
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Heterogeneity of DNA methylation status among alleles is observed in various cell types and is involved in epigenetic gene regulation and cancer biology. However, the individual methylation profile within each allele has not yet been examined at the whole-genome level. In the present study, we applied linkage disequilibrium analysis to the DNA methylation data obtained from whole-genome bisulfite sequencing studies in mouse germline and other types of cells. We found that the methylation status of 2 consecutive CpG sites showed deviation from equilibrium frequency toward concordant linkage (both methylated or both unmethylated) in germline cells. In the imprinting loci where methylation of constituent alleles is known, our analysis detected the deviation toward the concordant linkage as expected. In addition, we applied this analysis to the transitional zone between methylated and unmethylated regions and to the cells undergoing epigenetic reprogramming. In both cases, deviation to the concordant-linked alleles was conspicuous, indicating that the methylation pattern is not random but rather concordant within each allele. These results will provide the key to understanding the mechanism underlying allelic heterogeneity.
Introduction
DNA methylation is the most important epigenetic modification and is involved in cellular differentiation, fate determination, developmental plasticity, and environmental responses. The genome-wide pattern of methylation status is maintained through cell division and is thus inherited by the descendant cell population. While the methylation status of individual CpG sites is generally consistent among alleles, resulting in methylated or unmethylated CpG sites in all alleles, there are certain regions that show heterogeneity in methylation status among alleles. The methylation heterogeneity (intermediate methylationCitation1 or partially methylated domainCitation2) is reported in primordial germ cells under epigenetic reprogramming,Citation3 sperms,Citation4 embryonic stem cells,Citation5 induced pluripotent stem cells,Citation6 placental cells,Citation2 and other somatic cells under normal and disease conditions.Citation7–9 These regions are highly gene enriched and often interact with other epigenetic markers such as histone modifications.Citation1 In embryonic stem cells, allelic heterogeneity is involved in maintaining the bimodal states of gene expression.Citation5 Furthermore, allelic heterogeneity is frequently found in various types of cancer cells, and the possible relationship with aberrant gene expression in cancer cells is of particular interest.Citation9 While previous studies have defined allelic heterogeneity based mainly on the methylation levels of individual CpG sites, the underlying methylation pattern within each allele has not yet been examined. Accordingly, it is important to determine the linear methylation pattern of consecutive CpG sites within each allele.
In the present study, we introduce a novel approach for the analysis of the methylation pattern within each allele using bisulfite sequencing reads. Characterization of the methylation pattern underlying allelic heterogeneity will provide the basis for understanding the dynamics of heterogeneity, its biological significance, and its roles in epigenetic gene regulation.
Results and Discussion
Coefficient of linkage disequilibrium D
We used the published data of whole-genome bisulfite sequencing experiments on mouse embryonic germ cells, developing and mature oocytes, sperms, ES cells, and astrocytes.Citation3,10,11 To characterize the methylation pattern within each allele underlying heterogeneity, we focused on the methylation status of the 2 adjacent CpG sites (CpG pair) residing in a single sequencing read, which is derived from a single allele. We examined the linkage of methylation states in a CpG pair using a modified version of the coefficient of linkage disequilibrium, hereafter designated as D. The coefficient D corresponds to the deviation of the observed frequency of alleles from their equilibrium frequency. The range of D depends on the allele frequency, i.e., D has a maximum range when the allele frequency of each locus is 50%, whereas it becomes 0 when the frequency of one allele is 100%. With respect to the linkage of methylation status, D has maximum range when the methylation level is intermediate, whereas it becomes 0 when all alleles are fully methylated or unmethylated (). Because the region of allelic heterogeneity shows the intermediate methylation level, D is suitable for our analysis. Furthermore, the sign of D can be used to determine the type of linkage, i.e., both methylated or both unmethylated (concordant) or alternately methylated and unmethylated (discordant).
Figure 1. Coefficient of linkage disequilibrium D indicates the linkage between 2 adjacent CpG sites. (A) The methylation level and the range of D. The D has a maximum range at the intermediate methylation level. The sign of D corresponds to the concordant (red bars) or discordant (blue bars) linkage. (B) An example plot of methylation level and D in mouse embryonic stem cells at the genomic region chr1:134,200,000–134,600,000. Upper track: methylation levels of individual CpG sites (black). Lower track: D values of individual CpG pairs (red for positive and blue for negative values). Bottom track shows the gene annotation.
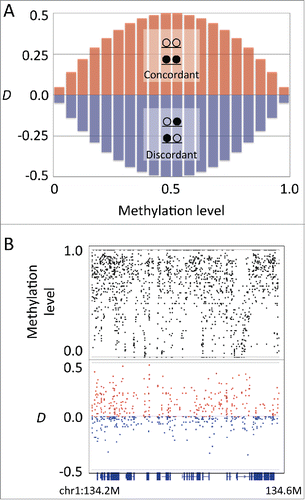
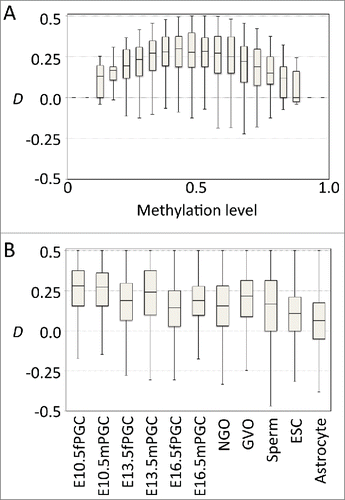
For the mouse reference genome (mm10), the total number of CpG pairs with the distance of <100 bp is 14,137,359, of which the median distance is 28 bp. We first examined the relationship between D and the methylation level of the CpG pairs (). In the examined germline cells, D generally showed positive values at intermediate methylation levels (0.25–0.75) and approached zero at higher or lower methylation levels (). The positive D values at the intermediate methylation levels indicate that the methylation status of CpG pairs at the regions of heterogeneity tend to show concordant linkage. This tendency was conspicuous in developing germline cells but not as evident in embryonic stem cells and adult astrocyte cells (), whereas the means were significantly different between all samples examined (. t-test). The embryonic germ cells undergo epigenetic reprogramming during which methylation levels change dramatically.Citation3 Thus, it is possible that positive D values observed in germline cells may be the result of global changes in methylation status. In contrast, methylation patterns are relatively stable in adult somatic cells. If the stochastic changes continuously accumulate on stable methylation patterns in somatic cells, it might be expected that D would approach equilibrium. Thus, the value of D close to zero observed in adult somatic cells indicates that epigenetic information carried by DNA methylation is continuously degraded along with cell division and differentiation.Citation12 Importantly, heterogeneity among alleles does not indicate stochastic methylation within each allele, i.e., the methylation pattern within each allele is not necessarily an interspersed mixture of methylated and unmethylated sites. In fact, the CpG pairs showing negative D values are rare and distributed sporadically, and there was no continuous region with highly discordant methylation pattern. The fact that positive D prevailed in the regions showing allelic heterogeneity indicates that the methylation pattern within each allele is generally concordant rather than stochastic.
Figure 2. Germline cells showed positive D values. (A) Boxplots of D at various methylation levels in male primordial germ cells at embryonic day 10.5. (B) Boxplot for D at intermediate methylation level (0.25–0.75) in various cell types (female and male primordial germ cells at embryonic days 10.5, 13.5, and 16.5, non-growing oocytes, germinal vesicle oocytes, sperm cells, embryonic stem cells, and astrocytes).
Heterogeneity in imprinting loci
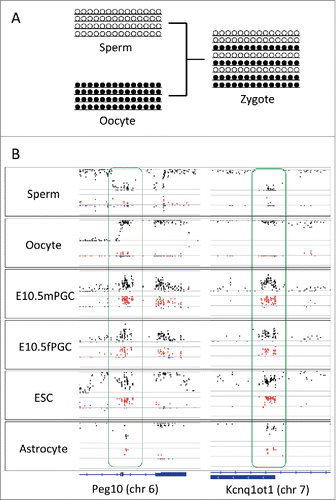
Since the sign of D corresponds to concordant or discordant linkage, we consider that D might be suitable for analyzing the linear pattern of methylation within each allele. To test this, we focused on the imprinting loci where maternal and paternal alleles show a distinct methylation status. In the region involved in the regulation of paternal expression, the methylation level is low in sperm-derived alleles and high in oocyte-derived alleles.Citation13–15 Accordingly, the sexually differential methylation pattern in gametes results in allelic heterogeneity in the zygote, which might be inherited by the descendant cells (). As expected, D was close to zero in both sperms and oocytes, reflecting their uniform methylation status in imprinting loci (). In contrast, the deviation to concordant linkage was conspicuous in descendant cells, such as ES cells and astrocytes, as well as in 10.5 embryonic day germ cells where methylation erasure had not yet completed (). The same tendency was additionally observed in several known imprinting loci (see Fig. S1). The result of D reflecting the linkage of methylation status indicates that D is suitable to infer the spatial pattern of methylation within alleles constituting heterogeneity.
Figure 3. D in known imprinting loci. (A) Schematic representation of allele constitutions at paternally imprinted loci for sperms, oocytes, and zygotes. Alleles in sperms and in oocytes are fully unmethylated and methylated, respectively. Alleles in zygotes are a mixture of unmethylated and methylated. (B) Plots of methylation levels (black) and D (red for positive and blue for negative values) at paternally imprinted loci (Peg10 and Kcnq1ot1) in sperms, oocytes, male and female primordial germ cells (embryonic day 10.5), embryonic stem cells, and astrocytes. While D is close to zero in sperms and oocytes, D becomes positive in primordial germ cells, embryonic stem cells, and astrocytes at imprinting loci. Located at the bottom is the track for gene annotation.
Heterogeneity in transitional zones
We then applied linkage disequilibrium analysis to various genomic regions showing heterogeneity. We first analyzed the transitional zone between fully methylated and fully unmethylated regions. In mouse oocytes, the transitional zone found at the boundary of a gene body often shows allelic heterogeneity.Citation10 There are 2 possible explanations for the methylation pattern underlying heterogeneity in the transitional zone: 1) methylation activity is stochastic in the transitional zone, resulting in each allele consisting of an interspersed mixture of methylated and unmethylated CpG sites, or 2) the transition between methylated and unmethylated regions occurs at a distinct boundary in each allele, whereas the boundary position differs among alleles (). In the former case, the methylation pattern in the transitional zone is expected to show no deviation (), whereas in the latter case, there is a deviation to concordant linkage (
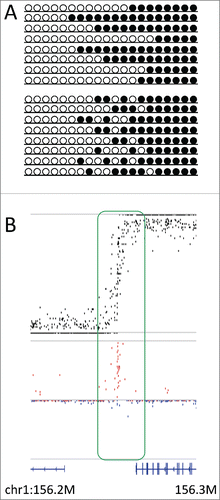
). Accordingly, we examined the deviation at the transitional zones found in the methylome of mouse germinal vesicle oocytes.Citation10,11 In almost all regions examined, the CpG pair in the transitional zone showed a clear deviation to concordant linkage (). These results indicate that methylation activity is not stochastic in the transitional zone, but the transitional boundaries differ among alleles. It is known that DNA methylation is found not only in promoter regions but additionally in the gene bodies. In addition, the boundary between methylated and unmethylated regions often corresponds to the boundary between the gene body and intergenic regions.Citation16 Furthermore, gene body methylation is associated with intragenic chromatin modifications such as H3K9me3.Citation17 Thus, the present finding that the transitional boundary differs among alleles may indicate that the boundary of gene body methylation differs among cells, possibly depending on the degree of histone modifications and/or activity of gene expression.
Figure 4. D in the transitional zone. (A) Two models explaining allelic heterogeneity in the transitional zone, i.e., boundary position is stochastic (upper panel) or methylation activity is stochastic (lower panel) at the transitional zone. (B) Plots showing the methylation level (upper panel, black) and D (lower panel, red for positive and blue for negative values) at the transitional zone (chr1:156,200,000–156,300,000), where methylation levels showed fluctuation around 0.5 and D showed conspicuous deviation to positive values. Bottom track shows the gene annotation.
Heterogeneity during reprogramming
We next examined the heterogeneity observed in the embryonic germ cells during the epigenetic reprogramming. Reprogramming of the DNA methylome consists of 2 phases, namely whole-genome demethylation and de novo methylation.Citation18–20 The allelic heterogeneity becomes most obvious at the period between these 2 phases.Citation3 Knowledge of the methylation pattern underlying allelic heterogeneity may assist in understanding the mechanism of de novo methylation acting in the epigenetic reprogramming. We considered 2 hypotheses: 1) de novo methylation progresses stochastically at individual CpG sites, resulting in the spatially interspersed methylation patterns in all alleles, or 2) de novo methylation occurs stochastically among alleles; however, once present, it propagates rapidly, resulting in the mixture of fully methylated and unmethylated alleles (). It is expected that deviation should not be observed in the former case (), whereas deviation to the concordant pattern may be observed in the latter case (
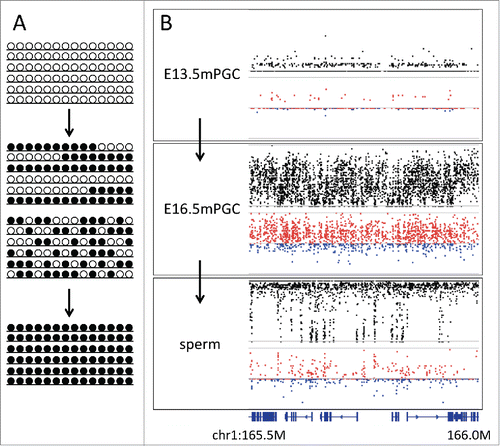
). We examined the deviation in male primordial germ cells (embryonic day 13.5 and 16.5) and in sperm cells, each corresponding to the progressive stages of reprogramming.Citation3 On day 13.5, no deviation in male primordial germ cells is observed when methylation is fully erased and reprogramming has not yet started (). In sperm cells where de novo methylation is completed and most of the CpG sites are fully methylated, only a small part showed heterogeneity with deviation to the concordant linkage (). When reprogramming was in progress and almost all regions showed allelic heterogeneity, clear deviation to the concordant linkage was observed (). These results support the latter hypothesis of allelic heterogeneity being caused by mixture of methylated and unmethylated alleles. It has been reported that in 16.5-day male primordial germ cells, DNA methyltransferases, Dnmt3a, Dnmt3b, and Dnmt3l show a burst of expression.Citation21 Thus, it is possible that allelic heterogeneity may reflect the heterogeneity in timing of their expression burst among the cell population. It is known that mouse primordial germ cells show heterogeneity in the status of differentiation and proliferation,Citation22 further supporting our hypothesis.
Figure 5. Changes in D during epigenetic reprogramming. (A) Schematic representation of changes in allelic methylation patterns during epigenetic reprogramming from erased (upper panel) to reprogrammed (lower panel). Middle panels show 2 models explaining allelic heterogeneity at the time of reprogramming, i.e., the mixture of fully methylated and unmethylated alleles or the spatially interspersed methylation patterns in all alleles. (B) Plots showing methylation level and D at the genomic region chr1:165,490,000 –166,000,000 in male primordial germ cells at embryonic days 13.5 and 16.5 and sperms. In 13.5 day germ cells, methylation is erased, while in sperms, reprogramming is completed. In 16.5 day germ cells, when methylation levels showed intermediate values, D tended to show positive values, thereby supporting the latter hypothesis. Bottom track shows the gene annotation.
Length of CpG pair and deviation D
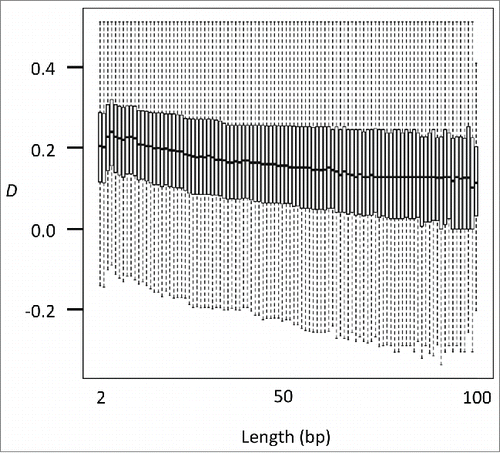
The deviation to concordant linkage observed in various biological conditions suggests that DNA methyltransferases do not act in a site-specific manner but rather act by simultaneously targeting a range of CpG sites. It is thus expected that if the distance between neighboring CpG sites increases, the degree of deviation to concordant linkage might decrease. To test this, we examined the relationship between the length of CpG pair and D (). As expected, a significant negative correlation was identified in all samples examined (. t-test for Pearson correlation coefficient), indicating that closer neighboring CpG sites are more likely to be simultaneously methylated. It has been suggested that DNA methyltransferases are recruited to the target position by interaction with various chromatin modifications and remodeling factors, resulting in the target region of methylation being of at least a nucleosome size.Citation23 Our results support the nucleosomal but not the site-specific action of DNA methyltransferases. A recent model further suggests that DNA methylation is not a static process but rather reflects the dynamics between the methylation and de-methylation processes.Citation23 Heterogeneity among regionally methylated and unmethylated alleles may be the consequence of the dynamic methylation and demethylation processes. Considered collectively, our approach to analyze the linkage between the methylation status of adjacent CpG sites has proven to be a powerful tool to understand the mechanism of the methylation processes and is possibly useful for application to various cancer and disease epigenomics studies.
Figure 6. D depends on the length of the CpG pair. Box plots show the relationship between the length of the CpG pair and D at an intermediate methylation level (0.25–0.75) in male primordial germ cells (embryonic day 16.5).
Methods
Read data
We used the read data from a whole-genome bisulfite sequencing experiment in mouse primordial germ cells, oocytes, sperm cells, embryonic stem cells, and astrocytes.Citation3,10,11 The sequence reads were downloaded from the NCBI short read archives (accessions: DRA000484, DRA000495, DRA000570, and DRA000607).
Mapping of bisulfite reads
The reads were mapped onto the mouse reference genome (mm10) and analyzed using the Bismark software.Citation24 CpG pairs with the distance being less than 100 bp on the mm10 reference were listed and used for subsequent analyses. For each sample, the methylation status of the CpG pair on a single sequencing read was extracted and summarized. We included only the CpG pairs with depths of 10–200.
Calculation of deviation D
For each CpG pair, both the methylation level and the coefficient of linkage disequilibrium D were calculated from the frequency of the 4 allele types using the equation, as described below (note ). In the present study, we defined D as the deviation for concordant alleles (
).
: Frequency of methylated concordant alleles
: Frequency of unmethylated concordant alleles
,
: Frequency of discordant alleles
: Methylation level of a CpG pair
: Expected frequency of concordant alleles
: Deviation of observed from expected for concordant alleles
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
KEPI_S_1115176.pdf
Download PDF (227.9 KB)Related Research Data
References
- Elliott G, Hong C, Xing X, Zhou X, Li D, Coarfa C, Bell RJA, Maire CL, Ligon KL, Sigaroudinia M, et al. Intermediate DNA methylation is a conserved signature of genome regulation. Nat Commun 2015; 6:6363; PMID:25691127; http://dx.doi.org/10.1038/ncomms7363
- Schroeder DI, Blair JD, Lott P, Yu HOK, Hong D, Crary F, Ashwood P, Walker C, Korf I, Robinson WP, et al. The human placenta methylome. Proc Natl Acad Sci 2013; 110:6037–42; PMID:23530188; http://dx.doi.org/10.1073/pnas.1215145110
- Kobayashi H, Sakurai T, Miura F, Imai M, Mochiduki K, Yanagisawa E, Sakashita A, Wakai T, Suzuki Y, Ito T, et al. High-resolution DNA methylome analysis of primordial germ cells identifies gender-specific reprogramming in mice. Genome Res 2013; 23:616–27; PMID:23410886; http://dx.doi.org/10.1101/gr.148023.112
- Jenkins TG, Aston KI, Trost C, Farley J, Hotaling JM, Carrell DT. Intra-sample heterogeneity of sperm DNA methylation. Mol Hum Reprod 2015; 21:313–9; PMID:25542834; http://dx.doi.org/10.1093/molehr/gau115
- Singer ZS, Yong J, Tischler J, Hackett JA, Altinok A, Surani MA, Cai L, Elowitz MB. Dynamic heterogeneity and DNA methylation in embryonic stem cells. Mol Cell 2014; 55:319–31; PMID:25038413; http://dx.doi.org/10.1016/j.molcel.2014.06.029
- Shao X, Zhang C, Sun M-A, Lu X, Xie H. Deciphering the heterogeneity in DNA methylation patterns during stem cell differentiation and reprogramming. BMC Genomics 2014; 15:978; PMID:25404570; http://dx.doi.org/10.1186/1471-2164-15-978
- Brocks D, Assenov Y, Minner S, Bogatyrova O, Simon R, Koop C, Oakes C, Zucknick M, Lipka DB, Weischenfeldt J, et al. Intratumor DNA methylation heterogeneity reflects clonal evolution in aggressive prostate cancer. Cell Rep 2014; 8:798–806; PMID:25066126; http://dx.doi.org/10.1016/j.celrep.2014.06.053
- Hutchinson JN, Raj T, Fagerness J, Stahl E, Viloria FT, Gimelbrant A, Seddon J, Daly M, Chess A, Plenge R. Allele-specific methylation occurs at genetic variants associated with complex disease. PLoS ONE 2014; 9:e98464; PMID:24911414; http://dx.doi.org/10.1371/journal.pone.0098464
- Wang Q, Jia P, Cheng F, Zhao Z. Heterogeneous DNA methylation contributes to tumorigenesis through inducing the loss of coexpression connectivity in colorectal cancer. Genes Chromosomes Cancer 2015; 54:110–21; PMID:25407423; http://dx.doi.org/10.1002/gcc.22224
- Kobayashi H, Sakurai T, Imai M, Takahashi N, Fukuda A, Yayoi O, Sato S, Nakabayashi K, Hata K, Sotomaru Y, et al. Contribution of intragenic DNA methylation in mouse gametic DNA methylomes to establish oocyte-specific heritable marks. PLoS Genet 2012; 8:e1002440; PMID:22242016; http://dx.doi.org/10.1371/journal.pgen.1002440
- Shirane K, Toh H, Kobayashi H, Miura F, Chiba H, Ito T, Kono T, Sasaki H. Mouse oocyte methylomes at base resolution reveal genome-wide accumulation of non-CpG methylation and role of DNA methyltransferases. PLoS Genet 2013; 9:e1003439; PMID:23637617; http://dx.doi.org/10.1371/journal.pgen.1003439
- Xu Z, Taylor JA. Genome-wide age-related DNA methylation changes in blood and other tissues relate to histone modification, expression and cancer. Carcinogenesis 2014; 35:356–64; PMID:24287154; http://dx.doi.org/10.1093/carcin/bgt391
- Ono R, Kobayashi S, Wagatsuma H, Aisaka K, Kohda T, Kaneko-Ishino T, Ishino F. A Retrotransposon-derived gene, PEG10, is a novel imprinted gene located on human chromosome 7q21. Genomics 2001; 73:232–7; PMID:11318613; http://dx.doi.org/10.1006/geno.2001.6494
- Mancini-DiNardo D, Steele SJS, Ingram RS, Tilghman SM. A differentially methylated region within the gene Kcnq1 functions as an imprinted promoter and silencer. Hum Mol Genet 2003; 12:283–94; PMID:12554682; http://dx.doi.org/10.1093/hmg/ddg024
- Ono R, Shiura H, Aburatani H, Kohda T, Kaneko-Ishino T, Ishino F. Identification of a Large Novel Imprinted Gene Cluster on Mouse Proximal Chromosome 6. Genome Res 2003; 13:1696–705; PMID:12840045; http://dx.doi.org/10.1101/gr.906803
- Lou S, Lee H-M, Qin H, Li J-W, Gao Z, Liu X, Chan LL, Lam VK, So W-Y, Wang Y, et al. Whole-genome bisulfite sequencing of multiple individuals reveals complementary roles of promoter and gene body methylation in transcriptional regulation. Genome Biol 2014; 15:408; PMID:25074712; http://dx.doi.org/10.1186/s13059-014-0408-0
- Hahn MA, Wu X, Li AX, Hahn T, Pfeifer GP. Relationship between gene body DNA methylation and intragenic H3K9me3 and H3K36me3 chromatin marks. PLoS ONE 2011; 6:e18844; PMID:21526191; http://dx.doi.org/10.1371/journal.pone.0018844
- Seisenberger S, Andrews S, Krueger F, Arand J, Walter J, Santos F, Popp C, Thienpont B, Dean W, Reik W. The dynamics of genome-wide DNA methylation reprogramming in mouse primordial germ cells. Mol Cell 2012; 48:849–62; PMID:23219530; http://dx.doi.org/10.1016/j.molcel.2012.11.001
- Hajkova P, Erhardt S, Lane N, Haaf T, El-Maarri O, Reik W, Walter J, Surani MA. Epigenetic reprogramming in mouse primordial germ cells. Mech Dev 2002; 117:15–23; PMID:12204247; http://dx.doi.org/10.1016/S0925-4773(02)00181-8
- Reik W, Dean W, Walter J. Epigenetic reprogramming in mammalian development. Science 2001; 293:1089–93; PMID:11498579; http://dx.doi.org/10.1126/science.1063443
- Okano M, Bell DW, Haber DA, Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for De novo methylation and mammalian development. Cell 1999; 99:247–57; PMID:10555141; http://dx.doi.org/10.1016/S0092-8674(00)81656-6
- Morita-Fujimura Y, Tokitake Y, Matsui Y. Heterogeneity of mouse primordial germ cells reflecting the distinct status of their differentiation, proliferation and apoptosis can be classified by the expression of cell surface proteins integrin α6 and c-Kit. Dev Growth Differ 2009; 51:567–83; PMID:21314674; http://dx.doi.org/10.1111/j.1440-169X.2009.01119.x
- Jeltsch A, Jurkowska RZ. New concepts in DNA methylation. Trends Biochem Sci 2014; 39:310–8; PMID:24947342; http://dx.doi.org/10.1016/j.tibs.2014.05.002
- Krueger F, Andrews SR. Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 2011; 27:1571–2; PMID:21493656; http://dx.doi.org/10.1093/bioinformatics/btr167