
ABSTRACT
Neuroblastoma is a very heterogeneous tumor of childhood. The clinical spectra range from very aggressive metastatic disease to spontaneous regression, even without therapy. Aberrant DNA methylation pattern is a common feature of most cancers. For neuroblastoma, it has been demonstrated both for single genes as well as genome-wide, where a so-called methylator phenotype has been described. Here, we present a study using Illumina 450K methylation arrays on 60 neuroblastoma tumors. We show that aggressive tumors, characterized by International Neuroblastoma Risk Group (INRG) as stage M, are hypermethylated compared to low-grade tumors. On the contrary, INRG stage L tumors display more non-CpG methylation. The genes with the highest number of hypermethylated CpG sites in INRG M tumors are TERT, PCDHGA4, DLX5, and DLX6-AS1. Gene ontology analysis showed a representation of neuronal tumor relevant gene functions among the differentially methylated genes. For validation, we used a set of independent tumors previously analyzed with the Illumina 27K methylation arrays, which confirmed the differentially methylated sites. Top candidate genes with aberrant methylation were analyzed for altered gene expression through the R2 platform (http://r2.amc.nl), and for correlations between methylation and gene expression in a public dataset. Altered expression in nonsurvivors was found for the genes B3GALT4 and KIAA1949, CLIC5, DLX6-AS, TERT, and PIRT, and strongest correlations were found for TRIM36, KIAA0513, and PIRT. Our data indicate that methylation profiling can be used for patient stratification and informs on epigenetically deregulated genes with the potential of increasing our knowledge about the underlying mechanisms of tumor development.
Abbreviations
| CpG | = | cytosine-phosphate-guanine |
| DMR | = | differentially methylated region |
| MNA | = | MYCN amplification |
| MVP | = | methylaton variable position |
| MYCN | = | v-myc avian myelocytomatosis viral oncogene neuroblastoma derived homolog |
| NB | = | neuroblastoma |
Introduction
Neuroblastoma (NB) is a pediatric extracranial solid tumor of the peripheral sympathetic nervous system showing vast clinical heterogeneity. The tumors are characterized by patterns of genetic aberrations with unbalanced translocations and focal changes in ploidy. The proto-oncogene MYCN is amplified in 20% of the primary tumors,Citation1 and is associated with a more aggressive disease.Citation2 Rare but recurrent somatic mutations have been identified in the anaplastic lymphoma receptor tyrosine kinase (ALK) gene in both sporadic and familial cases.Citation3-7 The International Neuroblastoma Risk Group (INRG) classification system classifies tumors into localized tumors (stage L1 and L2), metastatic tumors (M), and metastatic tumors with particular features in children younger than 18 months (MS).Citation8,9
Aberrant DNA methylation has emerged as an important feature of both development and progression in many cancers. Although the full scope of regulatory mechanisms exerted from DNA methylations remain to be elucidated, certain modes of the regulatory effect on expression patterns have been discovered. For instance the hypermethylation of CpG islands in promoter regions are generally associated with reduced transcriptional activity. Thus, this adds to yet another mechanism that can silence a tumor suppressor gene, apart from deletional events and mutational gene silencing, and thus contribute to induction of malignancy. However, not only gene promoter regions harbor deregulated methylation sites with transcriptional regulatory impact. In other malignancies, the combined analysis of global DNA methylation patterns and global transcription patterns have revealed that hypomethylation of intragenic methylation sites are associated with increased gene expression, but for some genes also the opposite is true. In neuroblastoma the transcriptional regulation of DNA methylation has also been shown to be valid for a smaller subset of genes.Citation10,11 Furthermore, genomic regions such as enhancers and silencers are known to have cis-regulatory importance but can be positioned distant from its target gene. Recently, species of non-coding RNAs have emerged as important regulators of cancer, and the dysregulation of these RNAs have been reported to be associated with regional methylation alterations in neuroblastoma.Citation12
Most analyses of DNA methylation in NB tumors have been performed on single genes or with limited sample sizes. Several genes in various cellular pathways (apoptosis, cell cycle, differentiation, invasion, and metastasis) have been identified as aberrantly methylated. The study by Alaminos et al. was one of the first to demonstrate that the clustering of NB tumors based on the methylation profile of 10 genes could divide NB into clinical risk groups.Citation13 Since then, genome-wide analyses of DNA methylation have revealed a DNA methylator phenotype in NB with poor prognosis, characterized by the methylation of a set of multiple CpG islands.Citation14 A study of 7 genes in 2 NB cell lines (tumorigenic LA1-55n and non-tumorigenic LA1-5s) by Yang et al. reports that tumorigenic properties can be inhibited by reversing epigenetic changes with a demethylating agent, suggesting that epigenetic aberrations contribute to the NB phenotype.Citation15
Recent advances in high-throughput technology in the field of epigenetics now enable large-scale studies using bisulfite modified DNA hybridized to DNA methylation arrays. We have previously used the Illumina HumanMethylation27 DNA analysis BeadChip to determine methylation profiles of NB primary tumors and identified aberrantly methylated genes that can be used as biomarkers.Citation16 Extending on that study we now used the Illumina 450K arrays, profiling more than 450,000 CpG sites, for a large set of NB tumors.
Results
Clustering the samples based on methylation data identifies a NB methylator phenotype
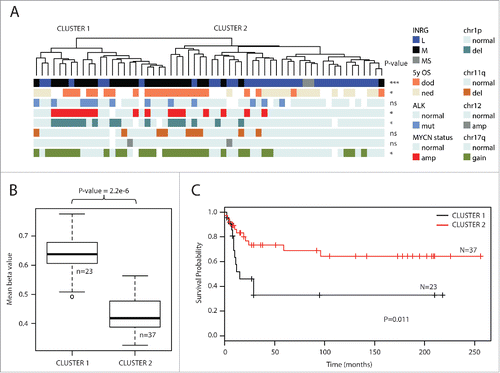
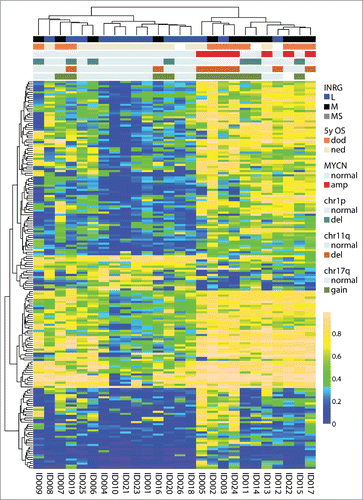
Genome-wide methylation analysis using the Infinium HumanMethylation450 BeadChips was performed on a set of 60 primary NB tumors. The sample set contained 17 tumors with MYCN amplification, 9 samples with 11q-deletion and 12 with ALK mutations, and is further described in Supplementary Table 1 and in . After quality control analysis and filtering of methylation data, a set of 364,774 probes remained for further analyses. To get an overview of the methylation patterns in the data, we applied unsupervised clustering of the 1% most variable probes (n=3,648). The analysis separated samples into 2 clusters () that discriminated also by INRG classification (P=0.00017), deletion of chromosome 1p (P=0.015) and on MYCN-amplification (MNA) (P=0.034) (). The two INRG MS cases in the cohort clustered together (). Furthermore, the probes that were used for clustering differed in mean methylation levels (, P=2.2×10−6, t-test), and in 5-year overall survival (OS) (P=0.014; ). The CpG site with largest difference in methylation β value between the 2 clusters was cg17878351, located in the gene CLIC5. As the samples separated most strongly based on INRG stage, we focused our further analyses on this grouping.
Figure 1. Top 1% most variable sites. (A) Clustering dendrogram from hierarchical clustering. Differences between clusters were tested with χ2 statistics (except for chr11q and chr12 that were tested with Fisher exact test due to low numbers). Abbreviations: amp, amplified; del, deletion; OS_5y, Overall survival 5 years; dod, dead of disease; ned, no evidence of disease (>60 months; samples with ned <60 months were blanked in display); chr1p, chromosome 1p; chr11q, chromosome 11q; chr12, chromosome 12; chr17q, chromosome 17q; ns, non-significant. (B) Boxplot presenting mean methylation level in cluster 1 compared with cluster 2 (P-value < 2.2×10−16, Student t-test). Upper and lower hinges of the box represent the 75th percentile and 25th percentile respectively; whiskers indicate the highest and lowest values that are not outliers; thick horizontal line within box, median. Open circles represent outliers. (C) Kaplan-Meier plot representing survival probability of cluster 1 and cluster 2. (P=0.011; log-rank test).
Sites with variable methylation are hypermethylated in INRG stage M tumors
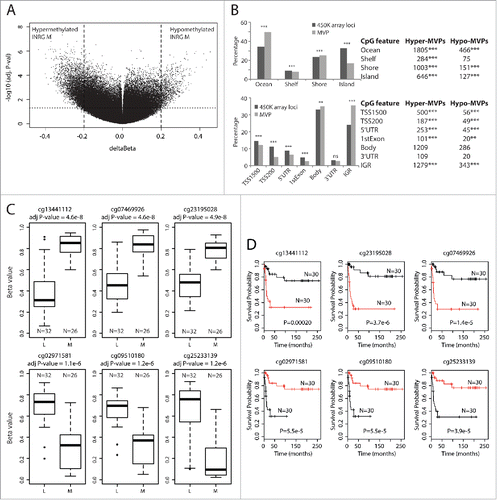
Using the ChAMP packageCitation17 we compared the groups of INRG metastatic (M, 26 samples) and INRG localized (L, 32 samples). In total, 37,891 probes gave an adjusted P-value below 0.05, and 4,557 of these probes showed a difference in β-value methylation between the INRG M and L groups that was larger than 0.2 (20%). We defined this set of probes as the Methylation Variable Positions (MVPs) ( and Supplementary Table 2). More than 80% of these MVPs were hypermethylated in INRG M (; 3,738 hyper- and 819 hypo-methylated). The genomic distribution of the MVPs in relation to CpG islands (CGI) was compared to the reference distribution of probes in relation to CGIs for the data set of 364,774 probes for each locational feature analyzed. MVPs were overrepresented in regions of low CpGs (“ocean”) and underrepresented in CGIs (P<0.001 for both, Pearson χ2 test). Also CGI shelves and shores showed a non-random distribution between the identified MVPs and the genomic CGI representation on the analyzed platform (; P<0.01 for shelves and shores, respectively). When the MVPs were divided into hyper- or hypo-MVPs, both the direction of distributional change and significance remained for probes located in oceans and CGI ( upper panel). In hyper-MVPs, shores were overrepresented compared to the reference distribution (P<0.0001, ), whereas localization of MVPs in shelves were underrepresented compared to reference (P<0.001, ). On the contrast, hypo-MVPs were underrepresented at shores compared to the reference (P<0.001; ).
Figure 2. INRG methylated variable positions. (A) Volcano plot showing the distribution between adjusted P-values and difference in β value (deltaBeta) from the ChAMP analysis of the 450K data of INRG M (n=26) and L (n=32) samples. Lines represent used cut-off values to identify the most hyper- and hypomethylated sites in INRG M (n=4557). (B) Distribution of hyper- and hypomethylated loci (MVP; methylated variable position) across CpG features (top) and gene regions (bottom). (C) The 3 INRG M hyper and hypo MVPs with smallest P-values. (D) Kaplan–Meier plots showing survival probability for subjects with above median methylation (red line; n=30), and below median methylation (black line; n=30) for the 3 INRG hyper, and hypo MVPs with lowest P-values. P-value of log-rank test is shown in each plot.
INRG differentially methylated sites are enriched in enhancer regions
When MVPs were mapped in relation to gene positions we found a significant overrepresentation of MVPs in intergenic regions (IGR; P<0.0001) and this significance remained also when subdividing into hyper- and hypo-methylated MVPs ( lower panel). An increased representation of MVPs in gene body (P<0.01) for MVPs was not seen when subdividing into hyper- and hypo-methylated MVPs. An overall underrepresentation in MVPs located in the first exon (P<0.0001, ) was present in both hyper- and hypo-MVPs (P<0.0001 and P<0.0012, respectively; ). Furthermore, there was an underrepresentation of MVPs in the upstream region of transcription start site (TSS) that remained when analyzed separately for in hyper- and hypo-MVPs, both in the region 200 bases upstream of TSS and in the region 1,500 bases upstream of TSS (P<0.0001 for all, lower panel). In line with this, there was an underrepresentation of MVPs in 5′ untranslated region (UTR). However, the representation of MVPs in 3′UTR was similar as in the reference data, both when hyper- and hypo-MVPs were analyzed together and separately. Both hypermethylated and hypomethylated MVPs were enriched in enhancer sites (P<0.0001), suggesting a regulatory function of the differential methylation.
Location of MVPs
The distributions of the 3 most discriminatory hyper-MVPs and 3 hypo-MVPs for INRG are shown in . The cg13441112 is located on chromosome region 1q32 within the cluster of complement factor H related genes. Its genomic feature is classified as intergenic, although its position can be intronic by alternative splicing of the nearby complement factor H-related 4 (CFHR4) gene. The INRG M hypermethylated cg07469926 and cg23195028 are located in gene bodies of the genes tripartite motif containing 36 (TRIM36; chr5q22.3) and protein phosphatase 2, regulatory subunit B, gamma (PPP2R2C; chr4p16.1), respectively. The three most selective hypomethylated probes were located in intergenic regions on chromosome region 1p36 (cg02971581 and cg09510180), and on chromosome 13 (cg25233139). Kaplan-Meier survival curves based on the methylation of these sites are shown in and Kaplan-Meier curves of INRG stage, 11q-deletion and MYCN-amplification in Supplementary Fig. 1A. The gene with the associated probe with largest difference in methylation β value between INRG M and L was the PIRT gene, harboring 3 of the top 5 hyper-MVP sites (Δβ of 0.4–0.45, all sites located in the TSS200 region).
We next explored the predictive capacity of using tumor methylation signatures for improved patient stratification for the 23 patients who died from the disease and the 18 patients with >60 months of no evidence of disease. We performed multivariate analysis (logit regression) using INRG stage and MNA status as predictors of outcome, which gave a predictive accuracy of AUC = 0.86. When using the methylation cluster and the 6 top discriminating CpG sites mentioned above, the predictive accuracy increased to 0.95 (P-value for difference between the models P<0.05). Combining the methylation factors with INRG stage and MNA status did not further increase the accuracy of the model (AUC = 0.96).
The 4,557 identified MVPs mapped to 1,516 genes; out of these, 1,265 genes were hypermethylated and 290 genes hypomethylated (Supplementary Table 2) in the INRG M group. The genes with the highest number of MVPs () were the hypermethylated genes protocadherin gamma subfamily A, 4 (PCDHGA4; chr5q31) and telomerase reverse transcriptase (TERT; chr5p15). PCDHGA4 is located in a cluster of multiple overlapping protocadherin genes and has previously been recognized as hypermethylated in NB.Citation14 The TERT gene had 24 MVPs, the majority of which were located in the gene body (). The gene list also contains the 2 adjacent genes DLX5 and DLX6-AS1, the latter one transcribes antisense of the DLX6 gene. Genes with most hypomethylated MVPs in INRG M included XKR4, KNDC1, ABLIM1, BSX, CYTL1, ITGBL1, KIAA0513, MOG, C1orf92, and CDH22 (). Genes with both hyper- and hypo-MVPs were also identified (n=39; detailed in Supplementary Table 3). The gene PR domain containing 16 (PRDM16), located at chromosome 1p36, a region frequently deleted in neuroblastoma, was the only gene showing at least 2 of each hyper- and hypo-MVPs.
Figure 3. Hypermethylated genes in INRG stage (M) compared to stage L. (A) Gene plot of the TERT gene. The TERT gene represented by a UCSC track (http://genome.ucsc.edu/),Citation32 showing TERT transcripts and positions of 450K probes (upper panel). Gene plot showing mean methylation levels of INRG M, and L for all probes on the Illumina 450K platform annotated to TERT gene (lower panel); (B) The 10 gene ontologies (GO) with lowest P-value from functional GO analysis using the DAVID software. Adj P-value; Benjamini adjusted P-value. (C) Chromosomal distribution of genes at INRG DMRs.
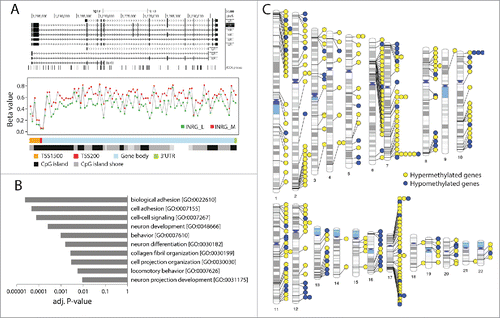
Table 1. Genes with highest number of hyper or hypo MVPs in INRG M group.
Gene ontology analysis identifies NB relevant terms
To evaluate the functions of the genes with associated MVPs we used gene ontology classification through the DAVID bioinformatics resource.Citation18 Genes containing either hyper- or hypo-methylated MVPs were submitted to default settings in DAVID. This analysis showed a representation of neuronal tumor relevant gene functions, such as cell adhesion, neuronal development, neuronal differentiation, and neuron projection development ().
Differentially methylated regions
To identify coherent chromosomal areas of aberrant methylation the dataset was analyzed for differentially methylated regions (DMRs). Using the default P-value of 0.01 in the ChAMP package we identified 459 DMRs (spanning 21–7,185 base pairs) distributed over 2,425 probes. One fourth (26%) of these probes was classified as MVPs in the above analysis. The DMRs represented 283 annotated genes out of which 204 genes were included among the identified MVP genes above. The DMRs with the lowest P-values are listed in (B3GALT4, KIAA1949, DLX6AS, HLA-DOA, intergenic region at 12q24.33, GABBR1). As we noted an uneven distribution among the chromosomes for the DMRs, we visualized the genomic locations of DMRs located to genes (). Interestingly, multiple DMRs were located at chromosomal regions 1p36 and 17q, 2 regions that frequently show copy number alterations in neuroblastoma, and on chromosome 6 several genes with DMRs were located in the MHC region.
Table 2. The strongest DMRs when comparing INRG groups.
Analysis of other prognostic factors in NB and overall survival
As a next step, we looked for methylation discriminating between samples with or without MYCN-amplification, 5-year OS, and 11q deletion. We applied the same strategy for MVP calling (volcano plots are shown in Supplementary Fig. 2). The genes with the highest number of MVPs in each comparison are listed in Supplementary Tables 4–6. Boxplots of the 3 probes with lowest P-values for both increased and decreased methylation are shown in Supplementary Figs. 1B–D for each of the 3 comparisons with and without MNA, with and without 11q deletion, and 5-year OS vs. dead of disease. The lowest P-value was found for the probe cg20678442 (chromosome region 1q44) that gave a complete separation for tumors with and without MNA (adjusted P=4.2×10−21, Supplementary Fig. 2B). The distributions of overlaps of genes with MVPs between all analyses (INRG, 5-year OS, MNA, and 11q) are shown in Supplementary Fig. 3 (Venn diagram), and the 102 genes with MVPs in all 4 analyses are listed in Supplementary Table 7. Only 8 probes were called MVPs in all 4 analyses and these were located in the genes HORMAD2 (2 probes), SUSD5, PPAPDC1A, NKPD1 and 2 intergenic probes located on chromosome 2 and 9 (Supplementary Fig. 4, Supplementary Table 8).
Non-CpG methylation separates NB patients
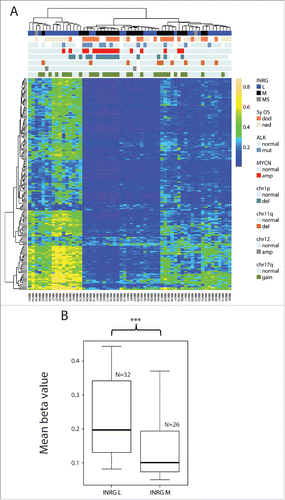
The Illumina arrays also contain 3,091 non-CpG sites, out of which 1,678 remained after filtering (see Materials & Methods). Cluster analysis of the 10% most variable sites (n=168) separated the patients into separate clusters, of which also here INRG was the strongest clinical variable (). For non-CpG sites, however, the highest methylation frequency was found in the INRG stage L (P=0.001; compared to INRG M, ).
Figure 4. Non-CpG methylation. (A) Hierarchical cluster analysis of most variable non-CpG sites (n=168 sites) defines 3 main groups of patients with different methylation frequency. (B) Boxplot presenting mean methylation level of non-CpG sites in INRG L (n=32) compared with INRG M (n=26).
Validation of the obtained methylation profiles using published data sets
For confirmation of the results we used a set of 26 primary NB tumors with INRG classification previously analyzed on the Illumina 27K platform.Citation11 Samples that were analyzed on both platforms showed, as expected, a strong correlation of β values for probes present on both platforms. The validation set contained 187 probes that overlapped with the 4,557 MVPs. We used unsupervised hierarchical clustering of these 187 probes measurements on the 27K platform. The generated clustermap separated both on INRG and survival (; 2-sided Fisher exact probability test P=0.045 and P=0.013, respectively). In addition, we used a recent published data set of 450K data from 35 NB tumors (GEO accession GSE54719) annotated with INSS stage.Citation19 Following unsupervised hierarchical clustering of the 4,557 MVPs in this dataset, the clusters separated on INSS (P-value=0.025 2-sided Fishers exact test of stages 1–3 vs. stage 4; Supplementary Fig 5A). The distribution of the 3 most top ranked hyper- and hypo-MVPs in this data set showed a similar trend as in our dataset (Supplementary Fig 5B).
Figure 5. Validation of the INRG separation from the 450K results with 27K data. 187 probes from the intersect that were on both the 450K and the 27K platform separated the samples on cluster 1 and cluster 2 (both on INRG and survival, 2-sided Fisher exact probability test P=0.045 and P=0.013, respectively). OS, overall survival; dod, dead of disease; ned, no evidence of disease; amp, amplified; del, deletion.
Expression of genes with differential methylation in prognostic groups
To further look into the biological relevance of the genes with identified MVPs, we used the R2: Genomics Analysis and Visualization Platform (http://r2.amc.nl). We used the Kocak set of 649 NB tumors (GEO ID: GSE45547).Citation20 Many of the genes identified in the current study also showed significant differences in gene expression when it comes to overall survival of the patients (Supplementary Fig 6). For example, the genes B3GALT4 and KIAA1949, identified as hypermethylated in INRG stage M showed lower gene expression in patients who died from their disease (Bonferroni corrected P-values 5.4 * 10−14 and 1.6 * 10−17, respectively). The gene DLX6AS1, which showed hypermethylation in the gene body (indicative of stable gene expression), showed high expression in patients who died from their disease (Bonferroni corrected P-values, 5.7 * 10−13). The CLIC5 gene, with the top site that discriminated the 2 methylation clusters, showed lower expression in non-survivors (Bonferroni corrected P-values, 2.1 * 10−5). We also verified that the same was true using other gene expression data sets (using the SEQC_NB data set composed of 498 samples, GEO ID: GSE62564). The PIRT gene, with the CpG sites with the largest difference in methylation value between INRG stage M and L, showed lower expression in non-survivors (Bonferroni corrected P-values, 3.3 * 10−22; SEQC_NB data set – gene not included in the Kocak set).
Correlation of DNA methylation and gene expression in neuroblastoma tumors
To further explore if the identified genes with differential methylation and differential expression were also correlated to each other in individual tumors, we used a recently published smaller set of NB tumors for which paired methylation and gene expression data was available for 19 samples (GEO accession numbers GSE54719 and GSE54720).Citation19 Correlation between DNA methylation and gene expression with nominal significance P<0.05 were found for 16 of 39 genes tested (Supplementary Table 9). The three strongest correlations were found for TRIM36 (rho=-0.83), KIAA0513 (rho=0.80), and PIRT (rho=-0.79); for TRIM36 and KIAA0513, the tested methylation site was located in the corresponding gene body; for PIRT, the methylation site was located within 200 bases upstream of the transcription start site (Supplementary Table 9 and Supplementary Fig. 5C).
Discussion
Current risk stratification for NB tumors is based on MYCN-amplification status and presence of chromosomal segmental breaks. Attempts have been made to improve classification based on methylation of single-genes, or genome-wide profiles.Citation16,21–26 The studies are often limited to small cohorts without validation sets. Here we present a larger study of 60 patients using Illumina methylation arrays, which profile 485,000 CpG sites; as validation, we use our previously published study using the 27,000 CpG site platform.Citation11 Cluster analysis identified 2 clusters with different methylation levels: the hypermethylated cluster had a mean methylation of 0.64 and the hypomethylated cluster had a mean methylation of 0.43. The patients in the hypermethylated cluster had a poorer survival than the other group of patients.
The methylation profiles separated the NB tumors into clinically relevant groups, of which INRG stage was the strongest clinical factor. Gene ontology analysis of the identified genes identified relevant terms as cell adhesion, neuronal development, neuronal differentiation, and neuron projection development. One of the top differentially methylated genes in INRG stage M was the telomerase reverse transcriptase gene TERT. Telomerase expression plays a role in cellular senescence, as it is normally repressed in postnatal somatic cells resulting in progressive shortening of telomeres. Deregulation of telomerase expression in somatic cells may be involved in oncogenesis. Gene expression of TERT is significantly higher in NB patients who die from their disease (R2: Genomics Analysis and Visualization Platform). Methylation of TERT has recently been proposed as a biomarker for risk stratification of childhood brain tumors, as hypermethylation of a region upstream of the transcription start site of TERT was found to be associated with tumor progression and poor prognosis.Citation27 A high-risk neuroblastoma subgroup defined by genomic rearrangements causing increased expression of TERT was recently presented.Citation28,29 In our study, TERT had the highest number of hypermethylated sites in INRG M (), suggesting altered epigenetic regulation, which is consistent with a high-risk TERT subgroup, although the genomic rearrangements are not known for our samples. TRIM36 was hypermethylated in INRG stage M and has lower expression in stage M tumors. Overexpression of TRIM36 has been described to decelerate the cell cycle and limit cell growthCitation28 and TRIM-NHL proteins are important regulators of developmental transitions, for example, by promoting differentiation and repressing cell growth and proliferation in stem and progenitor cells.Citation29 Also, the PIRT gene shows the same pattern in NB. PIRT functions as a regulatory subunit of the TRPV1 protein with roles in cell migration, cytoskeleton reorganization, and in neuronal guidance. TRPV1 could be important during neuronal differentiation as its expression is increased during all-trans-retinoic acid differentiation in NB cell lines.Citation30
As validation, we used a set of NB tumors previously analyzed on the Illumina 27K platformCitation11 and also here separate clusters were formed where the hypermethylated group of patients was associated with INRG stage M and a poor survival.
Our results fit well into and extend on previous studies. A hypermethylator phenotype linked to poor outcome has been suggested previously, which our study supports.Citation14 For single genes, for example the KRT19 gene has previously been suggested as a marker of clinical risk factors in NB and was also in the current study identified as differentially methylated in INRG stage M compared to stage L.Citation11,21 A recent study of a smaller number of NB tumors also reported on differences in non-CpG methylation where the presence of non-CpG methylation was mostly associated with tumors characterized by favorable clinicobiological features,Citation19 which the current study confirms.
Our data show that methylation profiles can separate NB tumors into relevant clinical groups. It also highlights genes that, in addition to the differential DNA methylation, also show differential gene expression of the respective gene, suggesting that these genes might have a role in tumor development or progression, which needs further functional studies to address.
Materials and methods
Patients and samples
A panel of 60 neuroblastoma tumors was used in this study (Supplementary Table 1). Ethical permission was granted by the local ethics committee.
DNA extraction and bisulfite modification
DNA extraction was carried out with the DNeasy Blood & Tissue Kit (Qiagen, Hilden, Germany), including a RNA removal step, according to the protocol provided by the supplier. The DNA was quantified with the Nanodrop and 1 ug was used for bisulfite modification using the EZ DNA methylation kit (D5001, Zymo Research, Orange, CA) according to the protocol provided with the modification step according to the recommendations for array processing of the samples. Control PCR reactions were carried out before array analysis to confirm successful modification of the DNA.
DNA methylation arrays
The bisulfite-modified DNA (500 ng) was applied to the Infinium HumanMethylation450 BeadChips (Illumina), which determine the methylation levels of 485,000 CpG sites.Citation31 After bisulfite treatment of the DNA samples, the cytosines in the CpG sites were genotyped as C/T polymorphisms according to the manufacturer's protocol. The fluorescence signals were measured from the BeadArrays using an Illumina BeadStation GX scanner. The fluorescence data were then analyzed using the GenomeStudio software (Illumina). The software assigns a score called a “β value” to each CpG site, which corresponds to the ratio between the fluorescence signal of the methylated allele (C) and the sum of the fluorescent signals of the methylated (C) and unmethylated (T) alleles. The data generated by the BeadStudio software was exported and further analyses were performed in the R programming environment.
The R-package ChAMPCitation17 was used for data preprocessing, normalization and comparison between groups. Singular value decomposition analysis was performed to identify confounding factors.
Positions on the X and Y chromosome were filtered away using ChAMP and we applied a filter for SNPs from the IMA package (https://www.rforge.net/IMA/); hereafter, a set of 364,774 probes remained for analyses. Probes with highest variation were identified using median absolute deviation. ChAMP was used to calculate probes that were differentially methylated between groups. We defined methylated variable positions (MVPs). We set the criteria for MVP as calling at significance of a Benjamini-Hochberg adjusted P<0.05 and a difference in β-value between groups larger than 0.2. Hierarchical clustering of samples was done with the heatmap function in the R NMF package using Euclidean distances. Samples classified as no-evidence-of-disease (NED) with less than 60 months follow-up time (n=19) were omitted from the survival analysis.
Analysis of differentially methylated regions (DMRs) was done using ChAMP. The web-based program Phenogram (http://visualization.ritchielab.psu.edu/) was used to map and visualize chromosomal positions of DMRs located to genes. When a DMR included more than one gene it was mapped to the gene with most probes or if equal to the gene with the strongest signal.
Differences between groups were tested with t-test, Pearson χ2 square test or by Fisher exact 2-tailed test as appropriate. Spearman rank correlation was used to test for correlation between expression and methylation. Log-rank test was used to compare survival distributions.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Supplemental_Datas.zip
Download Zip (3.1 MB)Acknowledgments
This work was supported by BioCARE - a National Strategic Research Program at University of Gothenburg, The Swedish Cancer Society, the Swedish Children's Cancer Society, the Harald och Greta Jeanssons Stiftelse, the Magnus Bergvall foundation, the Assar Gabrielsson foundation, the Ollie och Elof Ericsson Stiftelse, the Mary Béves Stiftelse för Barncancerforskning, Sven och Dagmar Saléns stiftelse, Lions Cancerfond Väst and the Royal Physiographic Society in Lund. HC was supported by the Swedish Research Council and the Wenner-Gren foundation.
References
- Carén H, Erichsen J, Olsson L, Enerbäck C, Sjöberg RM, Abrahamsson J, Kogner P, Martinsson T. High-resolution array copy number analyses for detection of deletion, gain, amplification and copy-neutral LOH in primary neuroblastoma tumors: Four cases of homozygous deletions of the CDKN2A gene. BMC Genomics 2008; 9:353; http://dx.doi.org/10.1186/1471-2164-9-353
- Carén H, Kryh H, Nethander M, Sjöberg RM, Träger C, Nilsson S, Abrahamsson J, Kogner P, Martinsson T. High-risk neuroblastoma tumors with 11q-deletion display a poor prognostic, chromosome instability phenotype with later onset. Proc Natl Acad Sci U S A 2010; 107:4323–8; http://dx.doi.org/10.1073/pnas.0910684107
- Carén H, Abel F, Kogner P, Martinsson T. High incidence of DNA mutations and gene amplifications of the ALK gene in advanced sporadic neuroblastoma tumours. Biochem J 2008; 416:153–9; http://dx.doi.org/10.1042/BJ20081834
- Mosse YP, Laudenslager M, Longo L, Cole KA, Wood A, Attiyeh EF, Laquaglia MJ, Sennett R, Lynch JE, Perri P, et al. Identification of ALK as a major familial neuroblastoma predisposition gene. Nature 2008; 455(7215):930–5; PMID:18724359
- Chen Y, Takita J, Choi YL, Kato M, Ohira M, Sanada M, Wang L, Soda M, Kikuchi A, Igarashi T, et al. Oncogenic mutations of ALK kinase in neuroblastoma. Nature 2008; 455:971–4; PMID:18923524; http://dx.doi.org/10.1038/nature07399
- George RE, Sanda T, Hanna M, Frohling S, Luther W, 2nd, Zhang J, Ahn Y, Zhou W, London WB, McGrady P, et al. Activating mutations in ALK provide a therapeutic target in neuroblastoma. Nature 2008; 455:975–8; PMID:18923525; http://dx.doi.org/10.1038/nature07397
- Janoueix-Lerosey I, Lequin D, Brugieres L, Ribeiro A, de Pontual L, Combaret V, Raynal V, Puisieux A, Schleiermacher G, Pierron G, et al. Somatic and germline activating mutations of the ALK kinase receptor in neuroblastoma. Nature 2008; 455:967–70; PMID:18923523; http://dx.doi.org/10.1038/nature07398
- Cohn SL, Pearson AD, London WB, Monclair T, Ambros PF, Brodeur GM, Faldum A, Hero B, Iehara T, Machin D, et al. The International Neuroblastoma Risk Group (INRG) classification system: an INRG Task Force report. J Clin Oncol 2009; 27:289–97; PMID:19047291; http://dx.doi.org/10.1200/JCO.2008.16.6785
- Monclair T, Brodeur GM, Ambros PF, Brisse HJ, Cecchetto G, Holmes K, Kaneko M, London WB, Matthay KK, Nuchtern JG, et al. The International Neuroblastoma Risk Group (INRG) staging system: an INRG Task Force report. J Clin Oncol 2009; 27:298–303; PMID:19047290; http://dx.doi.org/10.1200/JCO.2008.16.6876
- Sugito K, Kawashima H, Uekusa S, Yoshizawa S, Hoshi R, Furuya T, Kaneda H, Hosoda T, Masuko T, Ohashi K, et al. Identification of aberrant methylation regions in neuroblastoma by screening of tissue-specific differentially methylated regions. Pediatr Blood Cancer 2013; 60:383–9; PMID:22911660; http://dx.doi.org/10.1002/pbc.24282
- Carén H, Djos A, Nethander M, Sjöberg RM, Kogner P, Enström C, Nilsson S, Martinsson T. Identification of epigenetically regulated genes that predict patient outcome in neuroblastoma. BMC Cancer 2011; 11:66; http://dx.doi.org/10.1186/1471-2407-11-66
- Pandey GK, Mitra S, Subhash S, Hertwig F, Kanduri M, Mishra K, Fransson S, Ganeshram A, Mondal T, Bandaru S, et al. The risk-associated long noncoding RNA NBAT-1 controls neuroblastoma progression by regulating cell proliferation and neuronal differentiation. Cancer Cell 2014; 26:722–37; PMID:25517750; http://dx.doi.org/10.1016/j.ccell.2014.09.014
- Alaminos M, Davalos V, Cheung NK, Gerald WL, Esteller M. Clustering of gene hypermethylation associated with clinical risk groups in neuroblastoma. J Natl Cancer Inst 2004; 96:1208–19; PMID:15316056; http://dx.doi.org/10.1093/jnci/djh224
- Abe M, Ohira M, Kaneda A, Yagi Y, Yamamoto S, Kitano Y, Takato T, Nakagawara A, Ushijima T. CpG island methylator phenotype is a strong determinant of poor prognosis in neuroblastomas. Cancer Res 2005; 65:828–34; PMID:15705880
- Yang Q, Tian Y, Ostler KR, Chlenski A, Guerrero LJ, Salwen HR, Godley LA, Cohn SL. Epigenetic alterations differ in phenotypically distinct human neuroblastoma cell lines. BMC Cancer 2010; 10:286; PMID:20546602; http://dx.doi.org/10.1186/1471-2407-10-286
- Djos A, Martinsson T, Kogner P, Carén H. The RASSF gene family members RASSF5, RASSF6 and RASSF7 show frequent DNA methylation in neuroblastoma. Mol Cancer 2012; 11:40; PMID:22695170; http://dx.doi.org/10.1186/1476-4598-11-40
- Morris TJ, Butcher LM, Feber A, Teschendorff AE, Chakravarthy AR, Wojdacz TK, Beck S. ChAMP: 450k Chip Analysis Methylation Pipeline. Bioinformatics 2014; 30:428–30; PMID:24336642; http://dx.doi.org/10.1093/bioinformatics/btt684
- Dennis G, Jr., Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, Lempicki RA. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol 2003; 4:P3; PMID:12734009; http://dx.doi.org/10.1186/gb-2003-4-5-p3
- Gomez S, Castellano G, Mayol G, Sunol M, Queiros A, Bibikova M, Nazor KL, Loring JF, Lemos I, Rodriguez E, et al. DNA methylation fingerprint of neuroblastoma reveals new biological and clinical insights. Epigenomics 2015 Oct; 7(7):1137-53; PMID:25687459
- Kocak H, Ackermann S, Hero B, Kahlert Y, Oberthuer A, Juraeva D, Roels F, Theissen J, Westermann F, Deubzer H, et al. Hox-C9 activates the intrinsic pathway of apoptosis and is associated with spontaneous regression in neuroblastoma. Cell Death Dis 2013; 4:e586; PMID:23579273; http://dx.doi.org/10.1038/cddis.2013.84
- Decock A, Ongenaert M, Hoebeeck J, De Preter K, Van Peer G, Van Criekinge W, Ladenstein R, Schulte JH, Noguera R, Stallings RL, et al. Genome-wide promoter methylation analysis in neuroblastoma identifies prognostic methylation biomarkers. Genome Biol 2012; 13:R95; PMID:23034519; http://dx.doi.org/10.1186/gb-2012-13-10-r95
- Dreidax D, Bannert S, Henrich KO, Schroder C, Bender S, Oakes CC, Lindner S, Schulte JH, Duffy D, Schwarzl T, et al. p19-INK4d inhibits neuroblastoma cell growth, induces differentiation and is hypermethylated and downregulated in MYCN-amplified neuroblastomas. Hum Mol Genet 2014; 23:6826–37; PMID:25104850; http://dx.doi.org/10.1093/hmg/ddu406
- Hoebeeck J, Michels E, Pattyn F, Combaret V, Vermeulen J, Yigit N, Hoyoux C, Laureys G, De Paepe A, Speleman F, et al. Aberrant methylation of candidate tumor suppressor genes in neuroblastoma. Cancer Lett 2009; 273:336–46; PMID:18819746; http://dx.doi.org/10.1016/j.canlet.2008.08.019
- Buckley PG, Das S, Bryan K, Watters KM, Alcock L, Koster J, Versteeg R, Stallings RL. Genome-wide DNA methylation analysis of neuroblastic tumors reveals clinically relevant epigenetic events and large-scale epigenomic alterations localized to telomeric regions. Int J Cancer 2011; 128:2296–305; PMID:20669225; http://dx.doi.org/10.1002/ijc.25584
- Abe M, Watanabe N, McDonell N, Takato T, Ohira M, Nakagawara A, Ushijima T. Identification of genes targeted by CpG island methylator phenotype in neuroblastomas, and their possible integrative involvement in poor prognosis. Oncology 2008; 74:50–60; PMID:18544995; http://dx.doi.org/10.1159/000139124
- Carén H, Fransson S, Ejeskär K, Kogner P, Martinsson T. Genetic and epigenetic changes in the common 1p36 deletion in neuroblastoma tumours. Br J Cancer 2007; 97:1416–24; http://dx.doi.org/10.1038/sj.bjc.6604032
- Castelo-Branco P, Choufani S, Mack S, Gallagher D, Zhang C, Lipman T, Zhukova N, Walker EJ, Martin D, Merino D, et al. Methylation of the TERT promoter and risk stratification of childhood brain tumours: an integrative genomic and molecular study. Lancet Oncol 2013; 14:534–42; PMID:23598174; http://dx.doi.org/10.1016/S1470-2045(13)70110-4
- Miyajima N, Maruyama S, Nonomura K, Hatakeyama S. TRIM36 interacts with the kinetochore protein CENP-H and delays cell cycle progression. Biochem Biophys Res Commun 2009; 381:383–7; PMID:19232519; http://dx.doi.org/10.1016/j.bbrc.2009.02.059
- Tocchini C, Ciosk R. TRIM-NHL proteins in development and disease. Semin Cell Dev Biol 2015; 47–48:52–9; PMID:26514622
- El Andaloussi-Lilja J, Lundqvist J, Forsby A. TRPV1 expression and activity during retinoic acid-induced neuronal differentiation. Neurochem Int 2009; 55:768–74; PMID:19651168; http://dx.doi.org/10.1016/j.neuint.2009.07.011
- Bibikova M, Barnes B, Tsan C, Ho V, Klotzle B, Le JM, Delano D, Zhang L, Schroth GP, Gunderson KL, et al. High density DNA methylation array with single CpG site resolution. Genomics 2011; 98:288–95; PMID:21839163; http://dx.doi.org/10.1016/j.ygeno.2011.07.007
- Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D. The human genome browser at UCSC. Genome Res 2002; 12:996–1006; PMID:12045153; http://dx.doi.org/10.1101/gr.229102