
Abstract
Bisphenol A (BPA) is an endocrine disrupting chemical (EDC) that has been implicated as a potential carcinogen and epigenotoxicant. We have previously reported dose-dependent incidence of hepatic tumors in 10-month-old isogenic mice perinatally exposed to BPA. Here, we evaluated DNA methylation at 3 candidate genes (Esr1, Il-6st, and Stat3) in liver tissue of BPA-exposed mice euthanized at 2 time points: post-natal day 22 (PND22; n = 147) or 10-months of age (n = 78, including n = 18 with hepatic tumors). Additionally, DNA methylation profiles were analyzed at human homologs of murine candidate genes in human fetal liver samples (n = 50) with known liver tissue BPA levels. Candidate genes were chosen based on reported expression changes in both rodent and human hepatocellular carcinoma (HCC). Regions for bisulfite sequencing were chosen by mining whole genome next generation sequencing methylation datasets of both mice and human liver samples with known perinatal BPA exposures. One of 3 candidate genes, Stat3, displayed dose-dependent DNA methylation changes in both 10-month mice with liver tumors as compared to those without liver tumors and 3-week sibling mice from the same exposure study, implicating Stat3 as a potential epigenetic biomarker of both early life BPA exposure and adult disease in mice. DNA methylation profiles within STAT3 varied with liver tissue BPA level in human fetal liver samples as well, suggesting STAT3 may be a translationally relevant candidate biomarker. These data implicate Stat3 as a potential early life biomarker of adult murine liver tumor risk following early BPA exposure with early evidence of relevance to human health.
Introduction
Environmental epigenetics, or the study of the impact of environmental exposures on patterns of epigenetic regulation, seeks to identify future public health interventions that may reverse deleterious effects of the social, nutritional, and chemical environment to facilitate the prevention of exposure-linked disease. Environmental exposures that occur during early life development, in particular, have been shown to induce altered epigenetic programs that affect adult health outcomes.Citation1,2
We have previously reported a dose-dependent increase in hepatic tumors in 10-month mice that were perinatally exposed to one of 3 doses of the endocrine disrupting chemical (EDC) bisphenol A (BPA).Citation3 Notably, a significant increase in liver tumors was evident at 10-months of age, which represents early onset disease in mice. Furthermore, the classical sexual dimorphism of a higher tumor rate in males as compared to females was not present.Citation3 Developmental BPA exposure has also been linked to proliferative tissue changes in rat mammary glandsCitation4 and to frank prostate cancer in mice with human prostate stem cell xenografts supplemented with sex steroid hormones to mimic human male aging.Citation5 Additionally, these studies have identified epigenetic profiles linked to BPA dose and tissue alterations, supporting the hypothesis that epigenetic changes may mediate the relationship between early life BPA exposure and adult health outcomes, including cancer.Citation4,6
Epigenetic patterns consist of mitotically heritable marks that govern chromatin organization and gene expression, including DNA methylation, histone protein modifications, and non-coding RNA. These epigenetic profiles may serve as evidence of past exposure, risk factors for subsequent disease, and/or reversible targets of public health or clinical interventions.Citation1,7,8 Targeted epigenetic changes, including promoter hypermethylation of tumor suppressors and panels of microRNAs, are currently in use as diagnostic biomarkers in the laboratory and clinic for lung, colorectal, and prostate cancers.Citation9 Manikkam et al. report exposure-specific epigenetic signatures using epigenome-wide discovery approaches in sperm of mice exposed to endocrine disruptors.Citation10 Other studies have demonstrated the presence of stochastic and global epigenomic changes following environmental exposures in miceCitation1,11 and humans.Citation12 In order to facilitate translation of animal studies that identify epigenetic biomarkers of environmental exposure and disease risk to human health and disease prevention, studies must demonstrate epigenetic changes not only at a single time point but across the life-course, and validate with mechanistic and epidemiological studies.
To address concerns with past candidate gene epigenetic studies, here we developed and employed a novel method for detection of candidate epigenetic biomarkers. We chose translationally-relevant candidate genes with functional links to hepatocellular carcinoma (HCC) for analysis based on expression changes reported in the literature in both rodent and human liver tumors. Three candidate genes were chosen: estrogen receptor α (Esr1/ESR1)Citation13-16; interleukin 6 signal transducer (Il6st/IL6ST),Citation17-20 also known as gp130; and signal transducer and activator of transcription 3 (Stat3/STAT3).Citation18,21 These candidate genes are functionally linked. Increased production of the cytokine IL-6 initiates downstream STAT3 signaling.Citation18,22 STAT3 signaling is a key event in inflammation-driven liver carcinogenesis.Citation18 In fact, the classical sexual dimorphism in HCC incidence is thought to be due to the anti-inflammatory influence of endogenous estrogen via suppression of IL-6 production in an ERα-dependent manner.Citation23 BPA has been shown to increase expression of both IL-6 and STAT3 via both ERα and ERβ in human hepatoma Hep3b cells.Citation23 In order to identify short, defined regions for quantitative DNA methylation analysis within these candidate genes, we mined existing whole genome next generation sequencing DNA methylation data sets for windows that exhibited lability in DNA methylation profiles following perinatal BPA exposure in both miceCitation24 and humans (unpublished data, in review) (MethylPlex technology, Rubicon Genomics, Ann Arbor, MI). This approach allowed us to combine a traditional candidate gene approach, suitable for highly quantitative bisulfite sequencing on the Sequenom EpiTYPER MassARRAY platform, with epigenome-wide assessments of regions of altered methylation associated with early life BPA exposure, to identify candidate biomarkers linked to both exposure and disease.
Results
Of the 3 candidate genes profiled in mouse and human liver (), 2 (Esr1/ESR1 and Il-6st/IL-6ST) showed either no or sporadic association with tumor status or BPA dose level. However, Stat3 displayed dose-dependent DNA methylation changes in both 10-month mice with liver tumors as compared to those without liver tumors and in 3-week old sibling mice. DNA methylation in the human homolog, STAT3, also varied with liver tissue BPA level in human fetal liver samples. DNA methylation profiles for each candidate gene are presented in detail below, starting with comparisons in 10-month mice with and without tumors, proceeding to 3-week old mice, and finally to human homologs in fetal liver tissue.
Table 1. Post-natal day 22 (PND22) mouse liver samples (n = 147) and 10-month mouse liver samples (n = 78) for bisulfite sequencing
Candidate Gene DNA Methylation in Mice and Humans
Esr1/ESR1
Neither average nor site-specific percent DNA methylation at 2 CpG sites within murine Esr1 () in 10-month mice differed by tumor status, sex, or dose (Tables S1 and S2). No dose- or sex-based differences in average or site-specific DNA methylation were seen at Esr1 in PND22 mice (Table S3). Average and site-specific DNA methylation at 2 CpG sites in the human homolog ESR1 () in human fetal liver samples were not associated with either total or free BPA exposure (Table S4 and S5).
Table 2. Candidate gene regions for bisulfite sequencing
Il-6st/IL-6ST
Average percent DNA methylation at 3 CpG sites within murine Il-6st () in 10-month mice with tumors was ∼3% lower in the 50 mg group as compared to animals without tumors (P = 0.03) (Table S6), but no differences were seen by dose (Table S7). No differences in average methylation were seen in sibling PND22 mice (, Table S8). When methylation patterns were analyzed by individual CpG site, differences at CpG site 1 and CpG site 3 not seen in 10-month mice were evident in 3-week sibling mice. Approximately 9% hypermethylation at CpG site 1 was seen in the 50 µg group, as compared to control (50 µg: 81.1%, control: 71.9%, P = 0.04) (Table S8). CpG site 3 was approximately 5% hypomethylated as compared to control in the 50 ng group (50 ng: 79.3%, control: 84.0%, P = 0.04) (Table S8).
Average DNA methylation across 7 CpG sites in the human homolog IL-6ST () in human fetal liver samples did not differ by tertile of total (combined free and conjugated) liver tissue BPA (Table S9). When percent methylation was analyzed at individual CpG sites, approximately 4% hypomethylation at CpG site 5 was seen in the high tertile as compared to the medium tertile of total BPA (high BPA: 89.2%, medium BPA: 93.3%, P = 0.02) (Table S9). The high tertile of total BPA was also ∼5-6% hypomethylated at CpG site 7 as compared to the medium tertile (P = 0.01) and the low tertile (P < 0.01) (high BPA: 81.7%, medium BPA: 87.0%, low BPA: 88.0%) (Table S9). Similar to total BPA analyses, CpG site 7 was approximately 3-5% hypomethylated in the high tertile of free BPA as compared to the medium (P = 0.02) and low tertiles (P = 0.01) (high BPA: 82.6%, medium BPA: 86.0%, low BPA: 88.0%) (Table S10).
Stat3/STAT3
Average percent DNA methylation at 3 CpG sites within murine Stat3 (, , – see note in Figure legends) in 10-month mice with tumors was ∼7% lower in the 50 µg group (P = 0.01) as compared to animals without tumors (, Table S11). Site-specific analysis showed that CpG site 2 was ∼4% hypermethylated in the 50 mg group, as compared to control (P = 0.04) (, Table S12). No robust differences were observed in DNA methylation by sex (P = 0.06). Average percent DNA methylation at the same 3 CpG sites within murine Stat3 () was analyzed in sibling PND22 mice for evidence of early epigenetic changes with the potential to persist into adulthood. Following adjustment for sex and genotype, an approximately 2-3% decrease in average methylation were seen in the 2 highest dose groups (50 µg BPA/kg maternal diet: 83.5%, P<0.01; 50 mg BPA/kg maternal diet: 84.7%, P = 0.01) as compared to control (86.7%) (, Table S13). When methylation patterns were analyzed by individual CpG site, hypomethylation was noted at all 3 CpG sites sequenced. Approximately 2.5% hypomethylation at CpG site 1 was seen in the 50 µg group, as compared to control (50 µg: 85.3%, control: 87.9%, P = 0.03) (, Table S13). Approximately 2—3% hypomethylation was seen in the lowest and highest dose groups at CpG site 2 (50 ng: 85.4%, P = 0.01; 50 mg: 83.3%, P < 0.01) as compared to control (86.8%) (, Table S13). Similar differences (∼1-5% hypomethylation) were seen at CpG site 3 in all dose groups (50 ng: 84.8%, P < 0.01; 50 µg: 80.7%, P = 0.01; 50 mg: 83.9%, P < 0.01; control: 85.5%) (, Table S13). A small but statistically significant increase (∼1%) in average DNA methylation was noted in males as compared to females at Stat3 in PND22 mice (males: 85.9%, females: 84.5%, P = 0.01). This effect was not seen in bivariate models between sex and average methylation or in models containing interaction variables (sex*dose), indicating that no strong sex-based differences in methylation were present.
Figure 1. Stat3 methylation in 10-month mouse livers by dose and tumor status. (A) Average percent DNA methylation at Stat3 by dose group in 10-month mice perinatally exposed to 50 ng (n = 20), 50 µg (n = 21) or 50 mg (n = 18) BPA/kg maternal diet or a phytoestrogen-free control diet (n = 19). (B) Percent DNA methylation at Stat3 by tumor status in 10-month mice perinatally exposed to 50 ng (n = 20), 50 µg (n = 21) or 50 mg (n = 18) BPA/kg maternal diet or a phytoestrogen-free control diet (n = 19). *P <0.1, **P<0.05
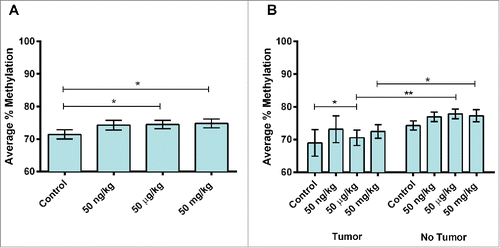
Figure 2. Site-specific Stat3 methylation in 10-month mouse livers by dose. (A) Average percent DNA methylation at Stat3 by dose group in 10-month mice perinatally exposed to 50 ng (n = 20), 50 µg (n = 21) or 50 mg (n = 18) BPA/kg maternal diet or a phytoestrogen-free control diet (n = 19). is the same as . These data are repeated for ease of comparison by the reader. (B) Percent DNA methylation at Stat3 by CpG site in 10-month mice perinatally exposed to 50 ng (n = 20), 50 µg (n = 21) or 50 mg (n = 18) BPA/kg maternal diet or a phytoestrogen-free control diet (n = 19). *P < 0.1, **P < 0.05.
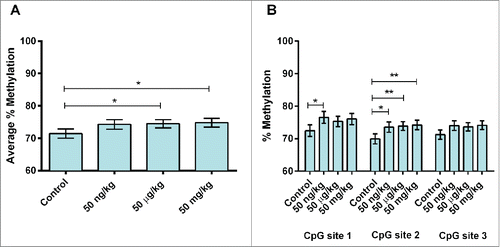
Figure 3. Site-specific Stat3 methylation in PND22 mouse livers by dose. (A) Average percent DNA methylation at Stat3 by dose group in post-natal day 22 (PND22) mice perinatally exposed to 50 ng (n = 48), 50 µg (n = 22) or 50 mg (n = 40) BPA/kg maternal diet or a phytoestrogen-free control diet (n = 37). is the same as . These data are repeated for ease of comparison by the reader. (B) Percent DNA methylation at Stat3 by CpG site in PND22 mice perinatally exposed to 50 ng (n = 48), 50 µg (n = 22) or 50 mg (n = 40) BPA/kg maternal diet or a phytoestrogen-free control diet (n = 37). *P < 0.1, **P < 0.05.
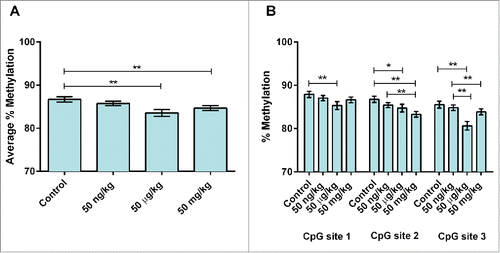
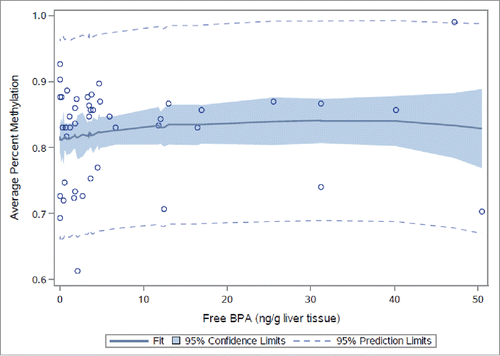
Neither average nor site-specific DNA methylation at 3 CpG sites in the human homolog of STAT3 () in human fetal liver samples differed by tertile of total (combined free and conjugated) liver tissue BPA (Fig. S1 and Table S14) or free BPA alone (Fig. S2 and Table S15). However, statistically significant associations with average percent DNA methylation were evident when total BPA (P = 0.01) and free BPA (P = 0.02) were modeled as continuous variables (Tables S14 and S15). Mean-centered squared variables [(centered total BPA)Citation2 and (centered free BPA)Citation2] were statistically significant when added to the models (P = 0.01 and P = 0.04, respectively), confirming a non-monotonic dose-response visually apparent at CpG site 1 (, Figs. S1 and S2). No differences in average percent DNA methylation were noted between sexes in models adjusting for total BPA (P = 0.22) or free BPA (P = 0.23).
Figure 4. STAT3 methylation in human fetal livers by continuous total BPA. Average percent DNA methylation at STAT3 by dose group in human fetal liver samples (n = 50) by continuous liver tissue total BPA.
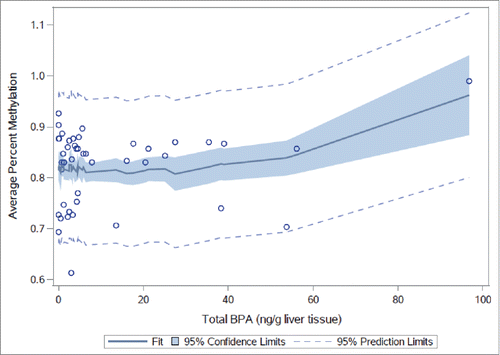
Figure 5. STAT3 methylation in human fetal livers by continuous free BPA. Average percent DNA methylation at STAT3 by dose group in human fetal liver samples (n = 50) by continuous liver tissue free BPA.
Candidate Gene Expression
Altered DNA methylation was noted at Stat3 in mice and STAT3 in human tissues. Transcript levels of both genes were analyzed to investigate a possible functional outcome linked to the observed epigenetic changes. Stat3 expression did not differ by perinatal BPA dose group in 10-month mice (Fig. S3). Tumor status was also not linked to Stat3 mRNA transcript level across dose groups or in a dose-dependent manner (Fig. S3). Endogenous reference genes did not differ by perinatal dose group (B2m P = 0.63, Gapdh P = 0.97; adjusted for sex) or tumor status (B2m P = 0.14, Gapdh P = 0.77), confirming their appropriate use as internal controls invariant to experimental exposure and outcome. A decrease in Stat3 expression with increasing perinatal BPA exposure was significant in PND22 mice (fold-change for 50 ng group 0.40, 50 µg group 0.23, 50 mg group 0.33; P = 0.05; adjusted for sex) (Fig. S4). Endogenous reference genes did not differ by perinatal dose group (B2M P = 0.80, Gapdh P = 0.80; adjusted for sex), confirming their appropriate use as internal controls.
Human fetal livers exhibited no change in STAT3 expression with continuous total or free BPA, following adjustment for sex and gestational age (Figs. S5 and S6). Gestational age was significant in fully adjusted models (total BPA P < 0.01, free BPA P < 0.01), but interaction variables indicated that the link between age and STAT3 expression was not dose-dependent (total BPA*gestational age P = 0.91, free BPA*gestational age). Human endogenous references genes did not differ by continuous total BPA (GAPDH P=0.60, UBC P = 0.52) or free BPA (GAPDH P = 0.38, UBC P = 0.16), even following adjustment for sex and gestational age, indicating chosen internal controls were invariant with exposure.
Discussion
Here, we show proof of principle for a novel method for detection of candidate epigenetic biomarkers associated with both early life exposure to the endocrine disruptor BPA and adult disease across the lifespan and across species. We focused this modified candidate gene study on Esr1, Il-6st, and Stat3, 3 candidate genes with reported expression differences in both rodent and human HCC. We investigated DNA methylation patterns within regions of these 3 genes that were empirically demonstrated to be labile following perinatal exposure to BPA in 3 sample groups: 10-month mice with and without liver tumors perinatally exposed to BPA, sibling PND22 mice from the same exposure study, and human fetal livers with known tissue BPA levels.
Tumor status was not associated with DNA methylation at Esr1 in 10-month mice. BPA was associated with hypomethylation in human fetal livers, indicating that Esr1 methylation may be altered by BPA exposure but is not linked to the previously observed liver tumor phenotype.Citation3 DNA methylation at human homolog ESR1 decreased with low level BPA exposure and increased with higher exposure, supporting a potentially non-linear epigenetic response to BPA. These results support Esr1 as a potential target of BPA-induced DNA methylation change, but do not implicate this candidate gene as a candidate biomarker of both exposure and disease in this study. Esr1 may exert its effects via DNA methylation changes outside of the region assayed or through other epigenetic or non-epigenetic changes not measured here. Alternatively, Esr1 may not be involved in the mechanism linking perinatal BPA exposure to liver tumors in these mice.
DNA methylation at the second candidate gene, Il-6st, was decreased in 50 mg-exposed mice with tumors and marginally increased in 50 ng-exposed mice with tumors, as compared to mice without tumors, supporting an interaction between dose and tumor status. DNA methylation at the same Il-6st locus in sibling PND22 mice showed site-specific hypomethylation in the 50 ng group at CpG site 3 and hypermethylation in the 50 µg group at CpG site 1. Taken together, data in the 10-month and PND22 groups at this locus indicate both the need for CpG site-level analyses to detect site-specific changes and the importance of subsequent testing to determine biological relevance of site-level changes. DNA methylation at the human homolog IL-6ST demonstrated hypomethylation with BPA exposure at specific CpG sites, but analysis of total and free BPA as continuous variables indicated no significant association between BPA exposure and average methylation at this locus. These data implicate Il-6st as a candidate epigenetic biomarker of exposure and disease in mice that may not be relevant to human BPA exposure.
The third candidate gene that we tested with this method, Stat3, displayed altered methylation with tumor status combined with altered methylation with increasing BPA exposure in 10-month mice, and altered methylation with increasing BPA exposure in 3-week sibling mice from the same exposure study, implicating Stat3 as a potential epigenetic biomarker of both early life BPA exposure and adult disease in mice. DNA methylation displayed non-monotonic dose-responses to continuous total and free liver tissue BPA level in human fetal liver samples, as well, suggesting STAT3 may be a translationally relevant candidate biomarker and supporting a growing body of literature on low dose effects of endocrine disruptors.Citation25 These data support STAT3 as a candidate biomarker for further testing, but do not support any statement of biological role or direct mechanistic link to either exposure or disease within the context of this study. However, STAT3 is a biologically feasible target for epigenetic shifts that impact liver tumor risk. IL-6 exerts its effects on hepatic function and HCC tumor progression in humans via activation of STAT3.Citation26 Further, a STAT3-dependent inflammatory feedback loop has been implicated in epigenetic control of transformation.Citation27 STAT3 has previously been implicated as a prognostic indicator of HCC progression in humans Citation28 and is currently being tested as a pharmacologic target in HCC therapy.Citation28,29 This transcription factor also plays a role in cellular differentiation. Temporarily increased expression of hormone-dependent STAT3 in primed human pluripotent stem cells stimulates cells to undergo an epigenetic shift to naïve-like pluripotency, which suggests a role in increasing the number of stem cell targets for mutational hits and eventual transformation.Citation30 In addition, a range of phytochemicals that have been shown to suppress STAT3 expression or signaling in vitro and in animal models,Citation31 implicating STAT3 as a viable target for future public health interventions.
We note that all 3 candidate genes chosen for analysis, Esr1/ESR1, Il-6st/IL-6ST, and Stat3/STAT3, exhibited lability in DNA methylation with BPA exposure, indicating our data mining approach is a successful strategy for distinguishing exposure-related epigenetic changes from those associated exclusively with disease status. However, BPA exposure was associated with both hypomethylation and hypermethylation, with no clear pattern with age or tumor status across candidate loci, supporting the need for validation of candidate epigenetic biomarkers to distinguish spurious findings from true biological markers and to elucidate the mechanisms surrounding the latter. In several comparisons, we note differential effects at specific CpG sites, highlighting the possibility for masking of true site-specific effects in studies analyzing average methylation levels only. However, the direction of site-specific effects was not consistent across age groups at the same locus, indicating that DNA methylation profiles may shift with age, and highlighting the need for functional validation of consistent and easily interpretable candidate biomarkers.
Our approach used agnostic epigenome-wide data mining to identify regions of variable DNA methylation of no predetermined genetic context. Four of 6 candidate gene region assays were located either within introns or at an exon-intron border, including Stat3 and STAT3 (). Studies that focus exclusively on DNA methylation of CpG islands within 5′ gene promoters disregard the potential biological roles of intergenic and intragenic epigenetic patterning, and also the fact that transcription commonly initiates within or between genes.Citation33-35 Intronic methylation has been shown to have functional relevance to human disease outcomes; differential methylation of gene variants within the second intron of KCNQ1 affects CTCF binding and confers risk for Beckwith-Wiedemann syndrome, an overgrowth disorder linked to epigenetic defects at imprinted genes.Citation36 Functional relevance of intronic methylation patterns in mice has been shown, as well. Intronic enhancer activity in the murine sf-1/ad4bp gene is correlated with both DNA methylation pattern and tissue-specific gene expression.Citation37 Rademacher et al. reported that the human genome contains at least 2,000 intronic CpG islands that are not present in the mouse genome, a subset of which show sequence similarities elsewhere in the human genome, suggesting that they arose via retrotransposition.Citation38 Transposable elements, such as the murine Avy metastable epiallele, have been previously implicated as targets for early environmental effects on the epigenome.Citation39 These data suggest that the human genome may contain a larger number of intronic epigenetic targets for environmental perturbation than rodents, indicating the importance of including intronic targets in studies evaluating the impact of environmental epigenetics on human health.
Methylation differences reported in this study are small as compared to those reported at the Avy locus in BPA-exposed animalsCitation40 and in human liver cancer pathology,Citation41 but are similar to effect sizes reported at other epigenetic loci in Avy mice exposed to BPA.Citation24,42 Further, several comparisons yielded p-values between 0.05 and 0.1, indicating that several additional data points may have reached our chosen threshold for statistical significant (P<0.05) given a larger sample size and therefore greater power to detect small effect sizes.
Expression differences were not observed at Stat3 or STAT3 in this study, but a 1% change in DNA methylation at the IGF2 DMR in humans exposed to environmental tobacco smoke has been previously linked to an approximate 2-fold increase in IGF2 transcription, indicating the potential functional relevance of small changes in DNA methylation.Citation43 Although combined expression levels of all Stat3 or STAT3 variants did not vary with murine BPA exposure in 10-month or PND22 mice, or with measured human liver tissue level in human fetal liver samples, respectively, these results do not definitively rule out the possibility of gene expression changes that may have occurred at a time point not measured here. Several studies suggest consideration of a broader range of potential functional outcomes of altered intronic methylation profiles. Tissue-specific intragenic methylation has been reported alternately to reduce or enhance transcriptional elongation efficiency,Citation44-48 which may affect ordering of subsequent cell signaling cascade activation. Intronic methylation may regulate relative expression of gene isoforms, rather than overall gene expression changes. Maunakea et al. utilized large-scale mapping of DNA methylation profiles in the human brain to demonstrate that tissue- and cell type-specific intragenic methylation can regulate alternative gene promoter activity and subsequent differential expression of alternative transcripts within a single cell type.Citation35 Gelfman et al. discovered that exon recognition relies on DNA methylation during co-transcriptional splicing and that alternative exons may be distinguished from constitutive exons by the presence of fewer methylated CpG sites.Citation49
The field of environmental epigenetics is currently in a period of discovery, analogous to the initial period of discovery in genetic linkage and association studies following the initial sequencing of the human genome. Genome-wide association studies and larger scale epidemiologic studies followed early individual-SNP candidate gene studies, increasing our understanding of epistatic effects and spurious results commonly reported in single gene analyses. The present study represents a high resolution, semi-agnostic method for identifying candidate epigenetic biomarkers, in contrast to wholly agnostic but lower resolution epigenome-wide approach. Our method is intended as a hypothesis-generating tool for selection of candidate biomarkers for further validation. In order to validate STAT3 as a biomarker of early life BPA exposure and possible early life risk indicator of increased susceptibility to one or more forms of liver tumors or other liver disease in adulthood, this candidate gene would need to be tested in larger epidemiological cohorts to determine its relevance to human health and possible use as a public health screening tool. Importantly, our results also highlight the need for life-course studies that follow model organisms or humans over one or more relevant life periods in order to identify early epigenetic changes that persist into adulthood.
Conclusion
Here, we demonstrate the utility of a novel method for detection of candidate epigenetic biomarkers of both exposure and disease. This two-pronged approach provides proof of principle for a novel method for identification of epigenetic biomarkers of exposure and outcome across the life-course and across species. First we demonstrate our method's ability to detect epigenetic changes associated with perinatal BPA exposure and liver tumor status in 10-month mice, as well as in 3-week sibling mice from the same exposure study, suggesting that alterations at these loci may serve as candidate early life epigenetic biomarkers for detection of exposure and estimation and potential mitigation of disease risk. Further, we include human fetal liver tissue characterized for BPA levels to demonstrate early translation of candidate biomarkers identified in this study. Our data support STAT3 as a translationally relevant candidate epigenetic biomarker of perinatal BPA exposure and adult liver tumors. However, this candidate biomarker requires validation via mechanistic studies to determine potential biological role(s) and epidemiological cohort studies to assess public health relevance. Future studies that combine in vivo animal models in controlled experimental environments with in vitro mechanistic and human epidemiological cohort validation will be a powerful approach for detection of epigenetic changes with definitive links to exposure and outcome with relevance to human health.
Materials and Methods
Mouse and human liver samples
Mice were obtained from a colony generated by backcrossing C3H/HeJ mice carrying the viable yellow Agouti (Avy) allele with C57BL/6J mice, followed by >200 generations of sibling mating.Citation39 The Avy strain is isogenicCitation39 and has been empirically confirmed to be genetically 93% C57BL/6J.Citation3 Offspring of virgin wild type (a/a) females and heterozygous (Avy/a) males were exposed during gestation and lactation to one of 3 doses of BPA (50 ng, 50 μg, or 50 mg BPA/kg maternal diet) or to a phytoestrogen-free control diet, as previously described.Citation3 Briefly, 6-week-old virgin wild type a/a female mice were randomly assigned to a phytoestrogen-free AIN-93G diet [TD.95092, with 7% corn oil instead of 7% soybean oil; Harlan Teklad, Madison, WI (composition available at http://www.harlan.com)] supplemented with one of 4 levels of BPA (0, 50 ng, 50 μg, or 50 mg /kg diet). The BPA was provided by the National Toxicology Program (NTP, Research Triangle Park, NC). After 2 weeks on the respective diet, 8-week-old virgin females were mated with Avy/a males and were impregnated within 5 d. We estimated that daily BPA exposure levels, based on a dam weighing 25 g and consuming 5 g chow daily, were 0, 10 ng, 10 μg, and 10 mg/kg body weight per day for diets containing BPA at 0, 50 ng, 50 μg, and 50 mg/kg of diet, respectively. We chose these diets to capture both mean and maximum human environmental exposures to BPA, which were reported by Vandenberg et al. Citation25 to range from 0.1 to 5 μg/kg body weight per day. In addition, BPA exposure within human relevant ranges was confirmed by direct measurement of BPA in liver tissue of a subset of exposed and control animals.Citation42 For example, total liver BPA measurements in animals fed the highest dose of BPA, 50 mg/kg chow, ranged from 9.5 to 870 μg/kg liver, which encompasses the maximum human environmental exposure indicated by human fetal liver measurements, ranging from below the limit of quantitation (LOQ) to 96.8 μg BPA/kg liver.Citation42 Anderson et al. Citation42 reported BPA concentrations of < LOQ–11.3 μg/kg liver (mean 2 μg/kg; median 0.6 μg/kg) and < LOQ–13 μg/kg liver (mean 2.8 μg/kg; median 0.3 μg/kg) in livers from mice fed diets containing BPA at 50 μg/kg chow and 50 ng/kg chow, respectively.Citation3
This mating scheme produced approximately 50% wild type (a/a) offspring and approximately 50% heterozygous (Avy/a) offspring. The majority of Avy/a mice were euthanized on post-natal day (PND) 22.Citation32 A subset of wild type (a/a) mice (approximately 1 male and 1 female per litter for each BPA diet group) were then housed with a same-sex Avy/a sibling and fed the standard phytoestrogen-free control diet until they were euthanized at 10 months of age, as described by Anderson et al. ().Citation50 During dissection of both PND22 and 10-month animals, liver tissue was collected and flash frozen in liquid nitrogen. Liver tissue from 10-month animals was evaluated for histopathological lesions, including benign and malignant tumors, as previously described by Weinhouse et al.Citation3 Grossly visible lesions from tumor-bearing mice were dissected by a board-certified veterinary pathologist (ILB). Animals used in this study were maintained in accordance with the Institute of Laboratory Animal Resources guidelines (ILAR 1996) and were treated humanely and with regard for alleviation of suffering. The study protocol was approved by the University of Michigan Committee on Use and Care of Animals.
In addition, human fetal liver tissue, gestational day 74-120, was analyzed in this study, to assess potential relevance of mouse results to human liver epigenetics. Human fetal liver tissue was obtained from the University of Washington Laboratory for the Study of Human Embryology (LSHE) fetal tissues bank (2R24 HD000836-47).Citation51 These clinical liver samples were taken from voluntary 1st and 2nd trimester pregnancy terminations, with appropriate consent from donors.Citation51 These samples meet criteria for IRB exemption for human subject research (UM IRB exemption: HUM00024929). No identifying information other than gestational age, race, and sex were provided to researchers.
Candidate gene assay design
In order to identify short regions (approximately 400–600 bp) for bisulfite sequencing of DNA methylation within these candidate genes, we mined whole genome next generation sequencing methylation datasets for regions of altered methylation (RAMs) that exhibited lability in DNA methylation profiles following perinatal BPA exposure in both miceCitation24 and humans [Faulk et al. 2015 in press] (MethylPlex technology, Rubicon Genomics, www.rubicongenomics.com). RAMs were defined as 150 bp windows that exhibited statistically significant differences in DNA methylation between exposure groups in mouse and human next-generation sequencing data sets. RAMs were identified by mining datasets with 100 bp reads that overlapped by 50 bp; regions within candidate genes that exhibited differential methylation (P < 0.05) in at least 2 consecutive windows (150 bp) in at least 2 of 3 comparisons tested (control vs. 50 µg BPA/kg maternal diet, control vs. 50 mg BPA/kg maternal diet, and 50 µg BPA/kg maternal diet vs. 50 mg BPA/kg maternal diet in mouse data; low BPA tertile vs. medium BPA tertile, low BPA tertile vs. high BPA tertile, and medium BPA tertile vs. high BPA tertile in human data) were chosen for bisulfite sequencing ().
Bisulfite sequencing of DNA methylation in candidate genes
Genomic DNA was isolated from 10-month (n = 78; ) and PND22 (n = 147; ) mouse liver tissue samples and from human fetal liver samples (n = 50) using buffer ATL (QIAGEN Inc., cat. no. 19076), proteinase K (QIAGEN Inc., cat. no. 19131), and RNase A (QIAGEN Inc., cat. no. 19101), followed by phenol-chloroform extraction and ethanol precipitation. DNA quality and concentration was assessed using a ND1000 spectrophotometer (NanoDrop Technology). Quantitative DNA methylation patterns at candidate biomarker genes were analyzed via sodium bisulfite treatment followed by sequencing using Sequenom EpiTYPER MassARRAY technology (technology now owned by Agena Bioscience). Sodium bisulfite treatment was performed using the QIAGEN EpiTect Bisulfite Kit (QIAGEN Inc., cat. no. 59104) with approximately 1 µg input genomic DNA. EpiTYPER assays were designed using the Sequenom EpiDesigner tool (www.epidesigner.com), and candidate primer sets were assessed for unique products in the bisulfite-converted mouse and human genomes using the BiSearch ePCR tool (http://bisearch.enzim.hu). PCR was carried out in 25 µl reactions using approximately 50 ng of bisulfite-converted DNA, HotStarTaq master mix (QIAGEN Inc., cat. no. 203443) and forward and reverse (0.1 pmol each) primers under the following conditions: 15 min at 95°C, 40 cycles of (30 s at 94°C, 30 s at annealing temperature (TA), 1 min at 72°C), 10 min at 72°C. Primer sequences for bisulfite sequencing are listed in .
Table 3. Primer sets for bisulfite sequencing.
mRNA expression analysis via qPCR
RNA was purified from 10-month mouse liver tissue samples (n = 78), a subset of PND22 mouse liver tissue samples with adequate quantity and quality of tissue (n = 38; n = 10 control, n = 18 50 µg BPA/kg diet; n = 10 50 mg BPA/kg diet), and human fetal liver samples (n = 50) using RNeasy Mini Kit (QIAGEN Inc., cat. no. 74106) or All-Prep Kit (QIAGEN Inc., cat. no. 80004). Briefly, approximately 10-15 mg of liver tissue was homogenized using 3 30-s bursts at 60 Hz in a TissueLyser II (QIAGEN Inc., cat. no. 85300). Crude lysates were processed on a QIAcube automated nucleic acid purification system (QIAGEN Inc., cat. no. 9001292) using manufacturer's kit instructions. Total RNA quality and concentration for human fetal liver samples was assessed using Agilent BioAnalyzer. Total RNA quality and concentration for mouse liver samples was assessed using a ND1000 spectrophotometer. For each sample, approximately 500 ng of total RNA was used with the iScript cDNA Synthesis Kit (Bio-Rad, cat. no. 170-8891) to generate cDNA for downstream real-time qPCR (RT-qPCR). Primer sequences for qPCR are listed in . Reference genes were empirically confirmed to be invariant across treatment groups during data analysis. All RT-qPCR primers were verified for product uniqueness using Primer BLAST (www.ncbi.nlm.hih.gov/tools/primer-blast/). Most primers span introns or exon-exon junctions to avoid amplification of genomic DNA. All primer sets were tested for PCR efficiency using 10-fold serial dilutions of a standard template and plotting of standard curves. Amplification of cDNA was carried out in 25 µl reactions using iQ SYBR Green Supermix (Bio-Rad, cat. no. 170-8880). Each reaction contained 1 µl of cDNA template and 100 nM primer concentration. All experimental and reference reactions were run in duplicate on a C100 Thermal Cycler with a CFX96 Real-Time head (Bio-Rad, cat. no. 185-1148). Each plate contained no template controls for each primer set, as well as 2 reference genes per species per assay (Gapdh and B2m for mouse samples, GAPDH and UBC for human samples). The average threshold cycle (C(t)) from duplicate reactions was calculated for individual genes using CFX Manager Version 1.6 software (Bio-Rad, cat. no. 184-5000). Expression values were normalized to endogenous reference genes by first averaging C(t) values for both reference genes for each sample and then subtracting the average reference value from the experimental C(t) value.
Table 4. Primer sets for mRNA expression analysis for Stat3, STAT3, and reference genes via qPCR.
Data Analysis
Analysis of bisulfite sequencing data in mouse liver samples
Hypothesized dose-dependent DNA methylation changes in 10-month mice with tumors, as compared to 10-month mice without tumors, as well as dose-dependent DNA methylation changes at the same epigenetic loci in sibling 3-week mice from the same exposure study were assessed using repeated measures mixed models (in SAS PROC MIXED). In the models, individual CpG sites were treated as repeated measures in order to account for lack of independence of multiple CpG sites at a single locus within an individual animal. Random intercepts for litter were included in the model to account for correlation of methylation for animals within litter. Fixed effects of the dependent variable assessed associations between dose and tumor status with average methylation at a locus. Dose variables were assessed on a natural scale, rather than an ordinal scale, to more closely model the true exposure and to facilitate practical interpretation of results. As natural scale dose variable coding has increased power to detect dose-based differences as compared to ordinal scale dose variable coding, results were confirmed with additional models run with ordinal scale dose variables, to facilitate easy comparison with a prior study using an ordinal scale dose variable for statistical analyses.Citation3
We tested for the presence of dose-dependent DNA methylation changes by tumor status in 10-month mouse liver samples (n = 78; ), both with and without liver tumors, using a total of 12 models, including 4 models per candidate gene (dependent variable as average DNA methylation): Model 1 contained 4 predictors, including a repeated measure variable for percent methylation at each CpG site, a natural scale dose predictor, a dichotomous tumor status variable, and an interaction variable (dose*tumor), to test for dose-dependent differences in average DNA methylation by tumor status; Model 2 included repeated CpG site predictor and dose predictor, and adjusted for both sex and tumor status to assess dose-dependent changes in average DNA methylation independent of tumor status; Model 3 included the previous predictors but contained an alternate interaction variable (dose*CpG site) to assess site-specific differences in DNA methylation; and the fourth model tested for a monotonic trend in DNA methylation with dose.
To investigate dose-dependent DNA methylation patterns in sibling PND22 mice from the same exposure study (n = 147; ), we ran a total of 9 final models on PND22 mouse liver samples, including 3 models per candidate gene (dependent variable as average DNA methylation): Model 1 contained 2 predictors, including a repeated measure variable for percent methylation at each CpG site and a natural scale dose predictor; Model 2 contained the previous 2 predictors and an additional interaction variable (dose*CpG site) to assess site-specific DNA methylation changes with dose; and Model 3 testing for monotonic trend in DNA methylation change with dose. Mixed models for PND22 mouse liver samples were adjusted for sex and genotype and included a random intercept to cluster data by litter. Genotype was included in the final models as a potential confounder after confirmation that an interaction variable (dose*genotype) was not significant at any candidate gene, indicating that DNA methylation did not vary across genotypes with exposure.
Analysis of bisulfite sequencing data in human fetal liver samples
Hypothesized dose-dependent DNA methylation changes in human fetal liver samples at human homologs of murine candidate genes were assessed using the 4 repeated measures mixed models described above, with individual CpG sites defined as repeated measures, in order to account for lack of independence of multiple CpG sites at a single locus within an individual. Similarly, fixed effects of the dependent variable assessed associations between dose and tumor status with average methylation at a locus. BPA exposure was coded both as a categorical variable (binned into tertiles) and a continuous variable, for both free and total BPA measurements. Free and conjugated BPA liver tissue measurements were performed in these samples by Nahar et al. (2013), as previously described. A total of 4 models were run for candidate genes ESR1 and IL-6ST (dependent variable average methylation). The first 2 models contained 4 predictors, including a repeated measure variable for percent methylation at each CpG site, and categorical dose predictor, one model each for total and free BPA measurements, as well as a dichotomous sex variable and continuous gestational age variable, to adjust for potential confounding. The second 2 models contained the same 4 predictors, albeit with continuous dose predictors, one model each for total and free BPA measurements. A total of 6 models were run on candidate gene STAT3. As continuous free and total BPA were significantly associated with average methylation at STAT3, 2 additional models were run containing quadratic dose terms [(centered free BPA)Citation2 and (centered total BPA),Citation2 respectively] to assess the presence of a parabolic, non-monotonic dose-response curve visually apparent at CpG site 1 with categorical BPA predictors.
Analysis of mRNA expression data
The average threshold cycle (C(t)) from duplicate reactions was calculated for individual genes using CFX Manager Version1.6 software (Bio-Rad). Expression values were normalized to endogenous reference genes to obtain ΔC(t) values by first averaging C(t) values for both reference genes for each sample and then subtracting the average reference value from the experimental C(t) value. Expression values for Stat3 or STAT3 were then assessed for association with perinatal BPA exposure (ΔΔC(t)) via simple linear regression models (PROC REG). Regression models for mouse samples were run separately for 10-month and PND22 mice, and were adjusted for sex and tumor status in the first and for sex and genotype in the second. Dose-dependent differences in expression of Stat3 in 10-month animals by tumor status were assessed by including an interaction variable (dose*tumor status) in subsequent models. Regression models for human samples were adjusted for sex and gestational age. BPA exposure was coded both as a categorical variable (binned into tertiles) and a continuous variable, for both free and total BPA measurements. Each endogenous reference gene was analyzed in separate linear regression models, to empirically confirm references to be invariant by BPA exposure. Regression coefficients (β coefficients) were used to calculate fold-change values [100(eˆβ-1)], with P-values <0.05 reported as statistically significant differences. All statistical analyses were performed in SAS v9.2.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
KEPI_A_1107694_Supplemental.pdf
Download PDF (695.1 KB)Acknowledgments
This work was supported by a pilot grant from the Michigan Nutrition Research Obesity Center, MNORC (P30 DK089503), as well as NIH grant R01 ES017524 and the Michigan Lifestage Environmental Exposures and Disease (M-LEEaD) NIEHS Center (P30 ES017885). Support for CW was provided by NIEHS Institutional Training Grant T32 ES007062. The authors thank Dr. Brisa Sanchez for assistance with statistical analysis, Dr. Olivia Anderson for generation of the mouse samples, Dr. Muna Nahar and Dr. Kurunthachalam Kannan for assistance with the human samples, Dr. Maureen Sartor for critical review of the manuscript, and the University of Washington Laboratory for the Study of Human Embryology (2R24 HD000836-47) for human tissue samples.
Related Research Data
References
- Dolinoy DC, Weidman JR, Waterland RA, Jirtle RL. Maternal genistein alters coat color and protects Avy mouse offspring from obesity by modifying the fetal epigenome. Environ Health Perspect 2006; 114:567-72; PMID:16581547; http://dx.doi.org/10.1289/ehp.8700
- Prins GS, Tang W-Y, Belmonte J, Ho S-M. Perinatal exposure to oestradiol and bisphenol A alters the prostate epigenome and increases susceptibility to carcinogenesis. Basic Clin Pharmacol Toxicol 2008; 102:134-8; PMID:18226066; http://dx.doi.org/10.1111/j.1742-7843.2007.00166.x
- Weinhouse C, Anderson OS, Bergin IL, Vandenbergh DJ, Gyekis JP, Dingman MA, Yang J, Dolinoy DC. Dose-dependent incidence of hepatic tumors in adult mice following perinatal exposure to bisphenol A. Environ Health Perspect 2014; 122:485-91; PMID:24487385; http://dx.doi.org/10.1289/ehp.1307449
- Dhimolea E, Wadia PR, Murray TJ, Settles ML, Treitman JD, Sonnenschein C, Shioda T, Soto AM. Prenatal exposure to BPA alters the epigenome of the rat mammary gland and increases the propensity to neoplastic development. PLoS One 2014; 9:e99800; PMID:24988533; http://dx.doi.org/10.1371/journal.pone.0099800
- Prins GS, Hu W-Y, Shi G-B, Hu D-P, Majumdar S, Li G, Huang K, Nelles JL, Ho S-M, Walker CL, et al. Bisphenol A promotes human prostate stem-progenitor cell self-renewal and increases in vivo carcinogenesis in human prostate epithelium. Endocrinology 2014; 155:805-17; PMID:24424067; http://dx.doi.org/10.1210/en.2013-1955
- Prins GS, Hu W-Y, Shi G-B, Hu D-P, Majumdar S, Li G, Huang K, Nelles JL, Ho S-M, Walker CL, et al. Bisphenol A promotes human prostate stem-progenitor cell self-renewal and increases in vivo carcinogenesis in human prostate epithelium. Endocrinology 2014; 155:805-17; PMID:24424067; http://dx.doi.org/10.1210/en.2013-1955
- Bassett SA, Barnett MPG. The Role of Dietary Histone Deacetylases (HDACs) Inhibitors in Health and Disease. Nutrients 2014; 6:4273-301; PMID:25322459; http://dx.doi.org/10.3390/nu6104273
- Shukla S, Meeran SM. Epigenetics of cancer stem cells: Pathways and therapeutics. Biochim Biophys Acta 2014; 1840:3494-502; PMID:25240776; http://dx.doi.org/10.1016/j.bbagen.2014.09.017
- Sandoval J, Peiró-Chova L, Pallardó FV, García-Giménez JL. Epigenetic biomarkers in laboratory diagnostics: emerging approaches and opportunities. Expert Rev Mol Diagn 2013; 13:457-71; PMID:23782253; http://dx.doi.org/10.1586/erm.13.37
- Manikkam M, Guerrero-Bosagna C, Tracey R, Haque MM, Skinner MK. Transgenerational actions of environmental compounds on reproductive disease and identification of epigenetic biomarkers of ancestral exposures. PLoS One 2012; 7:e31901; PMID:22389676; http://dx.doi.org/10.1371/journal.pone.0031901
- Weinhouse C, Anderson OS, Jones TR, Kim J, Liberman SA, Nahar MS, Rozek LS, Jirtle RL, Dolinoy DC. An expression microarray approach for the identification of metastable epialleles in the mouse genome. Epigenetics 2011; 6:1105-13; PMID:21829099; http://dx.doi.org/10.4161/epi.6.9.17103
- Dominguez-Salas P, Moore SE, Baker MS, Bergen AW, Cox SE, Dyer RA, Fulford AJ, Guan Y, Laritsky E, Silver MJ, et al. Maternal nutrition at conception modulates DNA methylation of human metastable epialleles. Nat Commun 2014; 5:3746; PMID:24781383; http://dx.doi.org/10.1038/ncomms4746
- Dai B, Geng L, Yu Y, Sui C, Xie F, Shen W, Zheng T, Yang J. Methylation patterns of estrogen receptor α promoter correlate with estrogen receptor α expression and clinicopathological factors in hepatocellular carcinoma. Exp Biol Med (Maywood) 2014; 239:883-90; PMID:24939822; http://dx.doi.org/10.1177/1535370214536651
- Lau MY, Han H, Hu J, Ji C. Association of cyclin D and estrogen receptor α36 with hepatocellular adenomas of female mice under chronic endoplasmic reticulum stress. J Gastroenterol Hepatol 2013; 28:576-83; PMID:23216077; http://dx.doi.org/10.1111/jgh.12084
- Liu W-H, Yeh S-H, Lu C-C, Yu S-L, Chen H-Y, Lin C-Y, Chen D-S, Chen P-J. MicroRNA-18a prevents estrogen receptor-alpha expression, promoting proliferation of hepatocellular carcinoma cells. Gastroenterology 2009; 136:683-93; PMID:19027010; http://dx.doi.org/10.1053/j.gastro.2008.10.029
- Kazantseva YA, Yarushkin AA, Pustylnyak VO. Dichlorodiphenyltrichloroethane technical mixture regulates cell cycle and apoptosis genes through the activation of CAR and ERα in mouse livers. Toxicol Appl Pharmacol 2013; 271:137-43; PMID:23684557; http://dx.doi.org/10.1016/j.taap.2013.05.008
- Bioulac-Sage P, Balabaud C, Zucman-Rossi J. Subtype classification of hepatocellular adenoma. Dig Surg 2010; 27:39-45; PMID:20357450; http://dx.doi.org/10.1159/000268406
- Rosell R, Bertran-Alamillo J, Molina MA, Taron M. IL-6/gp130/STAT3 signaling axis in cancer and the presence of in-frame gp130 somatic deletions in inflammatory hepatocellular tumors. Future Oncol 2009; 5:305-8; PMID:19374537; http://dx.doi.org/10.2217/fon.09.3
- Shi L, Feng Y, Lin H, Ma R, Cai X. Role of estrogen in hepatocellular carcinoma: is inflammation the key? J Transl Med 2014; 12:93; PMID:24708807; http://dx.doi.org/10.1186/1479-5876-12-93
- Umemura A, Park EJ, Taniguchi K, Lee JH, Shalapour S, Valasek MA, Aghajan M, Nakagawa H, Seki E, Hall MN, et al. Liver damage, inflammation, and enhanced tumorigenesis after persistent mTORC1 inhibition. Cell Metab 2014; 20:133-44; PMID:24910242; http://dx.doi.org/10.1016/j.cmet.2014.05.001
- Rebouissou S, Amessou M, Couchy G, Poussin K, Imbeaud S, Pilati C, Izard T, Balabaud C, Bioulac-Sage P, Zucman-Rossi J. Frequent in-frame somatic deletions activate gp130 in inflammatory hepatocellular tumours. Nature 2009; 457:200-4; PMID:19020503; http://dx.doi.org/10.1038/nature07475
- Shi L, Feng Y, Lin H, Ma R, Cai X. Role of estrogen in hepatocellular carcinoma: is inflammation the key? J Transl Med 2014; 12:93; PMID:24708807; http://dx.doi.org/10.1186/1479-5876-12-93
- Sekine Y, Yamamoto T, Yumioka T, Imoto S, Kojima H, Matsuda T. Cross-talk between endocrine-disrupting chemicals and cytokine signaling through estrogen receptors. Biochem Biophys Res Commun 2004; 315:692-8; PMID:14975756; http://dx.doi.org/10.1016/j.bbrc.2004.01.109
- Kim JH, Sartor MA, Rozek LS, Faulk C, Anderson OS, Jones TR, Nahar MS, Dolinoy DC. Perinatal bisphenol A exposure promotes dose-dependent alterations of the mouse methylome. BMC Genomics 2014; 15:30; PMID:24433282; http://dx.doi.org/10.1186/1471-2164-15-30
- Vandenberg LN, Ehrlich S, Belcher SM, Ben-Jonathan N, Dolinoy DC, Hugo ER, Hunt PA, Newbold RR, Rubin BS, Saili KS, et al. Low dose effects of bisphenol A: An integrated review of in vitro, laboratory animal, and epidemiology studies. Endocr Disruptors 2013; 1;e26490; http://dx.doi.org/10.4161/endo.26490
- Kao J-T, Feng C-L, Yu C-J, Tsai S-M, Hsu P-N, Chen Y-L, Wu Y-Y. IL-6, through p-STAT3 rather than p-STAT1, activates hepatocarcinogenesis and affects survival of hepatocellular carcinoma patients: a cohort study. BMC Gastroenterol 2015; 15:50; PMID:25908103; http://dx.doi.org/10.1186/s12876-015-0283-5
- Fleming JD, Giresi PG, Lindahl-Allen M, Krall EB, Lieb JD, Struhl K. STAT3 acts through pre-existing nucleosome-depleted regions bound by FOS during an epigenetic switch linking inflammation to cancer. Epigenetics Chromatin 2015; 8:7; PMID:25784959; http://dx.doi.org/10.1186/1756-8935-8-7
- Tai W-T, Chu P-Y, Shiau C-W, Chen Y-L, Li Y-S, Hung M-H, Chen L-J, Chen P-L, Su J-C, Lin P-Y, et al. STAT3 Mediates Regorafenib-Induced Apoptosis in Hepatocellular Carcinoma. Clin Cancer Res 2014; 20:5768-76; PMID:25248379; http://dx.doi.org/10.1158/1078-0432.CCR-14-0725
- Mohan CD, Bharathkumar H, Bulusu KC, Pandey V, Rangappa S, Fuchs JE, Shanmugam MK, Dai X, Li F, Deivasigamani A, et al. Development of a Novel Azaspirane That Targets the Janus Kinase-Signal Transducer and Activator of Transcription (STAT) Pathway in Hepatocellular Carcinoma in Vitro and in Vivo. J Biol Chem 2014; 289:34296-307; PMID:25320076; http://dx.doi.org/10.1074/jbc.M114.601104
- Chen H, Aksoy I, Gonnot F, Osteil P, Aubry M, Hamela C, Rognard C, Hochard A, Voisin S, Fontaine E, et al. Reinforcement of STAT3 activity reprogrammes human embryonic stem cells to naive-like pluripotency. Nat Commun 2015; 6:7095; PMID:25968054; http://dx.doi.org/10.1038/ncomms8095
- Arumuggam N, Bhowmick NA, Rupasinghe HPV. A Review: Phytochemicals Targeting JAK/STAT Signaling and IDO Expression in Cancer. Phytother Res 2015; 29(6):805-17; PMID:25787773
- Anderson OS, Nahar MS, Faulk C, Jones TR, Liao C, Kannan K, Weinhouse C, Rozek LS, Dolinoy DC. Epigenetic responses following maternal dietary exposure to physiologically relevant levels of bisphenol A. Environ Mol Mutagen 2012; 53(5):334-42
- Carninci P, Kasukawa T, Katayama S, Gough J, Frith MC, Maeda N, Oyama R, Ravasi T, Lenhard B, Wells C, et al. The transcriptional landscape of the mammalian genome. Science 2005; 309:1559-63; PMID:16141072; http://dx.doi.org/10.1126/science.1112014
- Carninci P, Sandelin A, Lenhard B, Katayama S, Shimokawa K, Ponjavic J, Semple CAM, Taylor MS, Engström PG, Frith MC, et al. Genome-wide analysis of mammalian promoter architecture and evolution. Nat Genet 2006; 38:626-35; PMID:16645617; http://dx.doi.org/10.1038/ng1789
- Maunakea AK, Nagarajan RP, Bilenky M, Ballinger TJ, D'Souza C, Fouse SD, Johnson BE, Hong C, Nielsen C, Zhao Y, et al. Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature 2010; 466:253-7; PMID:20613842; http://dx.doi.org/10.1038/nature09165
- Demars J, Shmela ME, Khan AW, Lee KS, Azzi S, Dehais P, Netchine I, Rossignol S, Le Bouc Y, El-Osta A, et al. Genetic variants within the second intron of the KCNQ1 gene affect CTCF binding and confer a risk of Beckwith-Wiedemann syndrome upon maternal transmission. J Med Genet 2014; 51:502-11; PMID:24996904; http://dx.doi.org/10.1136/jmedgenet-2014-102368
- Hoivik EA, Bjanesoy TE, Mai O, Okamoto S, Minokoshi Y, Shima Y, Morohashi K-I, Boehm U, Bakke M. DNA methylation of intronic enhancers directs tissue-specific expression of steroidogenic factor 1/adrenal 4 binding protein (SF-1/Ad4BP). Endocrinology 2011; 152:2100-12; PMID:21343250; http://dx.doi.org/10.1210/en.2010-1305
- Rademacher K, Schröder C, Kanber D, Klein-Hitpass L, Wallner S, Zeschnigk M, Horsthemke B. Evolutionary origin and methylation status of human intronic CpG islands that are not present in mouse. Genome Biol Evol 2014; 6:1579-88; PMID:24923327; http://dx.doi.org/10.1093/gbe/evu125
- Waterland RA, Jirtle RL. Transposable elements: targets for early nutritional effects on epigenetic gene regulation. Mol Cell Biol 2003; 23:5293-300; PMID:12861015; http://dx.doi.org/10.1128/MCB.23.15.5293-5300.2003
- Dolinoy DC, Huang D, Jirtle RL. Maternal nutrient supplementation counteracts bisphenol A-induced DNA hypomethylation in early development. Proc Natl Acad Sci U S A 2007; 104:13056-61; PMID:17670942; http://dx.doi.org/10.1073/pnas.0703739104
- Raggi C, Invernizzi P. Methylation and liver cancer. Clin Res Hepatol Gastroenterol 2013; 37:564-71; PMID:23806627; http://dx.doi.org/10.1016/j.clinre.2013.05.009
- Anderson OS, Nahar MS, Faulk C, Jones TR, Liao C, Kannan K, Weinhouse C, Rozek LS, Dolinoy DC. Epigenetic responses following maternal dietary exposure to physiologically relevant levels of bisphenol A. Environ Mol Mutagen 2012; 53(5):334-42; PMID: 22467340
- Murphy SK, Adigun A, Huang Z, Overcash F, Wang F, Jirtle RL, Schildkraut JM, Murtha AP, Iversen ES, Hoyo C. Gender-specific methylation differences in relation to prenatal exposure to cigarette smoke. Gene 2012; 494:36-43; PMID:22202639; http://dx.doi.org/10.1016/j.gene.2011.11.062
- Ball MP, Li JB, Gao Y, Lee J-H, LeProust EM, Park I-H, Xie B, Daley GQ, Church GM. Targeted and genome-scale strategies reveal gene-body methylation signatures in human cells. Nat Biotechnol 2009; 27:361-8; PMID:19329998; http://dx.doi.org/10.1038/nbt.1533
- Brenet F, Moh M, Funk P, Feierstein E, Viale AJ, Socci ND, Scandura JM. DNA methylation of the first exon is tightly linked to transcriptional silencing. PLoS One 2011; 6:e14524; PMID:21267076; http://dx.doi.org/10.1371/journal.pone.0014524
- Cokus SJ, Feng S, Zhang X, Chen Z, Merriman B, Haudenschild CD, Pradhan S, Nelson SF, Pellegrini M, Jacobsen SE. Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature 2008; 452:215-9; PMID:18278030; http://dx.doi.org/10.1038/nature06745
- Flanagan JM, Wild L. An epigenetic role for noncoding RNAs and intragenic DNA methylation. Genome Biol 2007; 8:307; PMID:17601356; http://dx.doi.org/10.1186/gb-2007-8-6-307
- Rauch TA, Wu X, Zhong X, Riggs AD, Pfeifer GP. A human B cell methylome at 100-base pair resolution. Proc Natl Acad Sci U S A 2009; 106:671-8; PMID:19139413; http://dx.doi.org/10.1073/pnas.0812399106
- Gelfman S, Cohen N, Yearim A, Ast G. DNA-methylation effect on cotranscriptional splicing is dependent on GC architecture of the exon-intron structure. Genome Res 2013; 23:789-99; PMID:23502848; http://dx.doi.org/10.1101/gr.143503.112
- Anderson OS, Peterson KE, Sanchez BN, Zhang Z, Mancuso P, Dolinoy DC. Perinatal bisphenol A exposure promotes hyperactivity, lean body composition, and hormonal responses across the murine life course. FASEB J 2013; 27(4):1784-92; PMID: 23345456
- Nahar MS, Liao C, Kannan K, Dolinoy DC. Fetal liver bisphenol a concentrations and biotransformation gene expression reveal variable exposure and altered capacity for metabolism in humans. J Biochem Mol Toxicol 2013; 27:116-23; PMID:23208979; http://dx.doi.org/10.1002/jbt.21459
- Vandenberg LN, Chahoud I, Heindel JJ, Padmanabhan V, Paumgartten FJR, Schoenfelder G. Urinary, circulating, and tissue biomonitoring studies indicate widespread exposure to bisphenol A. Cien Saude Colet 2012; 17:407-34; PMID:22267036; http://dx.doi.org/10.1590/S1413-81232012000200015