
Abstract
Oxidation of 5-methylcytosine by TET family proteins can induce DNA replication-dependent (passive) DNA demethylation and base excision repair (BER)-based (active) DNA demethylation. The balance of active vs. passive TET-induced demethylation remains incompletely determined. In the context of large scale DNA demethylation, active demethylation may require massive induction of the DNA repair machinery and thus compromise genome stability. To study this issue, we constructed a tetracycline-controlled TET-induced global DNA demethylation system in HEK293T cells. Upon TET overexpression, we observed induction of DNA damage and activation of a DNA damage response; however, BER genes are not upregulated to promote DNA repair. Depletion of TDG (thymine DNA glycosylase) or APEX1 (apurinic/apyrimidinic endonuclease 1), two key BER enzymes, enhances rather than impairs global DNA demethylation, which can be explained by stimulated proliferation. By contrast, growth arrest dramatically blocks TET-induced global DNA demethylation. Thus, in the context of TET-induction in HEK293T cells, the DNA replication-dependent passive mechanism functions as the predominant pathway for global DNA demethylation. In the same context, BER-based active demethylation is markedly restricted by limited BER upregulation, thus potentially preventing a disastrous DNA damage response to extensive active DNA demethylation.
Introduction
Although DNA methylation is a relatively stable epigenetic modification, both genome-wide and locus-specific demethylation events have been observed and play important roles in embryonic development and gene transcription regulation.Citation1,2 The ten-eleven translocation (TET) family proteins are DNA demethylases through consecutively converting 5-methylcytosine (5mC) into 5-hydroxymethylcytosine (5hmC), 5-formylcytosine (5fC), and 5-carboxylcytosine (5caC).Citation3-6 Because of the poor recognition of 5hmC, 5fC, and 5caC by maintenance DNA methyltransferase 1, TET proteins can induce DNA replication-dependent passive DNA demethylation.Citation7-9 TET proteins are also able to induce active DNA demethylation through the substitution of 5fC and 5caC for unmodified cytosine by thymine DNA glycosylase (TDG) initiated-base excision DNA repair (BER).Citation6,10-12 The physiological contexts of the passive and active demethylation pathways and their relative importance in TET-mediated DNA demethylation remain unclear.
TET proteins can induce not only locus-specific but also genome-wide DNA demethylation in physiological conditions.Citation10,13-16 During global demethylation in the paternal pronuclei of mouse zygotes, 5mC is oxidized by Tet3 into 5hmC, 5fC and 5caC, and all 3 products are relatively stable and undergo cell division-dependent dilution, pointing to the DNA replication-dependent passive demethylation pathway as the primary mechanism.Citation8,9,13,17 Similarly, a dramatic conversion of 5mC to 5hmC by Tet1 or Tet2 and a cell division-coupled dilution of 5hmC also occur in mouse primordial germ cells (PGCs), which is also evidence in favor of the passive demethylation pathway.Citation14 Considering that global DNA repair activation may stress the DNA repair machinery, the selection of the passive pathway rather than the BER-based active pathway for TET-induced global DNA demethylation may be essential to avoid deleterious genomic instability.Citation1,11 However, once a large amount of 5mC is converted to 5fC and 5caC, the intrinsic TDG and BER pathways are also believed to quickly trigger active demethylation. Therefore, a competition between passive and active demethylation may exist in TET-induced global demethylation, and the relative contribution of each pathway in global demethylation events is worth further investigation.
In this study, we used a tetracycline-controlled TET1 catalytic domain (TET1-CD) overexpression-induced global DNA demethylation system in HEK293T cells to evaluate the relative contributions of BER-based (active) vs. replication dependent (passive) demethylation. Our results unequivocally demonstrate that, for TET-induced global DNA demethylation in our model, the DNA replication-dependent passive pathway functions as the primary mechanism.
Results
Inducible TET-mediated global DNA demethylation in HEK293T
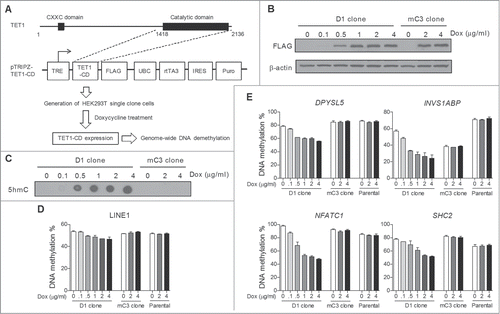
Our previous study showed that TET1-CD overexpression induces dramatic conversion of 5mC to 5hmC and concomitant massive DNA demethylation in HEK293T cells.Citation18 To develop an efficient model for studying the details of TET-induced global DNA demethylation, we constructed a tetracycline-controlled TET1-CD overexpression system in HEK293T cells. Lentiviral tetracycline-inducible expression vectors containing either wild type or catalytically mutant TET1-CD (referred to as mTET1-CD) were transduced into HEK293T cells (), and the overexpression of TET1-CD and mTET1-CD was documented after doxycycline treatment (Dox)(Figure S1A). As expected, Dox treatment also induced a significant increase of genomic 5hmC content in TET1-CD- but not mTET1-CD-transduced cells (Figure S1B). To ensure cellular homogeneity, we next derived single cell clones of transduced cells by limiting dilution, and D1 and mC3 cell clones were finally chosen based on their high and comparable induction of TET1-CD and mTET1-CD expression, respectively (Figure S1C). We verified Dox dose-dependent TET1-CD overexpression and 5hmC production in D1 cells (). Importantly, Dox treatment dose-dependently induced significant DNA demethylation in long interspersed nucleotide element-1 (LINE-1) in D1 clone cells but not mC3 clone (carrying mTET1-CD) or HEK293T parental cells (), confirming the successful induction of global DNA demethylation in D1 clone cells. Additionally, Dox treatment in D1 clone cells also induced DNA demethylation in 4 randomly selected genomic loci to a higher extent than in LINE-1 (). Thus, the D1 and mC3 cloned cells represent a useful platform to study TET-induced global DNA demethylation.
Figure 1. Construction of a tetracycline-inducible TET-mediated global DNA demethylation system in HEK293T cells. (A) Schematic description of constructing a tetracycline-inducible TET-mediated global DNA demethylation system. The entire system was established through 2 main steps: Cloning TET1-CD into the lentiviral pTRIPZ vector which is equipped with a Tet-On system for transgene overexpression in the presence of Dox, and transfecting pTRIPZ-TET1-CD into HEK293T cells and developing single cell clones. (B) Dox treatment dose-dependent induced expression of TET1-CD and mTET1-CD in D1 and mC3 clone cells, respectively. The cells were harvested 24 h after Dox treatment. (C) DNA dot blot assay showed that Dox treatment dose-dependently induced genomic 5hmC production only in D1 cells. The cells were harvested 24 h after Dox treatment. (D and E) Dox treatment dose-dependently induced DNA demethylation in LINE-1 (D) and 4 selected genomic loci (E) in D1 clone cells but not mC3 clone and HEK293T parental cells. The cells were treated with Dox for 3 days. Bisulfite-pyrosequencing was used for DNA methylation analysis. Error bars represent SD from 3 independent experiments.
DNA damage occurs during TET-induced global DNA demethylation
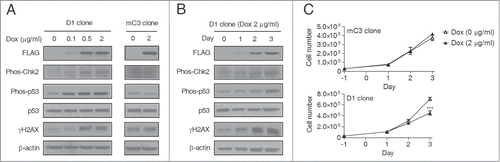
While passive demethylation does not induce a DNA damage response, 5fC and 5caC are excised by TDG followed by BER during active demethylation, and the intermediate apurinic/apyrimidinic (AP) sites and DNA single-strand breaks (SSBs) could theoretically quickly accumulate and induce a severe DNA damage response.Citation6,12 To distinguish these possibilities, we examined whether a DNA damage response is activated by global demethylation in HEK293T cells. Both CHK2 and TP53 are critical messengers of genome integrity and are involved in the DNA damage response signaling pathway.Citation19-21 As shown in , Dox-treated D1 but not mC3 clone cells showed increased phosphorylation of CHK2 (Thr68) and TP53 (Ser20), demonstrating activation of the DNA damage response signaling pathway. A significant production of γH2AX, which is a precise marker for DNA double-strand breaks (DSBs),Citation22 was also detected in Dox-treated D1 clone cells (), consistent with the fact that unrepaired SSBs can be converted into DSBs during DNA replication.Citation23 By examining the dynamic changes in phos-CHK2, phos-TP53, and γH2AX over 3 days of Dox treatment, we found progressive increases in all 3 proteins, suggesting the accumulation of DNA damage during such global demethylation (). We also found significant cell growth inhibition in Dox-treated D1 but not mC3 cells (), which may be explained by the abilities of phosphorylated CHK2 and TP53 to induce cell cycle arrest.Citation24 Taken together, the above studies show an accumulation of DNA damage during TET-induced global deme-thylation.
Figure 2. DNA damage and activation of a DNA damage response during TET-induced global DNA demethylation. (A) Western blot assay showed the activation of a DNA damage response in D1 but not mC3 cells. The cells were treated with 2 μg/ml Dox for 3 days. The marks for DNA damage response activation include phosphorylated CHK2 (Thr68) and TP53 (Ser20) and γH2AX. (B) Dynamic change of phosphorylated CHK2 (Thr68) and TP53 (Ser20) and γH2AX during 3 days of Dox treatment (2 μg/ml) in D1 clone cells. The cell lysates were prepared every day after Dox treatment. (C) Dox treatment (2 μg/ml, 3 days) induced cell growth inhibition in D1 but not mC3 clone cells. Error bars represent SD from 3 independent experiments. ***P < 0.001 compared with non-treatment control by Student's t test.
Lack of BER activation during TET-induced global DNA demethylation
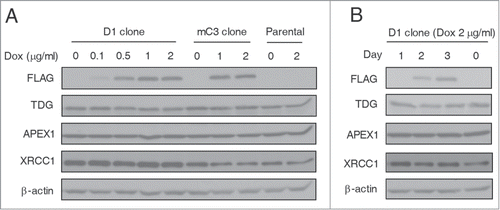
DNA repair genes can be upregulated by genotoxic stress to enhance DNA damage repair in mammalian cells.Citation25-27 We therefore asked whether BER genes are adaptively upregulated to enhance DNA damage repair and facilitate active demethylation after TET induction. As shown in , in D1 and mC3 clone cells and also in HEK293T parental cells, Dox treatment at all dosages did not induce significant changes in TDG and APEX1 (apurinic/apyrimidinic endonuclease 1), both of which are key players of BER.Citation28 Another BER pathway protein XRCC1 showed stable expression levels in D1 clone cells after treatment with Dox, and decreased expression in Dox-treated mC3 clone cells (where no DNA demethylation was induced). Considering the possibility that upregulation of these proteins may be transient, we further tested their dynamic expression over a period of 3 days and still did not detect any significant expression changes (). Thus, despite the accumulated DNA damage and the activation of DNA damage response signaling in TET-induced global demethylation, the critical BER genes are not adaptively upregulated, which may therefore restrict active demethylation after TET induction.
Figure 3. Lack of BER genes upregulation during TET-induced global DNA demethylation. (A) Western blot analysis showed no significant change of TDG, APEX1 and XRCC1 expression in D1 and mC3 clone cells and HEK293T parental cells after Dox treatment. All cells were harvested 3 days after Dox treatment at different dosages. (B) Constant expression levels of TDG, APEX1 and XRCC1 in D1 clone cells during 3 days of Dox treatment (2 μg/ml). The cell lysates were prepared every day after Dox treatment.
BER inhibition does not prevent TET-induced global DNA demethylation
We next directly investigated the extent to which BER contributes to TET-induced global DNA demethylation. As TDG initiates the active demethylation pathway by recognizing and cleaving 5fC and 5caC,Citation6,11,12 we inhibited TDG expression and examined its effect on DNA demethylation after TET induction (). siRNA-mediated knockdown of TDG did not affect genomic 5hmC content but significantly increased 5caC content in Dox-treated D1 clone cells (), confirming the cleavage of 5caC by TDG in TET-induced global demethylation. The induction of γH2AX by Dox treatment in D1 clone cells was also dramatically inhibited by TDG knockdown, which suggests that the DSBs are caused by the BER-based active demethylation pathway (). However, despite the changed relative distribution of 5mC oxidized derivatives (e.g., 5caC), we found by using bisulfite-pyrosequencing that TDG knockdown failed to decrease the demethylation extent in all studied genomic sites in Dox-treated D1 cells, even showing a small increase when compared with control knockdown (). Given that 5mC oxidized derivatives cannot be distinguished from 5mC by bisulfite assays, the appearance of unmodified cytosines truly reflect demethylation here. This finding strongly suggests that BER has a minimal contribution to the global DNA demethylation induced by TET1-CD overexpression. TDG knockdown also reversed the Dox-induced cell growth inhibition in D1 cells (), which suggests that the cell growth inhibition observed upon TET induction results from the DNA damage response rather than global demethylation (). Moreover, by promoting DNA-replication dependent passive demethylation, the rescued cell growth seems to also provide a reasonable explanation for the paradoxical finding of increased demethylation upon TDG knockdown.()
Figure 4. Inhibition of the BER pathway enhances TET-induced global DNA demethylation. (A) Western blot analysis showed siRNA-mediated TDG knockdown in D1 clone cells with or without Dox treatment (2 μg/ml, 3 days). (B) DNA dot blot assay showed that TDG knockdown increased genomic 5caC content but not 5hmC in D1 clone cells treated with Dox (2 μg/ml, 3 days). (C) Western blot analysis of γH2AX showed that TDG knockdown decreased DNA DSBs in D1 clone cells treated with Dox (2 μg/ml, 3 days). (D) TDG knockdown enhanced TET-induced DNA demethylation in D1 clone cells treated with Dox (2 μg/ml, 3 days). (E) TDG knockdown reversed cell growth inhibition in D1 clone cells treated with Dox (2 μg/ml, 3 days). The cell numbers were counted 3 days after Dox treatment and those of non-treated cells were set as 1. (F) Western blot analysis showed siRNA-mediated APEX1 knockdown in D1 clone cells with or without Dox treatment (2 μg/ml, 3 days). (G) APEX1 knockdown promoted TET-induced DNA demethylation in D1 clone cells treated with Dox (2 μg/ml, 3 days). Bisulfite-pyrosequencing was used for DNA methylation analysis. Error bars represent SD from 3 independent experiments. * P < 0.05, * *P < 0.01 by Student's t test.
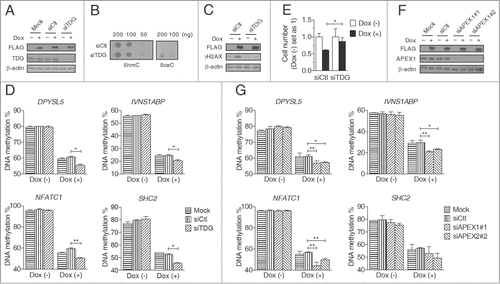
To further confirm these findings, we next performed knockdown of APEX1 which functions downstream of TDG and cleaves AP-sites in the BER pathway. Just like the TDG knockdown, APEX1 knockdown also paradoxically promoted TET-induced demethylation in all studied genomic sites (). Consistent with this, CRT0044876, a potent and selective APEX1 inhibitor,Citation29 also failed to inhibit DNA demethylation by TET induction (Fig. S2). Taken together, these data clearly indicate that in HEK293T cells, the BER-based active pathway is not the primary mechanism of TET overexpression-induced global DNA demethylation.
Cell growth arrest restrains TET-induced global DNA demethylation
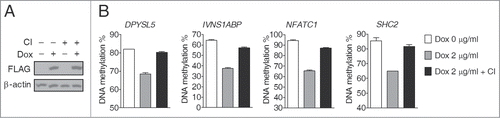
To directly determine the contribution of DNA replication to TET-induced global DNA demethylation, we investigated the effects of cell growth arrest. We initially used serum starvation and mimosine treatment, which, respectively, arrest cell cycle progression at the G0/G1 and G1/S phases,Citation30,31 but both markedly inhibited Dox-induced TET1-CD overexpression (data not shown). By contrast, contact inhibition-induced cell cycle arrest did not affect Dox-induced TET1-CD overexpression in D1 clone cells (). DNA demethylation in all studied genomic sites was largely inhibited in growth-arrested compared to non-growth-arrested cells (), clearly showing that TET induces global DNA demethylation in HEK293T cells predominately through the DNA replication-dependent passive pathway.
Figure 5. Inhibited TET-induced global DNA demethylation in growth-arrested cells induced by contact inhibition. (A) Western blot analysis showed that Dox treatment (2 μg/ml, 3 days) induced comparable TET1-CD expression levels in D1 clone cells under conditions of contact inhibition or not. (B) TET-induced DNA demethylation was blocked in D1 clone cells with contact inhibition. CI, contact inhibition; error bars represent SD from 3 independent experiments.
Discussion
In this paper, we successfully developed a tetracycline-controlled TET-induced global DNA demethylation system in HEK293T cells. We found that BER results in the accumulation of DNA damage during TET-induced global DNA demethylation but contributes minimally to DNA demethylation. The DNA damage response upon TET induction was minimized by the lack of BER gene upregulation, which results in a reduced cellular DNA repair capacity and may explain the low efficiency of the active demethylation pathway. By contrast, global demethylation was reversed by cell cycle arrest and enhanced by TDG knockdown, which reverses the effects of DNA damage on cellular growth. Thus, in our model, TET-induced global demethylation is primarily dependent on DNA replication and passive demethylation. It should be noted that bisulfite pyrosequencing cannot distinguish 5mC from its oxidized forms. Because of this inability, a change in the relative distribution of 5mC oxidized derivatives after TDG knockdown would not be detected by bisulfite pyrosequencing, which may therefore underestimate the degree of 5mC loss. Thus, technically, one might expect a low degree of pyrosequencing-measured demethylation after TDG knockdown, but we observed the opposite effect—knockdown of TDG slightly increased the net pyrosequencing-measured demethylation extent (likely through the rescue of cell proliferation inhibition and the resulting enhanced DNA replication-based passive pathway). Since only the conversion of 5mC (and its oxidized forms) to fully unmodified C is counted as demethylation by bisulfite pyrosequencing, any measured demethylation must be real. Therefore, our conclusion that TDG knockdown/BER inhibition does not prevent demethylation would not be affected by this technical issue.
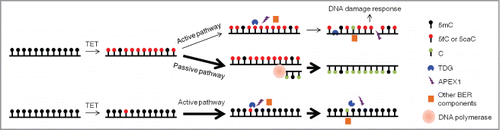
One drawback of our study is that the HEK293T cell model of TET-induced global demethylation does not necessarily mimic physiologically processes occurring in the paternal pronuclei of mouse zygotes and mouse PGCs.Citation13-15 In contrast to the pluripotent zygotes and PGCs, HEK293T cells are differentiated and present a dramatically distinct cellular environment. Additionally, TET1-CD-induced demethylation events have a different genomic distribution from those induced by full length TET1 or other TET family members,Citation18 and the level of TET1-CD expression in our model may also be not comparable to physiological levels. Despite these drawbacks, our findings still provide some insights into the physiological context and relative importance of active and passive pathways in TET-induced DNA demethylation. Consistent with those in the paternal pronuclei of mouse zygotes and mouse PGCs, the TET-induced global demethylation is proved again to be primarily through DNA replication-based passive pathway in the HEK293T cell model, suggesting the predominance of the passive pathway in all TET-induced global demethylation processes. By contrast, the reported locus-specific DNA demethylation events were always achieved through active demethylation,Citation10,16,32 Thus, passive and active demethylation pathways selectively function in TET-induced global and locus-specific DNA demethylation. To illustrate this, we propose the working model shown in . When TET proteins induce massive conversion of 5mC to 5hmC, 5fC and 5caC in genomic DNA, a small number of 5fC and 5caC sites are rapidly recognized and cleaved by TDG, but most of them are not successfully substituted with unmodified cytosines because of the easily saturated DNA repair capacity, thus leaving unrepaired DNA damage in various forms (including AP-sites, SSBs and DSBs). By contrast, the majority of 5hmC, 5fC, and 5caC sites are replaced with unmodified cytosines through DNA replication, which causes no DNA damage. Thus, selection of the passive pathway and suppression of the active pathway essentially maintains genomic stability during TET-induced global DNA demethylation.Citation1,11 On the other hand, when TET proteins oxidize 5mC only in specific and sparse loci, the cellular TDG-BER capacity is sufficient to demethylate the resultant 5fC and 5caC within a very short time. This preference for the active pathway allows cells (especially non-dividing cells) to rapidly respond to extracellular stimulation, such as the neuronal activity-induced gene expression in the adult brain.Citation10 Overall, because of their distinct capacity, efficiency and dependence on DNA replication, it is plausible that passive or active pathways are selectively utilized by TET proteins to achieve regulation of DNA methylation in different scales.
Figure 6. A model of demethylation pathway selection in TET-induced DNA demethylation. When TET proteins globally oxidize 5mC in genomic DNA (upper part), the BER-based active pathway is triggered but quickly becomes saturated mainly due to the lack of BER genes adaptive upregulation in response to DNA damage. Only a few 5fC and 5caC sites are cleaved and various types of DNA damage (e.g., AP-sites, SSBs) are produced as the end products. By contrast, through DNA replication, almost all 5hmC, 5fC, and 5caC sites are efficiently converted into unmodified cytosine at no risk of DNA damage. Thus, the DNA-replication dependent passive pathway is selected as the predominant way for TET-induced global DNA demethylation. However, when TET proteins only oxidize 5mC in specific genomic loci (lower part), the TDG-BER capacity is enough to quickly complete the whole active demethylation process at these specific loci before DNA replication starts. Thus, the BER-based active demethylation pathway efficiently underlies TET-induced locus-specific DNA demethylation.
The lack of BER upregulation during TET-induced global DNA demethylation in our HEK293T cell model reflects the sophisticated regulatory mechanisms of DNA repair genes in mammalian cells. DNA repair has to be tightly regulated in a temporal, spatial, and DNA lesion-appropriate fashion to optimize repair and also prevent unnecessary and deleterious alterations in the structure of DNA.Citation21,33 Only a few DNA repair genes have been found to be upregulated in response to DNA damage and extensively distributed DNA damage itself can block the transcription of DNA repair genes.Citation21 Thus, it is not entirely surprising that no BER genes were upregulated in our HEK293T cell model of global demethylation, even though the DNA damage response had been activated ( and ). Since TDG, APEX1 and XRCC1 were all kept at stable expression levels in our model, we also do not know which one serves as the limiting factor for increased active demethylation. Interestingly, Hajkova et al. reported significant activation of 3 BER genes (including Parp1, Ape1 and Xrcc1) during global DNA demethylation in mouse PGCs.Citation34 This upregulation of BER genes may reflect a unique mechanism in zygotes and PGCs to maintain genomic stability during epigenetic reprogramming and is obviously worthy of further study. On the other hand, post-translational modifications are also involved in the regulation of DNA repair. In contrast to other uracil DNA glycosylases, TDG requires a SUMO modification-induced conformational change for efficient dissociation from AP-sites.Citation35 In the case of extensive DNA damage, the SUMOylation system quickly becomes saturated and consequently leads to a low turnover of TDG and failed BER.Citation35,36 Thus, it is possible that a similarly saturated SUMOylation system and incomplete BER also contribute to the limited active mechanism in TET-induced global demethylation.
In summary, our study constructed a tetracycline-controlled TET-induced global demethylation model in HEK293T cells, where the DNA replication-dependent passive pathway functions as the primary mechanism as in physiological cell contexts, and furthermore, the BER-based active pathway is triggered but significantly restricted by a limited DNA repair capacity. These findings support a working model for pathway selection in TET-induced DNA demethylation and also have implications for understanding the global DNA demethylation in zygotes and PGCs.
Material and Methods
Inducible TET1-CD overexpression system
The open reading frame (ORF) of human TET1-CD was cloned from SY5Y cells, and inserted into the pIRES-hrGFP II vector, which contains a 3×FLAG tag (Stratagene) as previous described.Citation18 Catalytically mutant mTET1-CD (H1672Y, D1674A) was generated by site-directed mutagenesis.Citation4,18 TET1-CD-FLAG or mTET1-CD-FLAG was then cloned into the tetracycline inducible lentiviral vector pTRIPZ (Open Biosystems), which was initially designed for inducible shRNA expression. To make 2 unique restriction enzyme sites flanking the red fluorescent protein coding region in pTRIPZ vector, one AgeI site in the coding region was mutated by site-directed mutagenesis. Subsequently, (m)TET1-CD-FLAG ORF was transferred into the AgeI and MluI sites of a non-silencing pTRIPZ control vector. All constructs were verified by bi-directional sequencing.
To produce lentiviral particles, pTRIPZ-(m)TET1-CD-FLAG and package plasmids psPAX2 and pMD2.G (Addgene) were transfected into HEK293FT cells (Invitrogen) at a ratio of 1:1:1 using Lipofectamine→2000 Transfection Reagents (Invitrogen). The viral supernatant was collected 2 days after transfection and filtered with 0.45 µm filters (Millipore). HEK293T cells were then infected with each lentivirus supernatant in the presence of 8 µg/ml of polybrene (Sigma).
Transduced cells were subjected to one week of puromycin selection (1.5 µg/ml) and subcloned by limiting dilution. HEK293T cells were cultured in DMEM supplemented with 10% FBS. Overexpression of (m)TET1-CD was induced by Dox (Sigma, dissolved in sterile water) at indicated dosages. The Dox-containing medium was generally replaced every 2 days.
Western blot assay
Protein extraction was performed using RIPA buffer (Fisher) supplemented with 1×protease inhibitor cocktail solution (Roche). Lysates were fractionated by 10% or 12% SDS-PAGE and transferred to PVDF membranes (Millipore). The primary antibodies used included anti-FLAG (Cat#200471, Stratagene), anti-phos-CHK2 (Thr68) (Cat#2197, Cell signaling), anti-phos-TP53 (ser15) (Cat#9286, Cell signaling), anti-TP53 (sc-126, Santa Cruz), anti-γH2AX (ser139) (Cat#9718, Cell signaling), anti-TDG (GTX110473, GeneTex), anti-APEX1 (ab82, Abcam), anti-XRCC1 (Cat#2735, Cell signaling), and anti-ACTB (GTX109639, GeneTex), while secondary antibodies included HRP-conjugated labeled anti-rabbit and anti-mouse antibodies (GE Healthcare). The detection was performed using enhanced chemiluminescence (Amersham) and X-ray imaging film (Fisher).
DNA dot blot assay
Different amounts of genomic DNA samples diluted in 0.4 mM NaOH/10 mM EDTA were denatured at 100°C for 10 min, followed by rapid chilling on ice. Each denatured DNA (2 μl) was then spotted onto the positively charged nylon membrane (Roche), and the diameter of each dot was kept to < 4 mm. The membrane was air dried and subsequently rinsed in 2×SSC buffer (0.3 M NaCl, 30 mM Na3C6H5O7) followed by complete air dry. The membrane was then exposed to UV for 3 min with a UV transilluminator to immobilize the DNA. Lastly, the membrane was immunoblotted using 5hmC and 5caC antibodies (Active Motif) and HRP-conjugated anti-rabbit secondary antibody (GE Healthcare).
Reverse transcription-qPCR
RNA was isolated using TRIzol Reagent (Invitrogen) as per manufacturer's specifications. cDNA was synthesized using High-Capacity cDNA reverse transcription kit (Applied Biosystems) and tested by qPCR (Applied Biosystems 7500) using Power SYBR® Green PCR Master Mix (Applied Biosystems). PCR reaction comprised a 10 min activation step at 95°C, followed by 40 cycles of 95°C for 15 s, and 60°C for 1 min. The average threshold (Ct) was determined for each gene and normalized to β-actin as an internal normalization control. The primers used are listed in Table S1.
Cell growth curve
HEK293T cells (3 × 104 cells) from exponentially growing cultures were seeded in 6-well culture plate. Twenty-four hours later the medium was replaced with fresh growth medium with or without 2 μg/ml Dox. During the following 3 days the cell numbers were counted daily with the use of Z2 Cell and Particle Counter (Beckman Coulter).
Bisulfite-pyrosequencing
Bisulfite conversion of genomic DNA was done with EpiTect bisulfite kits (Qiagen) according to manufacturer's instructions. A two-step PCR for amplification was generally used as previously described.Citation37,38 Briefly, in the first step, 1 μl of each bisulfite-converted DNA sample was used in each reaction (25 μl total volume). In the second step, ∼0.1 μl of the 1st step PCR products was used as template, and the 5’ tailed forward or reverse primers and a biotinylated universal primer (5’-GGGACACCGCTGATCGTTTA-3’) were used to label PCR product. With the Pyrosequencing Vaccum Prep Tool (Biotage) the biotin-labeled DNA strands were captured by streptavidin sepharose beads (GE Healthcare). Then they were annealed to sequencing primers and sequenced by the PSQ HS 96 Pyrosequencing system (Biotage). The results were analyzed with Pyro Q-CpG Software (Qiagen) software. The primers used are listed on Table S1.
siRNA knockdown
Non-silencing control siRNA (SIC002), 2 TDG siRNA (SASI_Hs01_00108995 and SASI_Hs01_00108996), and 2 APEX1 siRNA (SASI_Hs01_00122789 and SASI_Hs01_00122791) were all purchased from Sigma. The cells were transfected with siRNA using LipofectamineTM RNAiMAX transfection reagent according to the manufacturer's instructions (Invitrogen). For each transfection, 75 pmol siRNA was diluted in 250 μl of OPTI-MEM® Reduced Serum Medium (Invitrogen), and then mixed with 4 μl Lipofectamine™ RNAiMAX Transfection Reagent (Invitrogen). To improve knockdown efficiency, a second transfection was performed 3 days after the first transfection.
Contact inhibition-induced growth arrest
HEK293T cells were seeded at a density of 2.5 × 106 cells per dish in 60 mm dishes and cultured for 3 days to get cell growth arrest. The medium was then replaced with fresh growth medium with or without 2 μg/ml Dox. The cells were further incubated for 3 days with the change of growth medium every day.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
1091145_Supplemental_Material.docx
Download MS Word (11.1 MB)Funding
This work was supported by National Institutes of Health grants CA158112 and CA100632. JPI is an American Cancer Society Clinical Research professor supported by a generous gift from the F. M. Kirby Foundation.
References
- Wu SC, Zhang Y. Active DNA demethylation: many roads lead to Rome. Nat Rev Mol Cell Biol 2010; 11:607-20; PMID:20683471; http://dx.doi.org/10.1038/nrm2950
- Bird A. DNA methylation patterns and epigenetic memory. Genes Dev 2002; 16:6-21; PMID:11782440; http://dx.doi.org/10.1101/gad.947102
- Ito S, Shen L, Dai Q, Wu SC, Collins LB, Swenberg JA, He C, Zhang Y. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science 2011; 333:1300-3; PMID:21778364; http://dx.doi.org/10.1126/science.1210597
- Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, Agarwal S, Iyer LM, Liu DR, Aravind L, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 2009; 324:930-5; PMID:19372391; http://dx.doi.org/10.1126/science.1170116
- Ito S, D'Alessio AC, Taranova OV, Hong K, Sowers LC, Zhang Y. Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature 2010; 466:1129-33; PMID:20639862; http://dx.doi.org/10.1038/nature09303
- He YF, Li BZ, Li Z, Liu P, Wang Y, Tang Q, Ding J, Jia Y, Chen Z, Li L, et al. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science 2011; 333:1303-7; PMID:21817016; http://dx.doi.org/10.1126/science.1210944
- Valinluck V, Sowers LC. Endogenous cytosine damage products alter the site selectivity of human DNA maintenance methyltransferase DNMT1. Cancer Res 2007; 67:946-50; PMID:17283125; http://dx.doi.org/10.1158/0008-5472.CAN-06-3123
- Inoue A, Shen L, Dai Q, He C, Zhang Y. Generation and replication-dependent dilution of 5fC and 5caC during mouse preimplantation development. Cell Res 2011; 21:1670-6; PMID:22124233; http://dx.doi.org/10.1038/cr.2011.189
- Inoue A, Zhang Y. Replication-dependent loss of 5-hydroxymethylcytosine in mouse preimplantation embryos. Science 2011; 334:194; PMID:21940858; http://dx.doi.org/10.1126/science.1212483
- Guo JU, Su Y, Zhong C, Ming GL, Song H. Hydroxylation of 5-methylcytosine by TET1 promotes active DNA demethylation in the adult brain. Cell 2011; 145:423-34; PMID:21496894; http://dx.doi.org/10.1016/j.cell.2011.03.022
- Wu H, Zhang Y. Mechanisms and functions of Tet protein-mediated 5-methylcytosine oxidation. Genes Dev 2011; 25:2436-52; PMID:22156206; http://dx.doi.org/10.1101/gad.179184.111
- Maiti A, Drohat AC. Thymine DNA glycosylase can rapidly excise 5-formylcytosine and 5-carboxylcytosine: potential implications for active demethylation of CpG sites. J Biol Chem 2011; 286:35334-8; PMID:21862836; http://dx.doi.org/10.1074/jbc.C111.284620
- Gu TP, Guo F, Yang H, Wu HP, Xu GF, Liu W, Xie ZG, Shi L, He X, Jin SG, et al. The role of Tet3 DNA dioxygenase in epigenetic reprogramming by oocytes. Nature 2011; 477:606-10; PMID:21892189; http://dx.doi.org/10.1038/nature10443
- Hackett JA, Sengupta R, Zylicz JJ, Murakami K, Lee C, Down TA, Surani MA. Germline DNA demethylation dynamics and imprint erasure through 5-hydroxymethylcytosine. Science 2013; 339:448-52; PMID:23223451; http://dx.doi.org/10.1126/science.1229277
- Iqbal K, Jin SG, Pfeifer GP, Szabo PE. Reprogramming of the paternal genome upon fertilization involves genome-wide oxidation of 5-methylcytosine. Proc Natl Acad Sci U S A 2011; 108:3642-7; PMID:21321204; http://dx.doi.org/10.1073/pnas.1014033108
- Klug M, Schmidhofer S, Gebhard C, Andreesen R, Rehli M. 5-Hydroxymethylcytosine is an essential intermediate of active DNA demethylation processes in primary human monocytes. Genome Biol 2013; 14:R46; PMID:23705593; http://dx.doi.org/10.1186/gb-2013-14-5-r46
- Wossidlo M, Nakamura T, Lepikhov K, Marques CJ, Zakhartchenko V, Boiani M, Arand J, Nakano T, Reik W, Walter J. 5-Hydroxymethylcytosine in the mammalian zygote is linked with epigenetic reprogramming. Nat Commun 2011; 2:241; PMID:21407207; http://dx.doi.org/10.1038/ncomms1240
- Jin C, Lu Y, Jelinek J, Liang S, Estecio MR, Barton MC, Issa JP. TET1 is a maintenance DNA demethylase that prevents methylation spreading in differentiated cells. Nucleic Acids Res 2014; 42:6956-71; PMID:24875481; http://dx.doi.org/10.1093/nar/gku372
- Matsuoka S, Rotman G, Ogawa A, Shiloh Y, Tamai K, Elledge SJ. Ataxia telangiectasia-mutated phosphorylates CHK2 in vivo and in vitro. Proc Natl Acad Sci U S A 2000; 97:10389-94; PMID:10973490; http://dx.doi.org/10.1073/pnas.190030497
- Shieh SY, Ahn J, Tamai K, Taya Y, Prives C. The human homologs of checkpoint kinases Chk1 and Cds1 (CHK2) phosphorylate TP53 at multiple DNA damage-inducible sites. Genes Dev 2000; 14:289-300.; PMID:10673501
- Christmann M, Kaina B. Transcriptional regulation of human DNA repair genes following genotoxic stress: trigger mechanisms, inducible responses and genotoxic adaptation. Nucleic Acids Res 2013; 41:8403-20; PMID:23892398; http://dx.doi.org/10.1093/nar/gkt635
- Kuo LJ, Yang LX. Gamma-γH2AX - a novel biomarker for DNA double-strand breaks. In Vivo 2008; 22:305-9; PMID:18610740
- Pascucci B, Russo MT, Crescenzi M, Bignami M, Dogliotti E. The accumulation of MMS-induced single strand breaks in G1 phase is recombinogenic in DNA polymerase β defective mammalian cells. Nucleic Acids Res 2005; 33:280-8; PMID:15647510; http://dx.doi.org/10.1093/nar/gki168
- Chehab NH, Malikzay A, Appel M, Halazonetis TD. CHK2/hCds1 functions as a DNA damage checkpoint in G(1) by stabilizing TP53. Genes Dev 2000; 14:278-88; PMID:10673500
- Cabelof DC, Raffoul JJ, Yanamadala S, Guo Z, Heydari AR. Induction of DNA polymerase β-dependent base excision repair in response to oxidative stress in vivo. Carcinogenesis 2002; 23:1419-25; PMID:12189182; http://dx.doi.org/10.1093/carcin/23.9.1419
- Ramana CV, Boldogh I, Izumi T, Mitra S. Activation of apurinic/apyrimidinic endonuclease in human cells by reactive oxygen species and its correlation with their adaptive response to genotoxicity of free radicals. Proc Natl Acad Sci U S A 1998; 95:5061-6; PMID:9560228; http://dx.doi.org/10.1073/pnas.95.9.5061
- Das A, Hazra TK, Boldogh I, Mitra S, Bhakat KK. Induction of the human oxidized base-specific DNA glycosylase NEIL1 by reactive oxygen species. J Biol Chem 2005; 280:35272-80; PMID:16118226; http://dx.doi.org/10.1074/jbc.M505526200
- Fortini P, Dogliotti E. Base damage and single-strand break repair: mechanisms and functional significance of short- and long-patch repair subpathways. DNA Repair (Amst) 2007; 6:398-409; PMID:17129767; http://dx.doi.org/10.1016/j.dnarep.2006.10.008
- Madhusudan S, Smart F, Shrimpton P, Parsons JL, Gardiner L, Houlbrook S, Talbot DC, Hammonds T, Freemont PA, Sternberg MJ, et al. Isolation of a small molecule inhibitor of DNA base excision repair. Nucleic Acids Res 2005; 33:4711-24; PMID:16113242; http://dx.doi.org/10.1093/nar/gki781
- Jackman J, O'Connor PM. Methods for synchronizing cells at specific stages of the cell cycle. Curr Protoc Cell Biol, 2001; 43:3.8.1-3.8.21.
- Watson PA, Hanauske-Abel HH, Flint A, Lalande M. Mimosine reversibly arrests cell cycle progression at the G1-S phase border. Cytometry 1991; 12:242-6; PMID:1903691; http://dx.doi.org/10.1002/cyto.990120306
- Okashita N, Kumaki Y, Ebi K, Nishi M, Okamoto Y, Nakayama M, Hashimoto S, Nakamura T, Sugasawa K, Kojima N, et al. PRDM14 promotes active DNA demethylation through the ten-eleven translocation (TET)-mediated base excision repair pathway in embryonic stem cells. Development 2014; 141:269-80; PMID:24335252; http://dx.doi.org/10.1242/dev.099622
- Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Molecular Cell 2010; 40:179-204; PMID:20965415; http://dx.doi.org/10.1016/j.molcel.2010.09.019
- Hajkova P, Jeffries SJ, Lee C, Miller N, Jackson SP, Surani MA. Genome-wide reprogramming in the mouse germ line entails the base excision repair pathway. Science 2010; 329:78-82; PMID:20595612; http://dx.doi.org/10.1126/science.1187945
- Hardeland U, Steinacher R, Jiricny J, Schar P. Modification of the human thymine-DNA glycosylase by ubiquitin-like proteins facilitates enzymatic turnover. EMBO J 2002; 21:1456-64; PMID:11889051; http://dx.doi.org/10.1093/emboj/21.6.1456
- Kunz C, Focke F, Saito Y, Schuermann D, Lettieri T, Selfridge J, Schar P. Base excision by thymine DNA glycosylase mediates DNA-directed cytotoxicity of 5-fluorouracil. PLoS Biol 2009; 7:e91; PMID:19402749; http://dx.doi.org/10.1371/journal.pbio.1000091
- Kroeger H, Jelinek J, Estecio MR, He R, Kondo K, Chung W, Zhang L, Shen L, Kantarjian HM, Bueso-Ramos CE, et al. Aberrant CpG island methylation in acute myeloid leukemia is accentuated at relapse. Blood 2008; 112:1366-73; PMID:18523155; http://dx.doi.org/10.1182/blood-2007-11-126227
- Colella S, Shen L, Baggerly KA, Issa JP, Krahe R. Sensitive and quantitative universal Pyrosequencing methylation analysis of CpG sites. Bio Techniques 2003; 35:146-50; PMID:12866414