
Abstract
Facioscapulohumeral muscular dystrophy is caused by incomplete epigenetic repression of the transcription factor DUX4 in skeletal muscle. A copy of DUX4 is located within each unit of the D4Z4 macrosatellite repeat array and its derepression in somatic cells is caused by either repeat array contraction (FSHD1) or by mutations in the chromatin repressor SMCHD1 (FSHD2). While DUX4 expression has thus far only been detected in FSHD muscle and muscle cell cultures, and increases with in vitro myogenic differentiation, the D4Z4 chromatin structure has only been studied in proliferating myoblasts or non-myogenic cells. We here show that SMCHD1 protein levels at D4Z4 decline during muscle cell differentiation and correlate with DUX4 derepression. In FSHD2, but not FSHD1, the loss of SMCHD1 repressor activity is partially compensated by increased Polycomb Repressive Complex 2 (PRC2)–mediated H3K27 trimethylation at D4Z4, a situation that can be mimicked by SMCHD1 knockdown in control myotubes. In contrast, moderate overexpression of SMCHD1 results in DUX4 silencing in FSHD1 and FSHD2 myotubes demonstrating that DUX4 derepression in FSHD is reversible. Together, we show that in FSHD1 and FSHD2 the decline in SMCHD1 protein levels during muscle cell differentiation renders skeletal muscle sensitive to DUX4.
Introduction
Facioscapulohumeral dystrophy (FSHD, OMIM 158900/158901) is characterized by progressive and often asymmetrical weakness of facial and upper extremity muscles typically starting in the second decade of life.Citation1 FSHD is one of the most common myopathies with a recently reported prevalence of 12:100,000 in the Dutch population and is inherited in an autosomal dominant fashion in most families.Citation2 FSHD is genetically linked to the polymorphic D4Z4 macrosatellite repeat array at 4q35, which in healthy subjects consists of 11-100 units, each of them being 3.3 kb in size and containing a copy of the DUX4 retrogene.Citation3-5
The prevailing FSHD disease model postulates that DUX4 is normally silenced in somatic cells by repeat mediated epigenetic repression and that failure of this mechanism leads to partial decompaction of the D4Z4 chromatin structure and the expression of DUX4 in skeletal muscle of FSHD individuals.Citation6 Stable DUX4 expression requires a specific genetic background of chromosome 4 that contains a polymorphic DUX4 polyadenylation signal (PAS) immediately distal to the D4Z4 repeat array.Citation7 Partial D4Z4 chromatin decompaction on a DUX4-PAS containing chromosome leads to the FSHD-specific pattern of DUX4 expression in myotube cultures with few myonuclei expressing relatively abundant amounts of DUX4 protein.Citation8 Expression of the germline double homeobox transcription factor DUX4 is harmful to skeletal muscle since it activates germline and early stem cell programs, and modulates inflammatory pathways eventually resulting in increased cell death.Citation9-11
In >95% of FSHD individuals (FSHD1) incomplete repression of DUX4 can be attributed to a contraction of the D4Z4 repeat array to a size of 1-10 units, whereas in the remainder of individuals with FSHD (FSHD2) the D4Z4 repeat arrays are in the normal size range.Citation5,12,13 The majority of FSHD2 individuals carry heterozygous mutations in the structural maintenance of chromosomes hinge domain 1 (SMCHD1) gene on chromosome 18.Citation14 SMCHD1 is a chromatin repressor that binds to D4Z4 and germline mutations in SMCHD1 result in reduced D4Z4 chromatin compaction and DUX4 expression.Citation14 Moreover, SMCHD1 was shown to act as a modifier of disease severity in FSHD1Citation15 and the functional consequences of SMCHD1 mutations, in combination with D4Z4 repeat size, determines the disease penetrance.Citation16
Several lines of evidence indicate that epigenetic derepression of the D4Z4 macrosatellite repeat array in FSHD results in DUX4 expression in skeletal muscle cells.Citation17,18 In somatic cells originating from healthy individuals, such as myoblasts, the D4Z4 repeat array is characterized by high, but inhomogeneous levels of CpG methylation and histone 3 lysine 9 trimethylation (H3K9me3).Citation18-22 In proliferating FSHD somatic cell cultures, a reduction of both CpG methylation and H3K9me3 at D4Z4 has been reported together with reduced cohesin and heterochromatin protein 1 γ (HP1γ) levels, presumably facilitating sporadic activation of DUX4.Citation18 In addition, members of the Polycomb Repressive Complex 2 (PRC2) and the histone modification deposited by the PRC2 complex, H3K27me3, were identified at D4Z4 in control and FSHD1 proliferating myoblasts.Citation17,18,23 The mechanism of PRC2 recruitment remains however to be determined. D4Z4 non-coding RNAs may have a role, since the endogenous RNAi pathway was recently demonstrated to facilitate DICER/AGO-dependent epigenetic silencing of the D4Z4 repeat arrays in muscle cells. Citation24,25, Citation26
Although DUX4 expression has repeatedly been shown to be increased in FSHD muscle cells upon in vitro differentiation,Citation8,27-29 data on the epigenetic regulation of the D4Z4 repeat array during this process is scarce. Nevertheless, extensive changes in the genome wide chromatin structure have been reported upon myogenic differentiation in mouse muscle cells. In particular, the Polycomb complexes were shown to be pivotal for the regulation of lineage commitment and differentiation specific genes.Citation30 Moreover, multi-nucleated myotubes more closely resemble the in vivo structural composition of muscle than proliferating myoblasts. Taken together, differentiated myotubes are likely a more relevant in vitro model to study the epigenetic requirements for DUX4 expression than proliferating myoblasts.
This prompted us to examine whether we could identify changes in D4Z4 chromatin regulation specific to differentiated muscle cells that might account for the susceptibility of this tissue to express DUX4 at higher levels than in myoblasts or many other tissues. Our study highlights the importance of SMCHD1 in regulating D4Z4 chromatin structure and DUX4 expression in muscle cells of both FSHD1 and FSHD2. Whereas a decline in SMCHD1 protein levels, and specifically its reduction at D4Z4, during skeletal muscle differentiation leads to DUX4 expression, ectopic expression of SMCHD1 in either FSHD1 or FSHD2 muscle cell cultures efficiently suppresses DUX4 expression. Moreover, in FSHD2 but not in FSHD1, the loss of SMCHD1 is accompanied by a gain in PRC2-mediated H3K27 trimethylation, likely as a compensatory but incomplete mechanism to silence DUX4 in skeletal muscle.
Results
DUX4 expression is induced upon myogenic differentiation and coincides with reduced SMCHD1 levels
Although DUX4 expression has been reported repeatedly in FSHD myotubes,Citation8,27,29 comprehensive data on the regulation of DUX4 expression in FSHD1 and in particular on FSHD2 muscle cells is scarce. Therefore, our first aim was to firmly establish that DUX4 expression increases with differentiation of FSHD1 and FSHD2 muscle cells. We cultured a large set of primary myoblasts derived from control, FSHD1 and FSHD2 individuals and harvested RNA from both proliferating myoblasts and differentiating myotubes (further referred to as myoblasts and myotubes). The differentiation status of the cultures was confirmed by myotube formation, as visualized by immunofluorescent labeling of myosin, and MYOG transcript levels (, Fig. S1 and S2). Multinucleated myofibers were observed in all conditions and differentiation was confirmed by the increase in MYOG levels in control, FSHD1, and FSHD2 muscle cell cultures.
Figure 1.(See previous page) DUX4 activation during myogenic differentiation coincides with SMCHD1 reduction. (A) Immunofluorescence microscopy analysis confirmed the typical DUX4 protein expression pattern in myosin positive, multinucleated myotubes derived from FSHD1 and FSHD2 individuals. Images were taken using a 200x magnification, scale bars are displayed. (B) Quantitative mRNA analysis in control (C), FSHD1 (F1), and FSHD2 (F2) primary myoblasts (B) and myotubes (T) showing increased levels of DUX4 expression upon differentiation. GAPDH and GUSB were used as reference genes, n indicates number of samples, error bars display SD, and significance was calculated using a 2-tailed Student t-test. (C) Quantitative mRNA analysis showed robust ZSCAN4 activation upon myogenic differentiation in FSHD myotubes. GAPDH and GUSB were used as reference genes, n indicates number of samples, error bars display SD, and significance was calculated using a 2-tailed Student t-test. (D) Western blot analysis showing reduced levels of SMCHD1 upon muscle cell differentiation in a primary muscle cell culture derived from a control individual. Myoblasts (B) were differentiated into myotubes (T) for 48 h. H3 serves as a control for equal protein loading, α-ACTIN serves as a control for the induction of myogenic differentiation. (E) Western blot analysis of a control derived primary fibroblast undergoing forced myogenesis by ectopic MyoD expression for 48, 72, or 94 h showed a decrease in SMCHD1 protein expression. TUBULIN serves as a loading control. (F) Duplicate RT-PCR analysis of DUX4 expression upon ectopic MyoD expression in a Control 1, FSHD1 1 or FSHD2 1 fibroblast (Table S2) revealed DUX4 activation in patient cells exclusively. Ectopic GFP expression was used as a control and GUSB serves as an internal PCR control. (G, H) Normalized ChIP qPCR analysis of SMCHD1 binding at D4Z4 in isogenic fibroblasts (F), myoblasts (B), and myotubes (T) derived from the same control individual (panel G) and 3 independent control derived myoblast – myotube pairs showed the highest SMCHD1 abundance in myoblasts with a further decrease during myogenesis. Error bars display SD in panel G and H and significance was calculated using a 2-tailed Student's test. NS = not significant; * = P <0.05; ** = P<0.005.
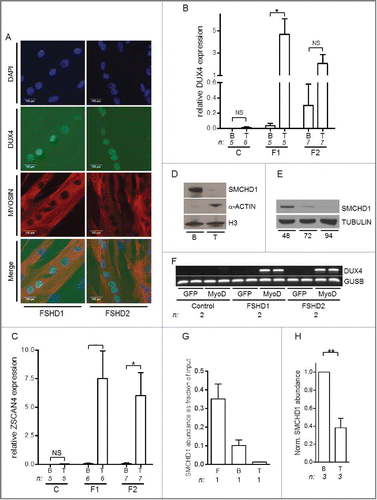
mRNA quantification by qRT-PCR confirmed the activation of DUX4 in FSHD1 and FSHD2 myotubes, whereas we could not reproducibly detect DUX4 transcripts in control samples (). While the increase of DUX4 expression during myotube formation in FSHD1 cells was statistically significant, FSHD2 cells were more variable. In FSHD2 cells, an increase of DUX4 during myogenesis can be observed; however, FSHD2 muscle cell cultures already show variable levels of DUX4 transcripts before differentiation, which may explain the non-significant effect (). Immunofluorescence microscopy on a subset of myotube cultures established the variegated pattern of DUX4 protein expression in both forms of the disease (, Fig. S2). We also quantified expression levels of the previously established DUX4 target gene ZSCAN4 and observed FSHD-specific induction during myoblast differentiation ().Citation9
Since we previously demonstrated that SMCHD1 is a D4Z4 chromatin repressor,Citation14 we examined SMCHD1 protein levels. Western blot analysis showed a reduction in SMCHD1 protein levels after control myoblast differentiation (), a phenomenon that could be reproduced by forced myogenic differentiation of control fibroblasts transduced with the myogenic transcription factor MyoD (). Upon forced myogenesis by MyoD transduction in 2 FSHD1 and 2 FSHD2 fibroblast cell lines we observed induction of DUX4, whereas DUX4 remained undetectable in 2 control samples treated with MyoD ( and Fig. S3). Employing ChIP-qPCR in control fibroblast, myoblast and myotube cultures from the same donor we observed higher density of SMCHD1 at qD4Z4, located around the transcriptional start site of DUX4 (Fig. S4),Citation18 in fibroblasts as compared to myoblasts, with a further decrease in myotubes (). Analysis of 3 additional independent pairs of control myoblast and myotube cultures confirmed the significant decrease in SMCHD1 levels at D4Z4 after myoblast differentiation (). Together, we confirm the FSHD-specific induction of DUX4 expression upon myogenic differentiation in a large set of FSHD1 and FSHD2 primary myotube cultures and upon MyoD-induced differentiation in FSHD fibroblasts. Moreover, we demonstrate that DUX4 expression negatively correlates with SMCHD1 levels at D4Z4 in myogenic cell cultures, specifically in differentiated myotubes.
SMCHD1 levels control DUX4 expression in somatic cells
To establish a causal relationship between SMCHD1 protein levels and DUX4 expression in myotubes and SMCHD1 protein levels, we conducted lentiviral shRNA-mediated depletion experiments in differentiating primary control myoblast cultures (Controls 2 and 8; Table S1) carrying an FSHD-permissive DUX4-PAS containing 4A161 allele with D4Z4 repeat sizes varying between 20 and 32 units. Depletion of SMCHD1 resulted in robust activation of DUX4 expression (, C, S5A), which is in agreement with our earlier observations.Citation14 This phenomenon of DUX4 activation seems to be rather specific for SMCHD1 since knocking down other known chromatin repressors of D4Z4 in parallel experiments using the same control myoblast cultures did not lead to DUX4 expression. Upon knockdown of SUV39H1, a histone methyltransferase previously shown to be involved in establishing the H3K9me3 modification at D4Z4Citation18 we did not observe a reduction in relative abundance of H3K9me3 at D4Z4 (, Fig. S5, S6B), nor the transcriptional activation of DUX4, in 2 independent control myotube cultures (). Similarly, independent depletion of cohesin proteins SMC3 or RAD21 by a similar strategy did not cause a consistent activation of DUX4 either under these conditions (). In support, we did not see significant changes in H3K9me3 levels or RAD21 levels between control and FSHD muscle cell cultures, in myoblasts or myotubes (Fig. S7 and S8).
Figure 2. (See previous page) SMCHD1, but not SUV39H1 and Cohesin, regulates DUX4 expression. Western blot confirmation of (A) SMCHD1, (B) SUV39H1, (D) RAD21, and E) SMC3 knockdown in control myotubes expressing the indicated lentiviral transduced shRNAs. Tubulin was used as a loading control. Representative blots of at least duplicate experiments are shown. (C, F) Standard gel electrophoresis analysis of DUX4 expression upon lentiviral knockdown of SMCHD1, SUV39H1, RAD21 and SMC3 in control myotubes. Only depletion of SMCHD1 resulted in reproducible activation of DUX4 transcription. Representative gel photos are shown of at least duplicate experiments. The PCR fragment visible in one RAD21 knockdown was sequenced and shown to be the product of an a-specific amplification. (G) Western blot analysis confirms a 2-3 fold increase of SMCHD1 expression upon its lentiviral transduction in 2 FSHD1 and 1 FSHD2 myotube cultures. GFP transduced myotubes served as a negative control. Numbers indicate normalized relative expression levels of SMCHD1 using Tubulin as a loading control followed by setting normalized SMCHD1 levels of GFP transduced samples (only expressing endogenous SMCHD1) to 1. (H) Expression levels of DUX4 were significantly reduced upon ectopic expression of SMCHD1 in 2 FSHD1 and 2 FSHD2 myotube cultures. Relative DUX4 expression was calculated for each sample by normalization to GUSB and GAPDH housekeeping genes. Bars show values of each samples adjusted to the expression value of GFP transduced sample as 1. (I) Expression levels of the DUX4 target genes RFPL2, ZSCAN4, and TRIM43 showed a significant reduction upon ectopic expression of SMCHD1 in 2 FSHD1 and 2 FSHD2 myotubes, concordant with decreased DUX4 protein expression. Expression levels were normalized as described for panel Figure 1A. For panel H, I: Error bars display SD and significance was calculated using a 2-tailed Student's t-test. All P <0.0005 indicated by ***.
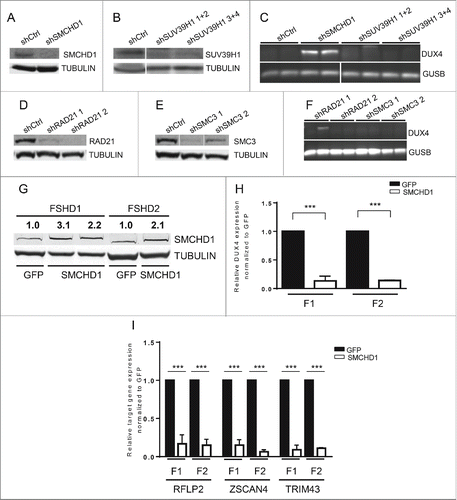
Since SMCHD1 levels decline at D4Z4 during myoblast differentiation, we next tested whether ectopic expression of SMCHD1 in FSHD myotubes can rescue the repression of DUX4. To this end, we transduced FSHD1 and FSHD2 myoblast cultures with SMCHD1 expressing lentiviruses and allowed the cells to form myotubes. In both FSHD1 and FSHD2 myotubes, 2-3-fold overexpression of SMCHD1, as determined by western blot analyses, resulted in a 70-90% reduction in DUX4 mRNA levels (). To confirm the silencing of DUX4, we analyzed the mRNA expression levels of 3 known downstream target genes of DUX4 protein. ZSCAN4, RFPL2, and TRIM43 expression levels decreased 70-90% upon overexpression of SMCHD1 (). To rule out that the lower DUX4 transcript levels were a consequence of reduced muscle cell differentiation, we measured MYOG transcript levels. MYOG increased similarly between the GFP expressing control and SMCHD1 expressing FSHD myotubes, with the exception of one FSHD2 myotube culture that showed substantially higher MYOG levels in the SMCHD1 expressing compared to control GFP expressing conditions (Fig. S9). This demonstrates that modest upregulation of SMCHD1 has no inhibiting effect on myogenic differentiation. In conclusion, DUX4 expression levels are strongly decreased upon moderate overexpression of SMCHD1, emphasizing its role as D4Z4 repressor in both genetic forms of the disease.
SMCHD1 reduction leads to PRC2 enrichment at D4Z4 in FSHD2 but not FSHD1
Since SMCHD1 was recently shown to associate with the repressive H3K27me3 modification at the inactive X chromosome,Citation31 we further determined the levels of H3K27me3 and the PRC2 complex at D4Z4 upon depletion of SMCHD1 in 2 independent control myotube cultures. SMCHD1 knockdown resulted in a 2-fold decrease in SMCHD1 levels at D4Z4 using 2 different SMCHD1 shRNA vectors () and increased H3K27me3 levels at D4Z4 (). Because of the increase in H3K27me3 at D4Z4 upon SMCHD1 depletion, we further analyzed the involvement of the PRC2 complex and observed a significant increase of the PRC2 protein SUZ12 at D4Z4 ().
Figure 3. SMCHD1 depletion at D4Z4 leads to increased H3K27me3 and PRC2 binding. (A) ChIP-qPCR analysis showed a statistically significant reduction of SMCHD1 at qD4Z4 upon its lentiviral knockdown in control myotubes with 2 independent shRNA constructs in 2 independent control myotube cultures. (B) ChIP-qPCR analysis of H3K27me3 at qD4Z4 upon SMCHD1 depletion showed a significant increase at qD4Z4. Enrichment values were normalized to H3 enrichment values. (C) Normalized ChIP-qPCR analysis of SUZ12 upon SMCHD1 depletion showed a statistically significant increase at qD4Z4 in 2 independent experiments performed on 2 control myoblast cultures. n indicates sample size, error bars indicate SD, and significance was tested with Student t-test. (D) ChIP-qPCR analysis of H3K27me3 at qD4Z4 showed a significant increase in FSHD2 myotubes compared to controls. Enrichment values were normalized to H3 enrichment values. (E) ChIP-qPCR analysis of SUZ12abundance showed a significant increase in FSHD2 myotubes at qD4Z4. On panel A, B, D, and E, n indicates sample size, error bars display SD, and significance was tested using a one way-ANOVA followed by Bonferroni multiple comparison test. *= P <0.05 **= P<0.005 ***= P<0.0005.
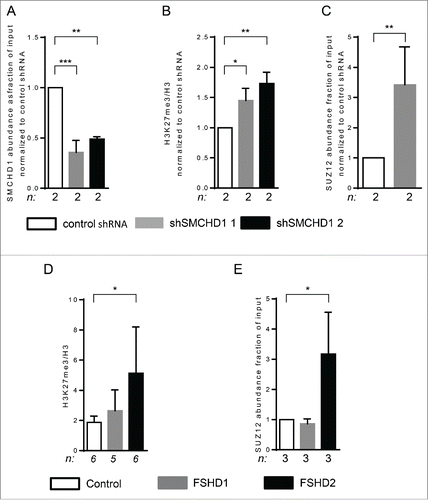
To validate these data derived from knock down experiments in control myotube cultures, we investigated the levels of PRC2 complex in primary control and FSHD myotubes. Previously, H3K27me3 levels at D4Z4 were reported to be similar in control and FSHD1 proliferating myoblast cultures at the DBE locus just proximal to qD4Z4 (Fig. S4).Citation24 We measured H3K27me3 levels at qD4Z4 and observed significantly higher levels of H3K27me3 in FSHD2 myotubes (), consistent with the increase we observed following knockdown of SMCHD1 (). Increased H3K27me3 levels were also observed in FSHD2 myoblasts (Fig. S10). Next, we quantified the levels of the PRC2 protein SUZ12 at D4Z4 and detected higher levels of SUZ12 at D4Z4 in FSHD2 samples (), all consistent with an increase in H3K27me3 through PRC2 recruitment at D4Z4.
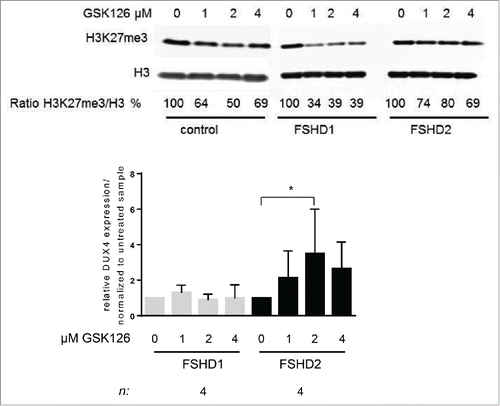
The decreased SMCHD1 activity in FSHD2 myotubes correlates with increased PRC2 levels at D4Z4. To test the repressive role of PRC2 at D4Z4, we inhibited PRC2 activity by treating a control, FSHD1, and FSHD2 muscle cell cultures with increasing concentrations of the EZH2 inhibitor GSK126.Citation32 After differentiation, total H3K27me3 levels were decreased in all cell cultures as shown by western blot analysis. DUX4 transcript levels increased significantly in FSHD2 samples treated at 2 μM GSK126, () suggesting that PRC2 is involved in the repression of D4Z4 in FSHD2, but not in FSHD1.
Figure 4. Treatment of FSHD2 myotube cultures with EZH2 inhibitor GSK126 increases DUX4 levels. (A) Western blot analysis of control, FSHD1, and FSHD2 myotube samples after treatment with 0, 1, 2, and 4 μM GSK126. Blots were probed with H3K27me3 antibody and H3 antibody as loading control. Shown are the reduced ratios of H3K27me3:H3 signal intensities in samples treated with GSK126 compared to the untreated sample as a result of EZH2 inhibition. (B) qRT-PCR analysis of DUX4 expression in FSHD1 and FSHD2 myotubes after GSK126 treatment shows that relative DUX4 transcript levels are significantly increased in FSHD2 samples treated with 2 μM GSK126 but not in FSHD1 myotubes. Graph shows the results of 4 FSHD1 and 4 FSHD2 cell lines. Results of control cell lines are not shown, DUX4 transcript was not detectable. Error bars show SD, n indicates number of independent cell lines, and significance was tested by 2-way ANOVA followed by Bonferroni multiple comparison test. *=P <0.05.
Discussion
Despite the recent advances in our understanding of common molecular disease mechanism in FSHD1 and FSHD2 and the consensus model of incomplete epigenetic repression of DUX4 in somatic cells, the key epigenetic changes at D4Z4 in FSHD1 and FSHD2, and the selective tissue involvement is only partly understood. FSHD is mainly a disease of skeletal muscle, suggesting that differentiated skeletal muscle might have an epigenetic state more permissive for DUX4 expression. In vitro, it was observed that DUX4 expression in FSHD muscle cells specifically increases upon myogenic differentiation. To date, however, most D4Z4 chromatin studies have focused on myoblast and non-myogenic cell cultures. The dynamics of the different epigenetic mechanisms involved in D4Z4 chromatin organization during muscle cell differentiation, and their causality to DUX4 expression, have not been studied in detail. Here we show that SMCHD1 protein levels decrease during muscle cell differentiation and correlate with DUX4 expression, possibly making differentiated muscle cells particularly prone to incomplete D4Z4 repression. Moreover we have identified the involvement of PRC2-mediated H3K27 trimethylation of D4Z4 selectively in FSHD2.
Thus far, most studies have been performed in FSHD1 muscle cell cultures, but we here firmly establish that in FSHD1 and FSHD2 DUX4 expression is strongly upregulated during muscle cell differentiation, emphasizing the importance of studying D4Z4 chromatin structure in differentiated muscle cells. Our observation of the decline in SMCHD1 levels during muscle cell differentiation may provide an explanation toward the susceptibility of muscle in expressing DUX4 and prompted us to further explore the role of SMCHD1 in somatic DUX4 repression. Depletion of SMCHD1 in control myotubes resulted in robust DUX4 expression. Conversely, 1.5-3-fold increases in SMCHD1 protein levels by ectopic expression in FSHD1 and FSHD2 myotubes led to a significant decrease in DUX4 levels and that of its target genes. This demonstrates that the derepression of DUX4 in FSHD muscle cells is a reversible process that can be rescued by increasing SMCHD1 levels.
SMCHD1 was recently shown to associate with H3K27me3 at the inactive X chromosome and we observed that loss of SMCHD1 at D4Z4 leads to an increase in PRC2 and H3K27me3 levels at D4Z4 in FSHD2. This strongly suggests that there is aberrant PRC2 regulation in FSHD2, but not FSHD1, myoblasts and myotubes. Since SMCHD1 has a role in the establishment and/or maintenance of CpG methylation, this observation consistent with recent studies showing an inverse relationship between CpG methylation and PRC2-mediated H3K27 methylation.Citation33 While the overall levels of some PRC2 complex proteins decline during murine myogenic differentiation,Citation34 our data suggest a local enrichment of PRC2 at D4Z4 in FSHD2 cells during this process.
Previous studies did not observe a difference in PRC2 and H3K27me3 at D4Z4 in FSHD. However, these studies were mostly restricted to FSHD1 myoblast cultures,Citation17,24 used primers that were not specific to chromosome 4,Citation17 or analyzed a single cell lineCitation35 which may explain this difference. Indeed, a recent study by Zeng et al. showed that although D4Z4 units are dispersed throughout the genome, epigenetically the D4Z4 repeat arrays on chromosomes 4 and 10 are distinctly regulated from their homologs.Citation35
Our data also identifies a difference between FSHD1 and FSHD2 in the epigenetic regulation of D4Z4. Depletion of SMCHD1 in control cells, mimicking FSHD2, leads to increased abundance of PRC2. The involvement of PRC2 in FSHD2 is further substantiated by the observation that GSK126 specifically increases DUX4 transcript levels in FSHD2, but not in FSHD1. Despite this epigenetic difference, SMCHD1 depletion and overexpression respectively leads to activation and repression of DUX4 in both FSHD1 and FSHD2 derived cells. It is unlikely that a selective involvement of a single contracted D4Z4 repeat array in FSHD1 creates a technical limitation in our ChIP approach (interrogating all 4 D4Z4 repeat arrays simultaneously) and explains the FSHD2-specificty of the involvement of PRC2 at D4Z4. Therefore, we conclude that although in both conditions DUX4 is derepressed, there are some unique epigenetic responses to repeat contraction or SMCHD1 malfunctioning in FSHD1 and FSHD2, respectively.
Increased PRC2 recruitment and H3K27me3 levels are generally associated with transcriptional repression.Citation36 However, in FSHD2 they are associated with the transcriptional derepression of the DUX4 locus. PRC2 recruitment to the derepressed D4Z4 region may reflect a rather effective failsafe mechanism to compensate for the loss of other repressive mechanisms. Indeed a compensatory repressive mechanism by PRC2 is supported by our observation of increased DUX4 transcript levels in FSHD2 myotubes in the presence of the EZH2 inhibitor GSK126. The specific nature of the trigger leading to the sporadic escape of repression typical for DUX4 remains unclear at this point and requires experimental approaches, which can dissect differences between single nuclei.
We could not confirm a role for SUV39H1 or the cohesin complex in DUX4 repression in a myogenic context: we did not observe different H3K9me3 or SMC3 levels in myotubes and knockdown of SUV39H1 and cohesin in myotubes also did not activate DUX4. Knockdown of SUV39H1 did not decrease H3K9me3 at D4Z4 in myotubes either. Previously, siRNA mediated knockdown of SUV39H1 led to reduced levels of H3K9me3 at D4Z4 in HeLa cells.Citation18 Similarly, reducing SUV39H1 activity in proliferating immortalized myoblasts by knock down or treatment of primary and immortalized myoblasts with Chaetocin decreased H3K9me3 levels and resulted in transcriptional derepression of DUX4.Citation35 The specificity of Chaetocin was recently challenged, however, as it might also inhibit the activity of other histone lysine methyltransferases.Citation37 Our data thus suggest that the regulation of this modification at D4Z4 is different between primary and immortalized myoblasts, and between proliferating and differentiating myoblasts, perhaps by involvement of additional methyltransferases such as G9a.Citation35 A recent report suggested a role for the telomere in the epigenetic regulation of D4Z4.Citation38 Since myoblasts are immortalized by telomerase expression, it will be important to comprehensively compare the epigenetic regulation of the D4Z4 repeat array between primary and immortalized myoblasts.
Together with earlier observations that SMCHD1 is a modifier of disease severity in FSHD1, our study suggests that D4Z4 derepression in FSHD1 and FSHD2 converges at SMCHD1. Contraction of D4Z4 in FSHD1 may lead to specific loss of SMCHD1 at the contracted allele, analogous to its interaction with telomeric repeats, which shows a non-linear positive correlation to telomere length.Citation39 All together, this emphasizes the crucial role of SMCHD1 in suppressing DUX4 activity in somatic cells and its decline during muscle cell differentiation provides a plausible explanation for the increased expression of DUX4 in skeletal muscle cells.
Materials and Methods
Culturing of human primary myoblast cell lines
Human primary myoblast cell lines were originating from the University of Rochester bio repository (http://www.urmc.rochester.edu/fields-center/). Muscle samples were obtained after subjects were consented under a protocol approved by the institutional review board at the University of Rochester. Myoblasts were cultured in DMEM/F-10 media (#31550, Gibco/Life Technologies, Bleiswijk, The Netherlands) supplemented with 20% heat inactivated fetal bovine serum (FBS #10270, Gibco/Life Technologies), 1% penicillin/streptomycin (#15140, Gibco/Life Technologies) and 10ng/ml rhFGF (#G5071, Promega, Leiden, The Netherlands) and 1μM dexamethasone (#D2915, Sigma-Aldrich, Zwijndrecht, The Netherlands) was added to the medium. Myoblasts were fused at 80% confluency by culturing them in DMEM/F-12 Glutamax media (#31331, Gibco/Life Technologies) containing 1% penicillin and streptomycin and 2% KnockOut serum replacement formulation (#10828, Gibco/Life Technologies) for 36 h. Human control fibroblast cell lines were maintained in DMEM/F-12, supplemented with 20% FBS, 1% penicillin and streptomycin, 10 mM HEPES (#15630#, Gibco/Life Technologies) and 1mM sodium pyruvate (##11360, Gibco/Life Technologies). Used cell lines, D4Z4 allele information, and experimental use are listed in Table S1.
RNA isolation, cDNA synthesis, and qRT-PCR
Myoblast and myotube samples were harvested for RNA isolation by adding QIAzol lysis reagent (#79306, Qiagen N.V., Venlo, The Netherlands). RNA was isolated by miRNeasy Mini Kit (#217004, Qiagen) including DNase treatment according to the manufacturer's instructions. cDNA was synthesized with RevertAid H Minus First strand cDNA Synthesis Kit (#K1632 Thermo Fischer Scientific Inc.., Waltham, MA) using 2μg template RNA and poly-dT primers. Gene specific cDNA products were quantified by qPCR in duplicate using SYBR green master mix supplemented with gene specific primers (Table S2) using the CFX96 system (Bio-Rad, Veenendaal, The Netherlands). qRT-PCR data were analyzed by Bio-Rad CFX manager version 3.0 (Bio-Rad) using GAPDH and GUSB as reference genes for every individual sample.
Transduction of primary cell cultures
Full length SMCHD1 was PCR amplified from cDNA and cloned into the pRRL-CMV lentiviral backbone containing a puromycin selection marker using standard cloning procedures and the obtained construct was verified by Sanger sequencing. shRNA constructs originating from the Mission shRNA library (MISSION shRNA library, TRC1 or TRC2; Sigma Aldrich) and are listed in Table S2. Constructs were used to generate lentiviral particles, and myoblast cell lines were transduced at 50% confluency and 24 h after transduction they were grown in media containing 0.5 μg/ml puromycin for selection. Differentiation to myotubes was induced by serum reduction at 80% confluency and cells were harvested after 36 h of differentiation. Transduction of fibroblasts to ectopically express MyoD1 and induce myogenesis was performed as described previously.Citation40
Chromatin immunoprecipitation
Histone ChIP studies were carried out as described beforeCitation19 using antibodies against H3 (ab1791, 2 μl/rxn, Abcam, Cambridge, UK), H3K9me3 (39161, 5 μl/rxn, Active Motif, Carlsbad, USA), H3K27me3 (#17-622, 5 μl/rxn, Merck-Millipore, Amsterdam Zuid-Oost, The Netherlands), and total IgG (5 μl/rxn, Merck-Millipore). Non-histone ChIPs were carried out as described before Citation14 using antibodies against SMCHD1 (ab31865, 5 μg/rxn, Abcam, Cambridge, UK) RAD21 (ab992, 5 μg/rxn, Abcam), SMC3 (ab9263, 5 μg/rxn, Abcam), SUZ12 (D39F6, 5 μg/rxn, Cell Signaling, Leiden, The Netherlands). All antibodies were qualified for ChIP application by the manufacturer. All ChIP-qPCR experiments were performed 2 independent times.
GSK126 treatment of primary myoblast cultures
Three control, 4 FSHD1, and 4 FSHD2 human primary myoblast cultures were grown to 50-60% confluency and culturing was continued in the presence of 0 μM, 1 μM, 4 μM and 8 μM GSK126 (#1346574-57-9, MedKoo Biosciences, North Carolina, USA) in proliferation media. GSK126 was dissolved in DMSO (#D2650, Sigma-Aldrich, St Louis, USA) and the different concentration of GSK126 was added in equal volumes of DMSO to the cells. Cultures shown as 0 μM GSK126 were treated only with DMSO. After 24 h culturing was continued in fusion media in the presence of the same concentration of GSK126 for another 24 h. RNA and protein samples were harvested as described above.
Western blot
Cell were directly lysed in NuPAGE LDS Sample Buffer (#NP0008, ThermoFisher Scientific) and loaded on Novex 4-12% Bis-Tris Protein Gels (#WT4121A, ThermoFisher Scientific) according to manufacturer's instructions. Running of the gel and transfer to Hybond nitrocellulose membranes (#10600048, GE Healthcare, Diegem, Belgium) were carried out using the Nupage Novex SDS-page gel system (ThermoFisher Scientific). Blots were blocked in 4% milk in PBS and incubated with antibodies against SMCHD1 (1:250), SUV39H1 (0.1 μg/ml, #07-958, Merck-Millipore), RAD21 (1:500), SMC3 (1:1000) and Tubulin (1:2000, T6199, Sigma-Aldrich). Detection and relative quantification were done using the Odyssey system (V3.0, LI-COR Biosciences, Lincoln, NE, USA), except for blots in , which were visualized by enhanced chemiluminescence (ECL).
Statistical analysis
All statistical analysis was done using GraphPad Prism 6 software and the exact method applied at different experiments is described in the figure legends.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
KEPI_S_1113798.zip
Download Zip (3.8 MB)Acknowledgments
The authors would like to thank all patients and their families for participating in our study. Lentiviral cloning and shRNA experiments were supported by Dr. Rob Hoeben and Martijn Rabelink, Department of Molecular and Cellular Biology, Leiden University Medical Center.
Funding
This study was supported with grants from the US National Institutes of Health (NIH) (National Institute of Neurological Disorders and Stroke (NINDS) P01NS069539, and National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS) R01AR045203 and R01AR066248, the Prinses Beatrix Spierfonds (W.OR12-20 and W.OR14-04), the Muscular Dystrophy Association (MDA; 217596), the Geraldi Norton and Eklund family foundation, the FSH Society, the FSHD Global Research Foundation, and Spieren voor Spieren.
Related Research Data
References
- Tawil R, van der Maarel SM, Tapscott SJ. Facioscapulohumeral dystrophy: the path to consensus on pathophysiology. Skelet Muscle 2014; 4:12; PMID:24940479; http://dx.doi.org/10.1186/2044-5040-4-12
- Deenen JC, Arnts H, van der Maarel SM, Padberg GW, Verschuuren JJ, Bakker E, Weinreich SS, Verbeek AL, van Engelen BG. Population-based incidence and prevalence of facioscapulohumeral dystrophy. Neurology 2014; 83:1056–9; PMID:25122204; http://dx.doi.org/10.1212/WNL.0000000000000797
- Gabriels J, Beckers MC, Ding H, De Vriese A, Plaisance S, van der Maarel SM, Padberg GW, Frants RR, Hewitt JE, Collen D, et al. Nucleotide sequence of the partially deleted D4Z4 locus in a patient with FSHD identifies a putative gene within each 3.3 kb element. Gene 1999; 236:25–32; PMID:10433963; http://dx.doi.org/10.1016/S0378-1119(99)00267-X
- Wijmenga C, Frants RR, Brouwer OF, Moerer P, Weber JL, Padberg GW. Location of facioscapulohumeral muscular dystrophy gene on chromosome 4. Lancet 1990; 336:651–3; PMID:1975852; http://dx.doi.org/10.1016/0140-6736(90)92148-B
- Wijmenga C, Hewitt JE, Sandkuijl LA, Clark LN, Wright TJ, Dauwerse HG, Gruter AM, Hofker MH, Moerer P, Williamson R, et al. Chromosome 4q DNA rearrangements associated with facioscapulohumeral muscular dystrophy. Nat Genet 1992; 2:26–30; PMID:1363881; http://dx.doi.org/10.1038/ng0992-26
- van der Maarel SM, Tawil R, Tapscott SJ. Facioscapulohumeral muscular dystrophy and DUX4: breaking the silence. Trends Mol Med 2011; 17:252–8; PMID:21288772; http://dx.doi.org/10.1016/j.molmed.2011.01.001
- Lemmers RJ, van der Vliet PJ, Klooster R, Sacconi S, Camano P, Dauwerse JG, Snider L, Straasheijm KR, van Ommen GJ, Padberg GW, et al. A unifying genetic model for facioscapulohumeral muscular dystrophy. Science 2010; 329:1650–3; PMID:20724583; http://dx.doi.org/10.1126/science.1189044
- Snider L, Geng LN, Lemmers RJ, Kyba M, Ware CB, Nelson AM, Tawil R, Filippova GN, van der Maarel SM, Tapscott SJ, et al. Facioscapulohumeral dystrophy: incomplete suppression of a retrotransposed gene. PLoS Genet 2010; 6:e1001181; PMID:21060811; http://dx.doi.org/10.1371/journal.pgen.1001181
- Geng LN, Yao Z, Snider L, Fong AP, Cech JN, Young JM, van der Maarel SM, Ruzzo WL, Gentleman RC, Tawil R, et al. DUX4 activates germline genes, retroelements, and immune mediators: implications for facioscapulohumeral dystrophy. Dev Cell 2012; 22:38–51; PMID:22209328; http://dx.doi.org/10.1016/j.devcel.2011.11.013
- Kowaljow V, Marcowycz A, Ansseau E, Conde CB, Sauvage S, Matteotti C, Arias C, Corona ED, Nunez NG, Leo O, et al. The DUX4 gene at the FSHD1A locus encodes a pro-apoptotic protein. Neuromuscul Disord 2007; 17:611–23; PMID:17588759; http://dx.doi.org/10.1016/j.nmd.2007.04.002
- Wallace LM, Garwick SE, Mei W, Belayew A, Coppee F, Ladner KJ, Guttridge D, Yang J, Harper SQ. DUX4, a candidate gene for facioscapulohumeral muscular dystrophy, causes p53-dependent myopathy in vivo. Ann Neurol 2011; 69:540–52; PMID:21446026; http://dx.doi.org/10.1002/ana.22275
- Gilbert JR, Stajich JM, Wall S, Carter SC, Qiu H, Vance JM, Stewart CS, Speer MC, Pufky J, Yamaoka LH, et al. Evidence for heterogeneity in facioscapulohumeral muscular dystrophy (FSHD). Am J Hum Genet 1993; 53:401–8; PMID:8328457
- van Deutekom JC, Wijmenga C, van Tienhoven EA, Gruter AM, Hewitt JE, Padberg GW, van Ommen GJ, Hofker MH, Frants RR. FSHD associated DNA rearrangements are due to deletions of integral copies of a 3.2 kb tandemly repeated unit. Hum Mol Genet 1993; 2:2037–42; PMID:8111371; http://dx.doi.org/10.1093/hmg/2.12.2037
- Lemmers RJ, Tawil R, Petek LM, Balog J, Block GJ, Santen GW, Amell AM, van der Vliet PJ, Almomani R, Straasheijm KR, et al. Digenic inheritance of an SMCHD1 mutation and an FSHD-permissive D4Z4 allele causes facioscapulohumeral muscular dystrophy type 2. Nat Genet 2012; 44:1370–4; PMID:23143600; http://dx.doi.org/10.1038/ng.2454
- Sacconi S, Lemmers RJ, Balog J, van der Vliet PJ, Lahaut P, van Nieuwenhuizen MP, Straasheijm KR, Debipersad RD, Vos-Versteeg M, Salviati L, et al. The FSHD2 gene SMCHD1 is a modifier of disease severity in families affected by FSHD1. Am J Hum Genet 2013; 93:744–51; PMID:24075187; http://dx.doi.org/10.1016/j.ajhg.2013.08.004
- Lemmers RJ, van den Boogaard ML, van der Vliet PJ, Donlin-Smith CM, Nations SP, Ruivenkamp CA, Heard P, Bakker B, Tapscott S, Cody JD, et al. Hemizygosity for SMCHD1 in Facioscapulohumeral Muscular Dystrophy Type 2: Consequences for 18p Deletion Syndrome. Hum Mutat 2015; 36:679–83; PMID:25820463; http://dx.doi.org/10.1002/humu.22792
- Bodega B, Ramirez GD, Grasser F, Cheli S, Brunelli S, Mora M, Meneveri R, Marozzi A, Mueller S, Battaglioli E, et al. Remodeling of the chromatin structure of the facioscapulohumeral muscular dystrophy (FSHD) locus and upregulation of FSHD-related gene 1 (FRG1) expression during human myogenic differentiation. BMC Biol 2009; 7:41; PMID:19607661; http://dx.doi.org/10.1186/1741-7007-7-41
- Zeng W, de Greef JC, Chen YY, Chien R, Kong X, Gregson HC, Winokur ST, Pyle A, Robertson KD, Schmiesing JA, et al. Specific loss of histone H3 lysine 9 trimethylation and HP1gamma/cohesin binding at D4Z4 repeats is associated with facioscapulohumeral dystrophy (FSHD). PLoS Genet 2009; 5:e1000559; PMID:19593370; http://dx.doi.org/10.1371/journal.pgen.1000559
- Balog J, Thijssen PE, de Greef JC, Shah B, van Engelen BG, Yokomori K, Tapscott SJ, Tawil R, van der Maarel SM. Correlation analysis of clinical parameters with epigenetic modifications in the DUX4 promoter in FSHD. Epigenetics 2012; 7:579–84; PMID:22522912; http://dx.doi.org/10.4161/epi.20001
- de Greef JC, Lemmers RJ, van Engelen BG, Sacconi S, Venance SL, Frants RR, Tawil R, van der Maarel SM. Common epigenetic changes of D4Z4 in contraction-dependent and contraction-independent FSHD. Hum Mutat 2009; 30:1449–59; PMID:19728363; http://dx.doi.org/10.1002/humu.21091
- Hartweck LM, Anderson LJ, Lemmers RJ, Dandapat A, Toso EA, Dalton JC, Tawil R, Day JW, van der Maarel SM, Kyba M. A focal domain of extreme demethylation within D4Z4 in FSHD2. Neurology 2013; 80:392–9; PMID:23284062; http://dx.doi.org/10.1212/WNL.0b013e31827f075c
- van Overveld PG, Lemmers RJ, Sandkuijl LA, Enthoven L, Winokur ST, Bakels F, Padberg GW, van Ommen GJ, Frants RR, van der Maarel SM. Hypomethylation of D4Z4 in 4q-linked and non-4q-linked facioscapulohumeral muscular dystrophy. Nat Genet 2003; 35:315–7; PMID:14634647; http://dx.doi.org/10.1038/ng1262
- Cabianca DS, Casa V, Bodega B, Xynos A, Ginelli E, Tanaka Y, Gabellini D. A long ncRNA links copy number variation to a polycomb/trithorax epigenetic switch in FSHD muscular dystrophy. Cell 2012; 149:819–31; PMID:22541069; http://dx.doi.org/10.1016/j.cell.2012.03.035
- Cabianca DS, Casa V, Gabellini D. A novel molecular mechanism in human genetic disease: a DNA repeat-derived lncRNA. RNA Biol 2012; 9:1211–7; PMID:23047063; http://dx.doi.org/10.4161/rna.21922
- Snider L, Asawachaicharn A, Tyler AE, Geng LN, Petek LM, Maves L, Miller DG, Lemmers RJ, Winokur ST, Tawil R, et al. RNA transcripts, miRNA-sized fragments and proteins produced from D4Z4 units: new candidates for the pathophysiology of facioscapulohumeral dystrophy. Hum Mol Genet 2009; 18:2414–30; PMID:19359275; http://dx.doi.org/10.1093/hmg/ddp180
- Lim JW, Snider L, Yao Z, Tawil R, Van Der Maarel SM, Rigo F, Bennett CF, Filippova GN, Tapscott SJ. DICER/AGO-dependent epigenetic silencing of D4Z4 repeats enhanced by exogenous siRNA suggests mechanisms and therapies for FSHD. Hum Mol Genet 2015; 24(17):4817–28; PMID:26041815; http://dx.doi.org/10.1093/hmg/ddv206
- Jones TI, Chen JC, Rahimov F, Homma S, Arashiro P, Beermann ML, King OD, Miller JB, Kunkel LM, Emerson CP, Jr., et al. Facioscapulohumeral muscular dystrophy family studies of DUX4 expression: evidence for disease modifiers and a quantitative model of pathogenesis. Hum Mol Genet 2012; 21:4419–30; PMID:22798623; http://dx.doi.org/10.1093/hmg/dds284
- Krom YD, Dumonceaux J, Mamchaoui K, den Hamer B, Mariot V, Negroni E, Geng LN, Martin N, Tawil R, Tapscott SJ, et al. Generation of isogenic D4Z4 contracted and noncontracted immortal muscle cell clones from a mosaic patient: a cellular model for FSHD. Am J Pathol 2012; 181:1387–401; PMID:22871573; http://dx.doi.org/10.1016/j.ajpath.2012.07.007
- Tassin A, Laoudj-Chenivesse D, Vanderplanck C, Barro M, Charron S, Ansseau E, Chen YW, Mercier J, Coppee F, Belayew A. DUX4 expression in FSHD muscle cells: how could such a rare protein cause a myopathy? J Cell Mol Med 2013; 17:76–89; PMID:23206257; http://dx.doi.org/10.1111/j.1582-4934.2012.01647.x
- Asp P, Blum R, Vethantham V, Parisi F, Micsinai M, Cheng J, Bowman C, Kluger Y, Dynlacht BD. Genome-wide remodeling of the epigenetic landscape during myogenic differentiation. Proc Natl Acad Sci U S A 2011; 108:E149–58; PMID:21551099; http://dx.doi.org/10.1073/pnas.1102223108
- Nozawa RS, Nagao K, Igami KT, Shibata S, Shirai N, Nozaki N, Sado T, Kimura H, Obuse C. Human inactive X chromosome is compacted through a PRC2-independent SMCHD1-HBiX1 pathway. Nat Struct Mol Biol 2013; 20:566–73; PMID:23542155; http://dx.doi.org/10.1038/nsmb.2532
- McCabe MT, Ott HM, Ganji G, Korenchuk S, Thompson C, Van Aller GS, Liu Y, Graves AP, Della Pietra A, 3rd, Diaz E, et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature 2012; 492:108–12; PMID:23051747; http://dx.doi.org/10.1038/nature11606
- Reddington JP, Perricone SM, Nestor CE, Reichmann J, Youngson NA, Suzuki M, Reinhardt D, Dunican DS, Prendergast JG, Mjoseng H, et al. Redistribution of H3K27me3 upon DNA hypomethylation results in de-repression of Polycomb target genes. Genome Biol 2013; 14:R25; PMID:23531360; http://dx.doi.org/10.1186/gb-2013-14-3-r25
- Juan AH, Kumar RM, Marx JG, Young RA, Sartorelli V. Mir-214-dependent regulation of the polycomb protein Ezh2 in skeletal muscle and embryonic stem cells. Mol Cell 2009; 36:61–74; PMID:19818710; http://dx.doi.org/10.1016/j.molcel.2009.08.008
- Zeng W, Chen YY, Newkirk DA, Wu B, Balog J, Kong X, Ball AR, Jr., Zanotti S, Tawil R, Hashimoto N, et al. Genetic and epigenetic characteristics of FSHD-associated 4q and 10q D4Z4 that are distinct from non-4q/10q D4Z4 homologs. Hum Mutat 2014; 35:998–1010; PMID:24838473; http://dx.doi.org/10.1002/humu.22593
- Simon JA, Kingston RE. Mechanisms of polycomb gene silencing: knowns and unknowns. Nat Rev Mol Cell Biol 2009; 10:697–708; PMID:19738629; http://dx.doi.org/10.1038/nrn2731
- Cherblanc FL, Chapman KL, Brown R, Fuchter MJ. Chaetocin is a nonspecific inhibitor of histone lysine methyltransferases. Nat Chem Biol 2013; 9:136–7; PMID:23416387; http://dx.doi.org/10.1038/nchembio.1187
- Stadler G, Rahimov F, King OD, Chen JC, Robin JD, Wagner KR, Shay JW, Emerson CP, Jr., Wright WE. Telomere position effect regulates DUX4 in human facioscapulohumeral muscular dystrophy. Nat Struct Mol Biol 2013; 20:671–8; PMID:23644600; http://dx.doi.org/10.1038/nsmb.2571
- Grolimund L, Aeby E, Hamelin R, Armand F, Chiappe D, Moniatte M, Lingner J. A quantitative telomeric chromatin isolation protocol identifies different telomeric states. Nat Commun 2013; 4:2848; PMID:24270157; http://dx.doi.org/10.1038/ncomms3848
- Yao Z, Fong AP, Cao Y, Ruzzo WL, Gentleman RC, Tapscott SJ. Comparison of endogenous and overexpressed MyoD shows enhanced binding of physiologically bound sites. Skelet Muscle 2013; 3:8; PMID:23566431; http://dx.doi.org/10.1186/2044-5040-3-8