832) and across all chromosomes revealing an overall genome-wide reduction of methylation in ALL (). Genomic distribution analysis showed that ∼90% of the methylated peaks were located within intronic and intergenic regions (). The distribution of methylation peaks relative to CpG islands (CGIs) revealed that 9,814 CGIs were methylated in HCB samples and 11,015 CGIs were methylated in ALL samples but the overwhelming majority of methylated peaks were present in regions of the genome not associated with CGIs ().
Figure 1. Genome-wide DNA methylation profiles in HCB and ALL. (A) Average read and alignment statistics. Reads were averaged across all individuals for HCB and ALL samples. The top of each bar represents the total number of reads for each category. Black bars: total reads; Dark gray bars: reads mapped; Light gray bars: unique reads. (B) Chromosome-wise methylation peaks. The X and Y chromosomes were excluded from analysis. (C) Genomic distribution of methylation peaks. TTS: transcription termination site. (D) Methylation peaks in CGI context.
Differentially methylated regions in ALL
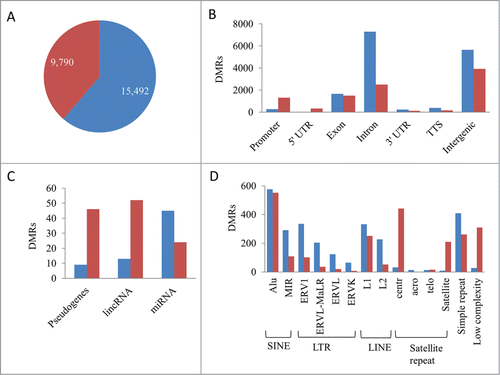
To determine methylation patterns distinct to ALL, differentially methylated regions (DMRs) between ALL and HCB samples with at least a 2-fold change and an FDR of ≤5% were identified (Table S2). A total of 15,492 regions lost methylation and 9,790 regions gained methylation in ALL compared to the normal HCB samples and the genomic distribution of loci harboring DMRs differed in the hypomethylated versus hypermethylated DMRs (). Hypermethylation was more prevalent in the 5′ regulatory regions of genes than hypomethylation. The majority of the DMRs coincided with intergenic and intronic genomic regions. DMRs have applicability as disease specific biomarkers and may also play regulatory roles in the expression of genes that are involved in the pathogenesis of ALL. To further elucidate the importance of DMRs, we sought to identify the DMRs with regulatory potential.
Figure 2. Differentially methylated regions in ALL. (A) Hypomethylated (blue) and hypermethylated (red) regions. (B) Genomic distribution of hypo- and hyper-methylated DMR. (C) DMRs associated with the 5′regulatory region of pseudogenes and non-coding RNA. (D) Intergenic DMRs associated with transposable elements and repeat sequences.
DMRs are associated with regulatory sequences: The promoters of protein coding genes harbor regulatory sequences required for the initiation of transcription. A total of 1,568 differentially methylated gene promoters were identified (corresponding to 1,252 hypermethylated genes and 240 hypomethylated genes) in ALL. To explore the association of DNA methylation and gene expression, MIRA-seq data and RNA-seq data were correlated. Sixty-two promoter DMRs were hypermethylated and downregulated in ALL and were significantly enriched for genes involved in the regulation of transcription and apoptosis, whereas 37 promoter DMRs were hypomethylated and upregulated and were significantly enriched for genes involved in GTPase activation, the regulation of cell proliferation, and those that play a role in protein complex assembly. Additionally, hypermethylated DMRs were identified in the promoters of 3 tumor suppressor genes, MTSS1, PAWR, and EXT1, and corresponded with a significant decrease in gene expression.
In addition to protein coding gene promoters, differential methylation was observed within 1,000 bp upstream or downstream of the TSS in non-coding RNAs and pseudogenes (). MicroRNAs (miRNA) are non-coding RNAs that regulate expression through imperfect base-pairing with the 3′UTR of multiple target genes. A total of 69 miRNAs were differentially methylated in ALL including miR-375, miR-196a, miR-3545, miR-9-1/2/3, miR-124-1/3, and miR-34b, which have been implicated in human malignancies.Citation13-15 RNA-seq libraries were prepared from poly(A) RNA and excluded the capture of miRNA; therefore, correlation studies between methylation and gene expression were not performed for miRNA. The regulatory potential of DMRs associated with miRNAs warrants further attention. Long intergenic non-coding RNAs (lincRNAs) are emerging as key regulators of numerous cellular processes and regulate the expression of multiple target genes. Differential methylation occurred in 65 lincRNAs. Of these, hypomethylation and upregulation was observed in AC002398.5, DIO3OS and LINC00642. Lastly, 55 pseudogenes were differentially methylated in ALL. No correlations between expression and promoter methylation was observed in the pseudogenes; however, pseudogenes, much like lincRNAs, have the potential to epigenetically regulate their parental genes and were further investigated.
It is well known that transposable element activities are often silenced by DNA methylation,Citation16 and that transcriptional activation of these elements results in transposable element mediated insertions and chromosomal rearrangements in many cancers.Citation17 Many of the intergenic DMRs were associated with transposable elements and repeat sequences (). Non-autonomous short interspersed nuclear elements (SINE) were the most abundantly present transposable element within the differentially methylated intergenic regions followed by long terminal repeat (LTR), autonomous long interspersed nuclear elements (LINE) and satellite repeats. Centromeric α satellite repeats were often hypermethylated in ALL, which may block CENP-A and result in centromere inactivation.
DMRs are associated with predicted regulatory sequence: Differential methylation predominately occurred in intergenic and intronic regions in ALL. One third of the intronic DMRs (3,341) were located within 150 base pairs of the 5′ or 3′ splice sites and could potentially alter appropriate splicing in ALL. To investigate whether the intergenic and intronic DMRs coincided with the location of regulatory enhancer elements, the sites for intergenic and intronic DMRs were overlaid with ENCODE ChIP-seq data for enhancer related histone marks (H3K4me1 and H3K27ac) in the GM12878 lymphoblastoid cell line. Overall, 765 intergenic and intronic DMRs overlapped with potential enhancer like regions (eDMR). Of these, 453 were hypomethylated and 312 were hypermethylated. Enhancer methylation has been shown to have a stronger association with gene deregulation than promoter methylation in cancer.Citation12 To investigate the association between enhancer methylation and gene expression in our data, lists were constructed of the nearest upstream and downstream gene to identify the potential target genes for each eDMR. A total of 81 genes exhibited significantly decreased expression in ALL that corresponded with hypermethylation of potential neighboring eDMRs, and 111 genes showed significantly increased expression that corresponded with eDMR hypomethylation (Table S3). Functional annotation clustering revealed that downregulated genes with eDMR hypermethylation included those involved in cell cycle processes, cell division, regulation of gene expression, cytoskeleton, and a large number of zinc finger proteins, whereas upregulated genes with eDMR hypomethylation included those involved in lymphocyte activation, cell migration, apoptosis, DNA replication, and DNA metabolic processes.
Gene body DMRs are associated with gene expression: Associations between gene body methylation and gene expression were also observed. Increasing gene body methylation along with promoter methylation has been shown to have a stronger repressive effect on gene expression during normal B-cell development than promoter methylation alone.Citation18 However, the effect of gene body methylation in the absence of promoter methylation is less clear. Both inverse and positive correlations between gene body methylation and gene expression were observed. Gene body hypermethylation and a significant decrease in expression was observed in 261 genes and included protein kinases (CDK5R1, NRBP1, LYN, NUAK2, PHKB, BLK, PRKAG2, MKNK2, SMG1, TRIO, GAK, PRKD2, ULK1, RIOK3, WNK4, MAP3K9, PDGFRA, NEK8, DCLK2, TLK2, LRRK1, CDC42BPB, CAMK1D), cell morphogenesis genes (CDK5R1, GDF7, ULK1, LAMA5, NR4A2, MAPK8IP3, SEMA3B, MYCBP2, NFATC1), lymphocyte differentiation genes (CHD7, IL7, CEBPG, HDAC9, FOXP1), chromatin modifiers (RSF1, CREBBP, BANP, ARID1B, UIMC1, CHD8, CHD7, WHSC1L1, PHF21A, TLK2, IRF4, HDAC9, RERE), and regulators of MAPK, JNK, JUN kinase activity. Conversely, gene body hypomethylation and a significant increase in expression was observed in 815 genes and included the DNA methyltransferases (DNMT3A and DNMT1), anti-apoptotic genes (IL2RB, PRDX2, BCL2L1, TCF7L2, DAPK1, AKT1, ATF5, BAX, TGM2, NOS3, THBS1, and MYO18A), and telomere organization genes (TERT and TNKS1BP1). Additionally, many genes showed positive correlations between methylation and expression. For example, several members of the protein tyrosine phosphatase family that regulate many cellular processes, such as cell growth, mitotic cycle, cellular differentiation, and malignant transformation, were upregulated and hypermethylated in ALL. Alternately, genes that play roles in B-cell activation were downregulated and hypomethylated in ALL.
B-cell development genes and epigenetic modifiers are aberrantly expressed in ALL
To investigate the deregulation of gene expression in ALL, genome-wide gene expression profiling of ALL patients and healthy precursor B-cells was performed using RNA-seq. A total of 3,700 genes were significantly upregulated in ALL vs. healthy samples and 2,734 genes were significantly downregulated (Table S4). Forty-three genes known to play roles in B-cell differentiation and activation were differentially expressed and may contribute to the pathogenesis of ALL (). The aberrant expression of epigenetic modifiers was also observed. The DNA methylation catalyzing enzymes DNMT1, DNMT3A, and DNMT3B were significantly upregulated in ALL. Conversely, 2 genes known to actively demethylate DNA,Citation19 TET2 and TET3, were significantly downregulated in ALL. In addition, 22 genes encoding histone proteins were significantly upregulated in ALL. Finally, the chromatin activating histone lysine acetyltransferases (MGEA5, CDYL, CREBBP, EP300, and NCOA3) were downregulated and the chromatin inactivating histone deacetylases (HDAC11 and SIRT2) were upregulated in ALL.
Table 1. Patient characteristics
Differential expression of transcripts with epigenetic regulatory functions
LincRNAs epigenetically regulate gene expression by a number of diverse mechanisms including recruitment of histone methyltransferases through polycomb repressor complex 2 to modify chromatin states,Citation20,21 and the differential expression of lincRNA has been shown to play critical roles in many diseases.Citation22,23 Differential expression analysis of lincRNAs in ALL patients compared to healthy controls revealed 197 lincRNAs were differentially expressed (Table S4). Among them, 104 lincRNAs were upregulated and 93 were downregulated in ALL.
Pseudogene transcripts play a significant role in cancer pathogenesis and are differentially expressed in different types of cancer.Citation24,25 The relationship between differentially expressed pseudogene transcripts and the expression of parent gene targets was diverse in our data (Table S5). In some instances, the upregulation of a pseudogene was associated with the downregulation of its parent gene. For example, the pseudogene GRK6P1 was upregulated and associated with downregulation of their parent gene GRK6. Interestingly, GRK6 phosphorylates the activated forms of G protein-coupled receptors (GPCRs) thereby instigating their deactivation. Further, the overexpression of GPCRs is known to contribute to cancer cell proliferation.Citation26 Thus, the upregulation of GRK6P1 may lead to the constitutive activation of GPCRs and contribute to the proliferation of cancer cells. Conversely, in other instances the downregulation of a pseudogene was associated with the upregulation of its parent gene. For example, the downregulation of AC007041.2, RP11-368P15.1 and KRT18P4 was associated with the upregulation of DRG1 and NDUFB3, and KRT18 respectively. KRT18 (cytokeratin 18) is involved in multiple cellular processes including apoptosis, mitosis, cell cycle progression, and cell signaling and is hypothesized to be involved in carcinogenesis through multiple signaling pathways.Citation27 Therefore, the pseudogene mediated upregulation of KRT18 may lead to the aberrant regulation of signaling pathways in ALL.
A positive correlation was also observed in which upregulated pseudogene transcripts were associated with upregulated parent gene transcripts and downregulated pseudogene transcripts were associated with downregulated parent gene transcripts. In these cases the pseudogene transcripts may upregulate their parent gene by competing with endogenous RNAs that share miRNA response elements,Citation28 or by competing for RNA binding proteins that degrade their parent gene and vice versa. In ALL, the upregulation of pseudogenes RP11-423H2.1, FAM86C2P, and HMGB1P41 was associated with the upregulation of their parent genes THOC3, FAM86A, and HMGB2. Previous studies have shown that HMGB2 is overexpressed in a variety of cancers and that there is a decline in the proliferation of cancer cells when siRNA is used to knockdown expression of HMGB2,Citation29,30 suggesting a putative role in the pathogenesis of ALL. Likewise, some pseudogenes were downregulated and their parent genes were also downregulated. Specifically, the downregulation of PABPC1P3 was associated with the downregulation of its parent gene, PABPC1, which encodes a poly(A) binding protein involved in stabilizing the 5′ cap of mRNA. The downregulation of PABPC1 has also been reported in esophageal cancer.Citation31 It is possible that the pseudogene mediated downregulation of PABPC1 results in unstable mRNA transcripts and contributes to the pathogenesis of ALL.
Discussion
On average, more than 50 million unique mapped MIRA-seq reads were generated providing genome-wide coverage of the methylome in 19 pediatric ALL patients. Importantly, these profiles were compared to healthy precursor B-cells isolated from umbilical cord blood, the normal counterparts of malignant precursor B-cells to identify DMRs. To determine the regulatory potential of DMRs, transcriptomes were also generated and differential expression was determined between ALL patients and normal controls. Previous studies in ALL have identified inverse correlations between gene expression and DNA methylation in CGIs, CGI shores and gene promoters.Citation32 In this study, 99% of DMRs associated with a CGI were hypermethylated in ALL; however, these only accounted for a small number of the total DMRs. In fact, more than 80% of DMRs were identified in intronic or intergenic regions and not within a CGI context. Since DMRs can be used as biomarkers and as targets for novel therapeutics, we sought to identify the most likely candidates with regulatory potential.
Strikingly, 70% of the intergenic DMRs were concomitant with functional regulatory elements including transposable elements, enhancers, transcription factor binding sites, ncRNA, and pseudogenes. Inverse and positive correlations between DNA methylation in regulatory regions and gene expression were observed. In addition, inverse and positive correlations were observed between gene body methylation and expression. The cause and effect of DNA methylation within gene bodies is not fully understood; however, mechanisms leading to faulty gene expression have been postulated including the regulation of transcriptional elongation,Citation33 cell-type specific selection of alternative promoters,Citation34 modulating alternative RNA splicing,Citation35 or defining alternative polyadenylation sites.Citation36
Genes that are regulated by DNA methylation and provide a selective growth advantage to cancer cells have been referred to as epi-driver genes.Citation37 The ability to weed out driver epi-mutations from passenger epi-mutations is crucial in the quest to delineate potential therapeutic targets from a multitude of passenger events. Integrated DNA methylation and gene expression analysis identified potential epi-driver genes including SYNE1 (cytokinesis), PTPRS (signaling molecule), PAWR (pro-apoptotic gene), HDAC9 (downstream target of KRAS), RGCC (cell-cycle regulator), and MCOLN2 (unknown function), which were hypermethylated in the 5′ regulatory region and downregulated in ALL. These genes have also been shown to be hypermethylated and/or downregulated in other malignancies,Citation38-40 indicating the potential for tumor suppressor activity and supporting the role of DNA methylation as a regulator of gene expression. Although the function of MCOLN2 is unclear, the B-cell lineage specific activator PAX5 regulates its expression, strongly implicating its involvement in early B-cell development.Citation41 Taken together, the downregulation of these genes due to DNA methylation may play important roles in the development of ALL.
Perhaps the most paramount finding of this study was the identification of potential regulatory enhancers (eDMR). In relation to this, potential epi-drivers regulated by DNA methylation of an eDMR were also identified. Three of the genes with hypermethylated promoter DMRs (SYNE1, PTPRS, and MCOLN2) also possessed a hypermethylated eDMR. In addition, LYN and TRAF3 were downregulated in ALL patients and associated with a hypermethylated eDMR. LYN plays an important role in the regulation of B-cell differentiation, proliferation, survival and apoptosis, and TRAF3 negatively regulates the activation of the NF-κB2 pathway in B-cells. Conversely, FLT1 and MELK were upregulated and associated with a hypomethylated eDMR. Both genes have previously been shown to be upregulated in cancer.Citation42,43 Furthermore, FLT1 has been shown play a role in the proliferation of tumor cells,Citation44 and suppression of MELK expression by siRNA has been shown to inhibit the growth of cancer cells. Therefore, the aberrant expression of these genes due to DNA methylation may provide a survival advantage to malignant cells and play a role in pediatric ALL.
In summary, novel differentially methylated regulatory regions and differentially expressed genes were identified that may contribute to the pathogenesis of ALL. As expected, genes associated with B-cell development and epigenetic modifier genes were differentially expressed. The de novo DNA methyltransferases (DNMT3A, DNMT3B) responsible for the establishment of DNA methylation patterns and chromatin inactivating deacetylase genes were upregulated, whereas TET1 and TET2, which are responsible for actively demethylating DNA and chromatin activating acetyltransferase genes were downregulated in ALL. The upregulation of methylating enzymes along with the downregulation of demethylating genes supports the theory that the loss of methylation is a passive event that occurs during DNA replication over multiple uncontrolled cell divisions.Citation45 Accordingly, the overall result of the aberrant expression of the epigenetic modifier genes observed in this study may effectively be the inactivation of key genes that contribute to ALL. In addition, pseudogenes and lincRNAs genes were also aberrantly expressed in ALL and have functional roles in epigenetic regulation through diverse mechanisms including behaving as antisense RNA, endo-siRNA, competing endogenous RNA, or competing for RNA-binding proteins to regulate their target genes. Moreover, for the first time, putative transcriptional enhancers were identified that were differentially methylated and associated with the expression of a neighboring gene. Importantly, these may be used as prospective biomarkers for ALL and/or as targets for novel therapeutic agents that can restore altered DNA methylation and gene expression back to the normal state with the ultimate goal of improving treatment therapies and patient outcomes.
Materials and Methods
Patient samples
De-identified patient samples were obtained under full ethical approval of the institutional review board at the University of Missouri. A total of 20 pre-B ALL patient samples () and pre-BI and pre-BII cells from 10 healthy individuals were used for this study. ALL patient samples contain at least 88% blasts. Normal control pre-BI and pre-BII cells were isolated from 10 human umbilical cord blood (HCB) samples as previously described.Citation46 Briefly, mononuclear cells were isolated by density gradient centrifugation using Ficoll-Paque PLUS (GE Healthcare Bio-Sciences AB; cat. no. 17–1440-03) followed by depletion of all non B-cells with biotin conjugated antibodies cocktail and anti-biotin monoclonal antibodies conjugated to magnetic beads using human B cell Isolation Kit (MACS Miltenyi Biotec; order no. 130-093-660). For the methylation studies, purified B-cells were fluorescently labeled with antibodies against cell surface antigen (CD19, CD34, CD45; BD Biosciences) specific to individual stages of B-cell development. Finally, the fluorescently labeled cells were sorted as pre-BI (CD19+/CD34−/CD45low) and pre-BII (CD19+/CD34−/CD45med). Because no regions of differential methylation were observed in pre-BI versus pre-BII cells, transcriptomes were generated for precursor B-cells which include both pre-BI and pre-BII subsets. To obtain this population of cells, purified B-cells were fluorescently labeled with antibodies against CD19 and IgM and precursor B-cells (CD19+/IgM−) were isolated by flow cytometry.
Table 2: Differentially expressed genes in ALL involved in B-cell development and epigenetic modifications
Antibodies
The following antibodies were used for flow cytometry and non B-cell isolation through column purification: BD PharMingen™ PE Mouse Anti-Human CD34 (BD Biosciences; cat. no. 560941); BD Pharmingen™ APC Mouse Anti-Human CD19 (BD Biosciences; cat. no. 555415); CD45 FITC (BD Biosciences, cat. no. 347463); BD Pharmingen™ PE Mouse Anti-Human IgM (BD Biosciences, cat. no. 555783); B cell Isolation kit (MACS Miltenyi Biotec; order no. 130-093-660).
MIRA-seq library preparation
Genomic DNA from ALL patient samples was isolated using DNeasy® Blood and Tissue Kit (Qiagen; cat. no. 69506) according to manufacturer's instructions. MIRA-seq libraries for normal precursor B-cells were prepared as previously described.Citation4 For ALL patient samples, 1.0 µg of DNA from each ALL patient was sonicated with alternating 30 seconds on/off intervals for a total of 9 minutes to generate 200- to 600-bp fragments. A small portion of sonicated DNA was run on 1% agarose gel to ensure the sonication accuracy. The remaining sonicated DNA fragments were concentrated and purified using the MinElute PCR purification kit (Qiagen; cat. no. 28004). Adaptor ligation to fragmented DNA followed by MIRA using MethylCollector™ Ultra kit (Active Motif; cat. no. 55005) was performed according to manufacturer's protocols and as previously described.Citation4 After size selection of enriched methylated DNA on 1% agarose gel, PCR amplification of recovered methylated DNA fragments was performed for 11 cycles and then purified with the MinElute gel extraction kit (Qiagen; cat no. 28604). In order to validate the enrichment of methylated DNA, end point PCR amplification of methylated SLC25A37- and unmethylated APC1- regions was performed with the following primer pairs: 5′-CCCCCTGGACGTCTGTAAG-3′ (forward) and 5′-GGCATCTGGTAGATGACACG-3′ (reverse) for SLC25A37, and 5′-ACTGCCATCAACTTCCTTGC-3′ (forward) and 5′-GCGGATTACACAGCTGCTTC C-3′(reverse) for APC1. Quantity and fragment analysis was performed using Qubit and Bioanalyzer before sequencing. Four high quality MIRA-seq libraries were multiplexed in 10nM concentrations and sequenced on the Illumina HiSeq 2000 (1×100 bp reads) at the DNA Core Facility, University of Missouri-Columbia.
Identification of methylated peaks and differentially methylated regions in ALL
MIRA-seq data processing and methylated peaks for individual samples were identified using MACS2 pipeline as previously described.Citation4 Briefly, following adaptor trimming, sequences were aligned to the human reference sequence (GRCh37 with SNP135 masked) with bowtie2 (version 2.1.0). Patient sample A32 had an insufficient numbers of reads and was excluded from subsequent analyses. Picard-tools (version 1.92) were used to remove duplicate reads from the BAM files. The resulting BAM files were indexed with SAMtools “index.” Methylated peaks were identified using MACS2 (version 2.0.10.20130712)Citation47 with default parameters. Unified peak locations across the samples were created using bedtools (version 2.17.0). Individual sample was assigned a peak when their own peak overlapped with the unified peaks. ALL and HCB peaks were included if the peak was present in at least 17 biological replicates. Differentially methylated regions (DMRs) between the ALL and control precursor B-cells isolated from HCB were identified as described previously.Citation4 The coverage depth for each sample was analyzed and any sample with insufficient depth (saturation correlation < 0.90) was omitted from further analyses. Following normalization of data using a CpG coupling factor-based method,Citation48 DMRs were identified. Initially regions of interest (ROIs) were determined based on read counts within 100 bp windows with a 300 bp overlaps (expected fragment size of 400 bp). Non-specific filtering was performed by discarding the ROIs with modest signal representation (<20 mean counts across all samples). Differentially methylated regions were identified from the remaining ROIs using the edgeR package called via the MEDIPS package in R/Bioconductor. The ROIs with <5% false discovery rate (FDR; Benjamini-Hochberg) and at least a fold2- change were identified as a DMRs. ROIs immediately adjacent to one another were combined into a single DMR. Hyper- and hypomethylated ROIs were merged separately so that only putatively consistent ROIs were combined. The reported log fold change for merged DMRs is the maximum log2 fold change for any of its constituent ROIs. All MIRA-seq data were deposited in NCBI Sequence Read Archive (Accession SRP058314).
Annotation and enhancer prediction
Methylated peaks and differentially methylated regions were annotated with HOMER (Hypergeometric Optimization of Motif EnRichment), version 4.3, using the default setting to identify genomic locations.Citation49 The X and Y chromosomes were excluded from the analysis as the genders of individual normal samples were unknown. CpG island positional information from the University of California Santa Cruz (UCSC) table browser was used to determine the position of methylation peaks within a CpG island context. The genomic locations of enhancers were identified based on the enrichment of histone H3 lysine 4 monomethylation (H3K4me1) and histone H3 lysine 27 acetylation (H3K27ac) modifications in the GM12878 cell line (lymphoblastoid) available from ENCODE.
RNA-seq library preparation and data analysis
RNA samples were also obtained from the pre-B ALL patients (20 samples) utilized in the MIRA-seq assays and from normal precursor B-cells isolated from HCB (8 samples). RNA sequencing libraries were constructed with the NEBNext® Ultra™ Directional RNA Library Prep Kit for Illumina® (New England Biolabs; cat. no. E7420) and sequenced on the Illumina HiSeq 2000 (1×100 bp reads) at the University of Missouri DNA Core Facility. The reads were preprocessed to remove poor quality reads of <20 using FastX toolkit (http://hannonlab.cshl.edu/fastx_toolkit/). Reads were aligned to hg19 using Tophat (v2.0.13) with default settings. Differential gene expression between ALL and healthy precursor B-cells were determined using Cufflinks with default parameters (version 2.2.1).Citation50 The read counts along with FPKM values and their variances were calculated by cuffdiff 2 and the log fold change and p-value was calculated for each gene. Multiple testing corrections using Benjamini-Hochberg was also performed (q-value). The same cutoffs for FDR and fold change used in the analysis of methylated ROIs were used to determine differential expression. All functional annotations were performed using the Database for Annotation, Visualization and Integrated Discovery (DAVID) v6.7.Citation51 All RNA-seq data were deposited in NCBI Sequence Read Archive (Accession SRP058414).
Disclosure of Potential Conflicts of Interest
Dr. J Wade Davis is currently employed by AbbVie. The remaining authors declare no conflict of interest.
1078050_Supplemental_Material.zip
Download Zip (3.5 MB)Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
Funding
This work was supported by grants from the NIH (CA132784) and the Bryan Thomas Campbell Foundation (KH Taylor).
References
- National cancer institute report. (http://www.cancer.gov/cancertopics/pdq/treatment/childALL/HealthProfessional/page1#Reference1.3).
- Hystad ME, Myklebust JH, Bø TH, Sivertsen EA, Rian E, Forfang L, Munthe E, Rosenwald A, Chiorazzi M, Jonassen I, et al. Characterization of early stages of human B cell development by gene expression profiling. J Immunol 2007; 179:3662-71; PMID:17785802; http://dx.doi.org/10.4049/jimmunol.179.6.3662
- van Zelm MC, van der Burg M, de Ridder D, Barendregt BH, de Haas EF, Reinders MJ, Lankester AC, Révész T, Staal FJ, van Dongen JJ. Ig gene rearrangement steps are initiated in early human precursor B cell subsets and correlate with specific transcription factor expression. J Immunol 2005; 175:5912-22; PMID:16237084; http://dx.doi.org/10.4049/jimmunol.175.9.5912
- Almamun M, Levinson BT, Gater ST, Schnabel RD, Arthur GL, Davis JW, Taylor KH. Genome-wide DNA methylation analysis in precursor B-cells. Epigenetics 2014; 9:1588-95; PMID:25484143; http://dx.doi.org/10.4161/15592294.2014.983379
- Hodges E, Molaro A, Dos Santos CO, Thekkat P, Song Q, Uren PJ, Park J, Butler J, Rafii S, McCombie WR, et al. Directional DNA methylation changes and complex intermediate states accompany lineage specificity in the adult hematopoietic compartment. Mol Cell. 2011; 44:17-28; PMID:21924933; http://dx.doi.org/10.1016/j.molcel.2011.08.026
- Berdasco M, Esteller M. Aberrant epigenetic landscape in cancer: how cellular identity goes awry. Dev Cell. 2010; 19:698-711; PMID:21074720; http://dx.doi.org/10.1016/j.devcel.2010.10.005
- Figueroa ME, Chen SC, Andersson AK, Phillips LA, Li Y, Sotzen J, Kundu M, Downing JR, Melnick A, Mullighan CG. Integrated genetic and epigenetic analysis of childhood acute lymphoblastic leukemia. J Clin Invest 2013; 123:3099-3111; PMID:23921123; http://dx.doi.org/10.1172/JCI66203
- Feinberg AP, Vogelstein B. Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature 1983; 301:89-92; PMID:6185846; http://dx.doi.org/10.1038/301089a0
- Liang P, Song F, Ghosh S, Morien E, Qin M, Mahmood S, Fujiwara K, Igarashi J, Nagase H, Held WA. Genome-wide survey reveals dynamic widespread tissue-specific changes in DNA methylation during development. BMC Genomics 2011; 12:231; PMID:21569359; http://dx.doi.org/10.1186/1471-2164-12-231
- Ji H, Ehrlich LI, Seita J, Murakami P, Doi A, Lindau P, Lee H, Aryee MJ, Irizarry RA, Kim K, et al. Comprehensive methylome map of lineage commitment from haematopoietic progenitors. Nature 2010; 467:338-42; PMID:20720541; http://dx.doi.org/10.1038/nature09367
- Xu J, Pope SD, Jazirehi AR, Attema JL, Papathanasiou P, Watts JA, Zaret KS, Weissman IL, Smale ST. Pioneer factor interactions and unmethylated CpG dinucleotides mark silent tissue-specific enhancers in embryonic stem cells. Proc Natl Acad Sci U S A. 2007; 104:12377-82; PMID:17640912; http://dx.doi.org/10.1073/pnas.0704579104
- Aran D, Sabato S, Hellman A. DNA methylation of distal regulatory sites characterizes dysregulation of cancer genes. Genome Biol. 2013; 14:R21; PMID:23497655; http://dx.doi.org/10.1186/gb-2013-14-3-r21
- Chim CS, Wan TS, Wong KY, Fung TK, Drexler HG, Wong KF. Methylation of miR-34a, miR-34b/c, miR-124-1 and miR-203 in Ph-negative myeloproliferative neoplasms. J Transl Med 2011; 9:197; PMID:22082000; http://dx.doi.org/10.1186/1479-5876-9-197
- Wang LQ, Kwong YL, Kho CS, Wong KF, Wong KY, Ferracin M, Calin GA, Chim CS. Epigenetic inactivation of miR-9 family microRNAs in chronic lymphocytic leukemia - implications on constitutive activation of NFκB pathway. Mol Cancer 2013;12:173; PMID:24373626; http://dx.doi.org/10.1186/1476-4598-12-173
- Chatterton Z, Morenos L, Saffrey R, Craig J, Ashley D, Wong N. DNA methylation and miRNA expression profiling in childhood B-cell acute lymphoblastic leukemia. Epigenomics 2010; 2:697-708; PMID:22122053; http://dx.doi.org/10.2217/epi.10.39
- Maksakova IA, Mager DL, Reiss D. Keeping active endogenous retroviral-like elements in check: the epigenetic perspective. Cell Mol Life Sci. 2008; 65:3329-47; PMID:18818875; http://dx.doi.org/10.1007/s00018-008-8494-3
- Lee E, Iskow R, Yang L, Gokcumen O, Haseley P, Luquette LJ 3rd, Lohr JG, Harris CC, Ding L, Wilson RK, et al. Landscape of somatic retrotransposition in human cancers. Science 2012; 337:967-71; PMID:22745252; http://dx.doi.org/10.1126/science.1222077
- Lee ST, Xiao Y, Muench MO, Xiao J, Fomin ME, Wiencke JK, Zheng S, Dou X, de Smith A, Chokkalingam A, et al. A global DNA methylation and gene expression analysis of early human B-cell development reveals a demethylation signature and transcription factor network. Nucleic Acids Res 2012; 40:11339-51; PMID:23074194; http://dx.doi.org/10.1093/nar/gks957
- Pastor WA, Aravind L, Rao A. TETonic shift: biological roles of TET proteins in DNA demethylation and transcription. Nat Rev Mol Cell Biol 2013; 14:341-56; PMID:23698584; http://dx.doi.org/10.1038/nrm3589
- Nagano T, Fraser P. No-nonsense functions for long noncoding RNAs. Cell 2011; 145:178-81; PMID:21496640; http://dx.doi.org/10.1016/j.cell.2011.03.014
- Wang KC, Chang HY. Molecular mechanisms of long noncoding RNAs. Mol Cell. 2011 Sep 16; 43:904-14; http://dx.doi.org/10.1016/j.molcel.2011.08.018
- Gupta RA, Shah N, Wang KC, Kim J, Horlings HM, Wong DJ, Tsai MC, Hung T, Argani P, Rinn JL, et al. Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature 2010; 464:1071-76; PMID:20393566; http://dx.doi.org/10.1038/nature08975
- Trimarchi T, Bilal E, Ntziachristos P, Fabbri G, Dalla-Favera R, Tsirigos A, Aifantis I. Genome-wide mapping and characterization of Notch-regulated long noncoding RNAs in acute leukemia. Cell 2014; 158:593-606; PMID:25083870; http://dx.doi.org/10.1016/j.cell.2014.05.049
- Han L, Yuan Y, Zheng S, Yang Y, Li J, Edgerton ME, Diao L, Xu Y, Verhaak RG, Liang H. The Pan-Cancer analysis of pseudogene expression reveals biologically and clinically relevant tumour subtypes. Nat Commun. 2014; 5:3963; PMID:24999802
- Xiao-Jie L, Ai-Mei G, Li-Juan J, Jiang X. Pseudogene in cancer: real functions and promising signature. J Med Genet 2015; 52:17-24; PMID:25391452; http://dx.doi.org/10.1136/jmedgenet-2014-102785
- Dorsam RT, Gutkind JS. G-protein-coupled receptors and cancer. Nat Rev Cancer 2007; 7:79-94; PMID:17251915; http://dx.doi.org/10.1038/nrc2069
- Weng YR, Cui Y, Fang JY. Biological functions of cytokeratin 18 in cancer. Mol Cancer Res 2012; 10:485-93; PMID:22452884; http://dx.doi.org/10.1158/1541-7786.MCR-11-0222
- Poliseno L, Salmena L, Zhang J, Carver B, Haveman WJ, Pandolfi PP. A coding-independent function of gene and pseudogene mRNAs regulates tumour biology. Nature 2010; 465:1033-8; PMID:20577206; http://dx.doi.org/10.1038/nature09144
- Sharma A, Ray R, Rajeswari MR. Overexpression of high mobility group (HMG) B1 and B2 proteins directly correlates with the progression of squamous cell carcinoma in skin. Cancer Invest 2008; 26:843-51; PMID:18798064; http://dx.doi.org/10.1080/07357900801954210
- Kwon JH, Kim J, Park JY, Hong SM, Park CW, Hong SJ, Park SY, Choi YJ, Do IG, Joh JW, et al. Overexpression of high-mobility group box 2 is associated with tumor aggressiveness and prognosis of hepatocellular carcinoma. Clin Cancer Res 2010; 16:5511-21; PMID:20851854; http://dx.doi.org/10.1158/1078-0432.CCR-10-0825
- Takashima N, Ishiguro H, Kuwabara Y, Kimura M, Haruki N, Ando T, Kurehara H, Sugito N, Mori R, Fujii Y. Expression and prognostic roles of PABPC1 in esophageal cancer: correlation with tumor progression and postoperative survival. Oncol Rep 2006; 15:667-671; PMID:16465428
- Busche S, Ge B, Vidal R, Spinella JF, Saillour V, Richer C, Healy J, Chen SH, Droit A, Sinnett D, et al. Integration of high-resolution methylome and transcriptome analyses to dissect epigenomic changes in childhood acute lymphoblastic leukemia. Cancer Res 2013; 73:4323-36; PMID:23722552; http://dx.doi.org/10.1158/0008-5472.CAN-12-4367
- Lorincz MC, Dickerson DR, Schmitt M, Groudine M. Intragenic DNA methylation alters chromatin structure and elongation efficiency in mammalian cells. Nat Struct Mol Biol 2004; 11:1068-75; PMID:15467727; http://dx.doi.org/10.1038/nsmb840
- Maunakea AK, Nagarajan RP, Bilenky M, Ballinger TJ, D'Souza C, Fouse SD, Johnson BE, Hong C, Nielsen C, Zhao Y, et al. Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature 2010; 466:253-57; PMID:20613842; http://dx.doi.org/10.1038/nature09165
- Maunakea AK, Chepelev I, Cui K, Zhao K. Intragenic DNA methylation modulates alternative splicing by recruiting MeCP2 to promote exon recognition. Cell Res 2013; 23:1256-69; PMID:23938295; http://dx.doi.org/10.1038/cr.2013.110
- Wood AJ, Schulz R, Woodfine K, Koltowska K, Beechey CV, Peters J, Bourc'his D, Oakey RJ. Regulation of alternative polyadenylation by genomic imprinting. Genes Dev 2008; 22:1141-46; PMID:18451104; http://dx.doi.org/10.1101/gad.473408
- Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA Jr, Kinzler KW. Cancer genome landscapes. Science. 2013; 339:1546-58; PMID:23539594; http://dx.doi.org/10.1126/science.1235122
- Nagai MA, Gerhard R, Salaorni S, Fregnani JH, Nonogaki S, Netto MM, Soares FA. Down-regulation of the candidate tumor suppressor gene PAR-4 is associated with poor prognosis in breast cancer. Int J Oncol 2010; 37:41-49; PMID:20514395; http://dx.doi.org/10.3892/ijo_00000651
- Okudela K, Mitsui H, Suzuki T, Woo T, Tateishi Y, Umeda S, Saito Y, Tajiri M, Masuda M, Ohashi K. Expression of HDAC9 in lung cancer-potential role in lung carcinogenesis. Int J Clin Exp Pathol 2014; 7:213-20; PMID:24427341
- Vlaicu SI, Cudrici C, Ito T, Fosbrink M, Tegla CA, Rus V, Mircea PA, Rus H. Role of response gene to complement 32 in diseases. Arch Immunol Ther Exp (Warsz) 2008; 56:115-22; PMID:18373239; http://dx.doi.org/10.1007/s00005-008-0016-3
- Valadez JA, Cuajungco MP. PAX5 is the transcriptional activator of mucolipin-2 (MCOLN2) gene. Gene 2015; 555:194-202; PMID:25445271; http://dx.doi.org/10.1016/j.gene.2014.11.003
- Van Limbergen EJ, Zabrocki P, Porcu M, Hauben E, Cools J, Nuyts S. FLT1 kinase is a mediator of radioresistance and survival in head and neck squamous cell carcinoma. Acta Oncol 2014; 53:637-45; PMID:24041258; http://dx.doi.org/10.3109/0284186X.2013.835493
- Alachkar H, Mutonga MB, Metzeler KH, Fulton N, Malnassy G, Herold T, Spiekermann K, Bohlander SK, Hiddemann W, Matsuo Y, et al. Preclinical efficacy of maternal embryonic leucine-zipper kinase (MELK) inhibition in acute myeloid leukemia. Oncotarget 2014; 5:12371-82; PMID:25365263
- Lichtenberger BM, Tan PK, Niederleithner H, Ferrara N, Petzelbauer P, Sibilia M. Autocrine VEGF signaling synergizes with EGFR in tumor cells to promote epithelial cancer development. Cell 2010; 140:268-79; PMID:20141840; http://dx.doi.org/10.1016/j.cell.2009.12.046
- Wild L, Flanagan JM. Genome-wide hypomethylation in cancer may be a passive consequence of transformation. Biochim Biophys Acta. 2010 Aug; 1806(1):50-7.
- Almamun M, Schnabel JL, Gater ST, Ning J, Taylor KH. Isolation of precursor B-cell subsets from umbilical cord blood. J. Vis. Exp 2013; 74:1-9.
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol 2008; 9:R137.
- Down TA, Rakyan VK, Turner DJ, Flicek P, Li H, Kulesha E, Gräf S, Johnson N, Herrero J, Tomazou EM, et al. A Bayesian deconvolution strategy for immunoprecipitation-based DNA methylome analysis. Nat Biotechnol 2008; 26:779-85; PMID:18612301; http://dx.doi.org/10.1038/nbt1414
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 2010; 38:576-589; PMID:20513432; http://dx.doi.org/10.1016/j.molcel.2010.05.004
- Trapnell C, Hendrickson DG, Sauvageau M, Goff L, Rinn JL, Pachter L. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat Biotechnol 2013;31:46-53; PMID:23222703; http://dx.doi.org/10.1038/nbt.2450
- Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID Bioinformatics Resources. Nature Protoc 2009; 4:44-57; http://dx.doi.org/10.1038/nprot.2008.211