
ABSTRACT
Based on the methylation status of 5 single CpG sites, a novel epigenetic classification of chronic lymphocytic leukemia (CLL) was recently proposed, classifying CLL patients into 3 clinico-biological subgroups with different outcome, termed memory like CLL (m-CLL), naïve like CLL (n-CLL), and a third intermediate CLL subgroup (i-CLL). While m-CLL and n-CLL patients at large corresponded to patients carrying mutated and unmutated IGHV genes, respectively, limited information exists regarding the less defined i-CLL group. Using pyrosequencing, we investigated the prognostic impact of the proposed 5 CpG signature in a well-characterized CLL cohort (135 cases), including IGHV-mutated and unmutated patients as well as clinically aggressive stereotyped subset #2 patients. Overall, we confirmed the signature's association with established prognostic markers. Moreover, in the presence of the IGHV mutational status, the epigenetic signature remained independently associated with both time-to-first-treatment and overall survival in multivariate analyses. As a prime finding, we observed that subset #2 patients were predominantly classified as i-CLL, probably reflecting their borderline IGHV mutational status (97–99% germline identity), though having a similarly poor prognosis as n-CLL patients. In summary, we validated the epigenetic classifier as an independent factor in CLL prognostication and provide further evidence that subset #2 is a member of the i-CLL group, hence supporting the existence of a third, intermediate epigenetic subgroup.
Introduction
Epigenetic alterations such as aberrant DNA methylation are implicated in the development of all types of cancer, including hematological malignancies.Citation1 In chronic lymphocytic leukemia (CLL), previous reports have revealed an altered methylation status of the promoter regions in a number of genes, e.g., ZAP70, LPL, KIBRA, HOXA, TWIST, and BTG4, also shown to correlate with clinical outcome and proposed as prognostic markers.Citation2-13 In recent years, taking advantage of high-density microarrays, the global methylation patterns have been investigated in major prognostic subgroups, such as favorable-prognostic IGHV-mutated (M-CLL) and poor-prognostic IGHV-unmutated CLL (U-CLL).Citation14-16 These studies demonstrated differential genome-wide methylation profiles between M-CLL and U-CLL, which were notably stable over time and highly similar within different CLL compartments,Citation14,16 thus implying a different cellular origin for these 2 subgroups and that epigenetic changes occur as early leukemogenic events.Citation16
More recently, whole-genome bisulfite sequencing and 450K methylation microarrays were applied to investigate larger number of CLL samples as well as normal B-cells from various stages of B-cell differentiation.Citation15 Based on similarities in DNA methylation signatures to normal B-cells, CLL samples were classified into 3 epigenetic subgroups with differential clinical outcome, i.e., naive B-cell-like CLL (n-CLL), memory B-cell-like CLL (m-CLL), and intermediate CLL (i-CLL).Citation15 In a subsequent study by Queiros et al., a B-cell epigenetic signature, based on the methylation pattern of 5 single CpG sites, i.e., cg00869668 (SCARF1), cg11472422 (B3GNTL1), cg17014214 (CTBP2), cg09637172 (TNF), and cg03462096 (chromosome 14 intergenic region), was proposed to classify CLL patients into the aforementioned epigenetic subgroups with high accuracy.Citation17 Although the epigenetic subgrouping of CLL patients generally corresponds well to the IGHV mutational status, i.e. most n-CLL and m-CLL patients express unmutated or mutated IGHV-genes, respectively, it remains to be clarified if the intermediate group merely represents a mix of outliers from the 2 other subgroups or may constitute a separate epigenetic entity.
In the current study, we aimed to validate the epigenetic classification in a well-characterized CLL cohort (135 cases) to further investigate its prognostic strength, particularly in relation to other established prognostic markers.Citation17 We mainly included M-CLL and U-CLL patients but also patients belonging to the clinically aggressive subset #2 (IGHV3-21/IGLV3-21). These latter patients are particularly interesting since they display poor clinical outcome despite mainly carrying mutated IGHV genes and were recently suggested, though based on few cases, to have an intermediate epigenetic signature.Citation18-21 Using pyrosequencing, we could confirm the clinical utility of the proposed epigenetic model, as it remained as an independent factor predicting both overall survival (OS) and time-to-first-treatment (TTFT), even in the presence of the IGHV gene mutational status. In addition, we observed that almost all subset #2 cases displayed a methylation signature similar to the i-CLL group, hence supporting the actual existence of a third epigenetic subgroup.
Results
Epigenetic classification and clinico-biological characteristics of the cohort
Methylation values for the 5 CpGs sites were determined by pyrosequencing in 135 CLL patients (Supplementary Table 2) and subjected to epigenetic classification using the support vector machine (SVM) model as recently described by Queiros et al.Citation17 Fifty-seven samples (42.2%) were classified as m-CLL, 29 samples (21.5%) grouped as i-CLL, while 49 samples (36.3%) carried the n-CLL signature. Similar to the recent report, 46/49 (94%) samples within the n-CLL category carried unmutated IGHV genes [average 99.6% germline identity (GI)], while 54/57 m-CLL cases (95%) showed mutated IGHV genes (average 93.2% GI).Citation17 The i-CLL cases were predominantly IGHV mutated (23/29, 79.3%) and displayed an intermediate mutational status (average 96.8% GI; ). Notably, 11/12 subset #2 cases were classified as i-CLL, while the remaining case was classified as m-CLL. The major clinico-biological characteristics of the 3 epigenetic subgroups are presented in . In brief, m-CLL cases predominantly carried favorable prognostic genomic aberrations [52/57 displayed no recurrent genomic aberration or carried isolated del(13q)], while there was an enrichment of del(11q) among both n-CLL and i-CLL cases (29.8% and 29.6%, respectively, ). In contrast to Queiros et al., we noted a higher proportion of i-CLL cases harboring del(11q) (29.6% vs. 9.1%, P = 0.10), and detected a lower frequency of SF3B1 mutations in i-CLL cases (6.8% vs. 21.7%, P = 0.12).
Figure 1. Epigenetic classification and hierarchical clustering of CLL patients. (A) Distribution of IGHV germline identity levels in the 3 epigenetic subgroups. (B) Bricks plot showing the methylation level of the of 5 CpG sites in 3 epigenetic subgroups. (C) Hierarchical clustering of the samples based on the methylation pattern of the 5 CpG signature.
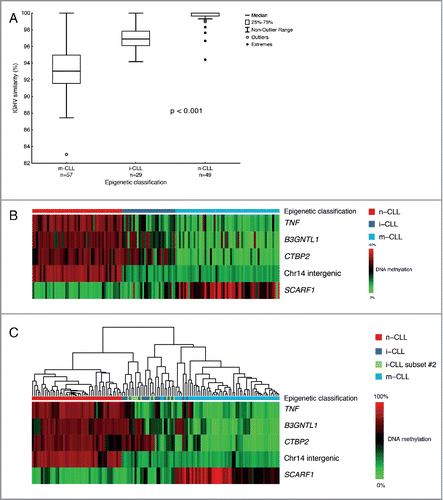
Table 1. Main clinico-biological characteristics of the 3 epigenetic subgroups (n = 135).
Methylation status of the 5 CpG sites and hierarchical clustering
Of the investigated CpGs, TNF, B3GNTL1, CTBP2, and chromosome 14 intergenic region (cg03462096) were hypermethylated in the n-CLL group, whereas the same regions were hypomethylated in the m-CLL group (). Vice versa, the CpG associated with SCARF1 was hypomethylated in n-CLL and hypermethylated in the m-CLL group. In contrast, patients classified as i-CLL displayed a considerably more heterogeneous methylation pattern for all investigated sites ().Citation17
We then performed hierarchical clustering of the samples based on the methylation status of the 5 CpG signature genes in order to assess similarities between samples in the different epigenetic classes. Without any exceptions, the m-CLL and n-CLL cases clustered separately creating 2 major branches (). In contrast, the i-CLL patients displayed an intermediate pattern, clustering to both major branches. Additionally, subset #2 cases did not cluster as one distinct group but instead displayed the same heterogeneous methylation profiles clustering together with either m-CLL or n-CLL cases ().
Clinical impact of epigenetic classification
As expected, m-CLL patients showed the most favorable prognosis (median survival not reached), while n-CLL displayed the worst OS (median 82 months) and the i-CLL group had an intermediate survival (median 110 months, ). Notably, subset #2 patients, although classified as i-CLL cases, demonstrated a shorter OS similar to n-CLL, borderline significantly different from the other i-CLL cases (P = 0.05, ).
Figure 2. Kaplan-Meier survival curve analysis for overall survival (OS) and time-to-first-treatment (TTFT) in epigenetic subgroups. (A) Impact of epigenetic classification on OS in 3 epigenetic subgroups. (B) Impact of epigenetic classification on OS when sub-classifying subset #2 and non-subset #2 i-CLL. (C) Impact of epigenetic classification on TTFT in 3 epigenetic subgroups. (D) Impact of epigenetic classification on TTFT when sub-classifying subset #2 and non-subset #2 i-CLL.
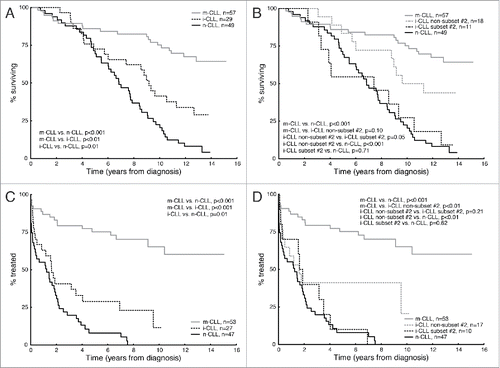
Similar findings were seen for TTFT; n-CLL patients had the shortest TTFT (median 15 months) while m-CLL patients showed the longest TTFT (median not reached) (). Again, i-CLL cases displayed an intermediate TTFT (median 19.5 months), significantly different from n-CLL cases (P = 0.01, ). When subdividing the i-CLL group, only non-subset #2 i-CLL was found to be significantly different from n-CLL (P<0.01, ), while subset #2 cases had similar TTFT as n-CLL.
We next assessed the prognostic impact of the epigenetic signature in the context of other established markers, including age at diagnosis, unmutated IGHV genes, TP53 abnormalities, and NOTCH1/SF3B1 mutations using multivariate Cox regression models. NOTCH1 and SF3B1 mutations were grouped together due to their low frequency in the present series. Notably and in contrast to the previous study, the epigenetic classifiers n-CLL and i-CLL individually remained as an independent prognostic marker in multivariate analysis for both OS (n-CLL: HR = 2.36, P = 0.037; i-CLL: HR = 2.31, P=0.015) and TTFT (n-CLL: HR = 3.67, P = 0.003; i-CLL: HR = 3.90, P > 0.001) in the presence of IGHV mutational status ().
Table 2. Multivariate Cox regression analysis of epigenetic classification and established molecular prognostic markers for overall survival and time-to-first-treatment in CLL.
Discussion
During the last decades, clinical and biological research has dramatically advanced the understanding of CLL and has revealed a plentitude of prognostic factors, including the more recently described DNA methylation-based markers, which can assist in identifying patients with higher risk of progression within this clinically very heterogeneous disease. Taking advantage of the genome-wide epigenetic profiles of normal B cells in different stages of the maturation process and based on similarities to these, Kulis et al. recently suggested that CLL patients can be grouped into 3 distinct epigenetic entities, namely n-CLL, i-CLL, and m-CLL.Citation15 Taking this approach a step further, the same group extracted 5 individual CpG sites, corresponding to TNF, SCARF1, B3GNTL1, CTBP2, and an intergenic region on chromosome 14, and could, based on the methylation profile of these sites, classify CLL patients with high accuracy into these 3 biological groups according to the proposed B-cell epigenetic signature.Citation15,17
The current study was designed to validate this novel epigenetic classification model and to investigate its association with other established prognostic markers in a well-characterized population-based CLL cohort, also including poor-prognostic subset #2 cases. Using pyrosequencing to assess the methylation status of each of the 5 CpG sites and applying the SVM model, we classified all 135 CLL patients into n-CLL (36%), i-CLL (22%), and m-CLL (42%) based on their DNA methylation profile.Citation17 Although the proportion of cases and the clinico-biological characteristics for each epigenetic subgroup correlated well with the initial report, particularly for the m-CLL and the n-CLL groups, we did note some differences in our cohort compared to the reported study. Among i-CLL patients in the present cohort, a notably higher proportion carried del(11q), while, in addition, the same epigenetic group showed a markedly lower frequency of poor-prognostic SF3B1 mutations. These differences, although notable, were not statistically significant and may be due to the relatively small number of i-CLL cases in both studies and need further clarification in larger cohorts.
A novel observation in our study was that almost all subset #2 cases (11/12) were classified as i-CLL. Subset #2 patients have previously been shown to be enriched for both del(11q) as well as mutations in SF3B1.Citation22 In fact, in this study, SF3B1 mutations in the i-CLL group were exclusively found in subset #2 cases. Therefore, differences in the number of subset #2 cases included, may also account for the observed inconsistencies between the 2 studies. Nevertheless, our data indeed support that subset #2 cases are a member of the i-CLL group,Citation21 which is in line with the intermediate IGHV mutational status observed both in subset #2 cases and non-subset #2 cases,Citation20 and provides evidence that the i-CLL group corresponds to a ‘true’ group and not merely a mix of ‘left-over’ cases. Hence, according to the methylation profiles, it can be speculated that the i-CLL group might have a different ontogenetic derivation compared to the other subgroups, which is also reflected by their borderline somatic hypermutation levels.
We next used the epigenetic classification model to study clinical outcome. In agreement with the previous publication, the different epigenetic subgroups showed significantly different clinical outcome both in terms of OS and TTFT (). Interestingly, when studying the impact on subset #2 cases, which are known to have very poor prognosis, we found these cases to resemble n-CLL for both OS and TTFT ().Citation22 Although relatively few cases were studied, it thus appears that subset #2 patients belong to the i-CLL group from a biological perspective, but not from a prognostic point of view. These results are in line with reports supporting that subset #2 is a distinct entity in terms of clinical behavior, remaining clinically aggressive independently of other well-established prognostic markers such as IGHV gene mutational status.Citation20 Therefore, the epigenetic classification should be further complemented, at least for the i-CLL group, with information regarding assignment to subset #2 prior to prognostication. However, larger cohorts need to be studied in order to validate this finding.
Finally, we took advantage of multivariate analysis to assess the prognostic impact of the epigenetic signature model in the context of other established markers. Although there is a clear correlation between the epigenetic signature model and IGHV mutational status as evidenced in both studies, we found, in contrary to the report by Queiros et al., which did not include IGHV mutational status,Citation17 the epigenetic signature model as an independent prognostic marker both for OS and TTFT in our cohort. Hence, this model could potentially be used to identify patients with mutated IGHV genes, subset #2 cases excluded, who would suffer from poor clinical outcome.
All together, we validated the recent findings that the epigenetic classification of CLL is associated with clinico-biological characteristics and clinical outcome. We also report that the epigenetic classification remained as an independent factor in multivariate analysis, supporting the clinical utility of this model in predicting both TTFT and OS. Additionally, we demonstrate that the poor-prognostic subset #2 cases are predominantly classified as i-CLL, although in terms of OS and TTFT they follow the more clinically aggressive n-CLL cases. Future studies are now warranted in order to standardize and harmonize the methodology before considering it for routine practice.
Materials and methods
Patients
The study cohort includes 135 CLL patients from the Swedish part of a Scandinavian population-based case-control study.Citation23 All cases were diagnosed and classified according to the recently revised iwCLL criteria and displayed a typical CLL immunophenotype.Citation24 The median time between diagnosis and sample collection was 3 months, and the samples contained median 90% tumor cells (range, 64–98%) and were collected before the administration of any treatment. The median age at diagnosis was 63.3 y (range, 38–75 y) with a male to female ratio of 2:1. Sixty-nine patients carried mutated IGHV genes and 54 patients unmutated IGHV genes, while 12 patients were assigned to stereotyped subset#2 (IGHV3-21/IGLV3-21).Citation25 Median follow-up for overall survival (OS) was 9.5 y (range 0.6–15.1 y) while median follow-up for time-to-first-treatment (TTFT) was 2.6 y (range, 0.0–15.0 y). Major clinico-biological parameters are summarized in . Informed consent was obtained according to the Helsinki declaration and the study was approved by the local ethics review committee.
Assessment of biological markers
Genomic DNA was extracted from peripheral blood mononuclear cells (PBMCs) using the QIAamp DNA mini kit (QIAGEN, Hilden, Germany) according to the manufacture's protocol. PCR amplification and sequence analysis of IGHV-IGHD-IGHJ rearrangement was performed as previously describedCitation25,26 CD38 expression was determined by flow cytometry as previously reported.Citation27 High-resolution genomic screening was performed using 250K SNP-arrays to detect recurrent genomic aberrations [del(13q), trisomy 12, del(11q), and del(17p)]; data was extracted and analyzed as previously described.Citation28 Mutational screening was performed for the following genes: NOTCH1: targeted analysis for del7544-45/p.P2514Rfs*4; TP53: exons 4–9; SF3B1: exons 14–16 as previously detailed.Citation29
Pyrosequencing assay
Bisulfite conversion of 250 ng of genomic DNA was performed using the EZ DNA Methylation-GoldTM Kit (Zymo Research Corporation, CA, USA) according to manufacturer's protocol and quantitative methylation analysis of the 5 prognostic CpG sites was performed using the PyroMark™ Q24 instrument (QIAGEN, Sweden). Briefly, bisulfite-converted DNA was PCR amplified using forward and biotinylated reverse primers for the 5 individual CpGs located in the promoter region of SCARF1 (cg00869668), in the gene body of B3GNTL1 (cg11472422), CTBP2 (cg17014214) and TNF (cg09637172), and in a chromosome 14 intergenic region (cg03462096).Citation17 The primer sequences are provided in Supplementary Table 1. The amplified PCR products were run on 2% agarose gel to check for the correct fragment size. Immobilization of PCR product was performed on Sepharose beads coated with streptavidin (GE Healthcare, Sweden). Bound PCR products were denatured to a single strand in a pyrosequencing vacuum workstation. Single-stranded PCR products attached to streptavidin beads were released into the pyrosequencing plate containing annealing buffer complemented with 0.3 µM sequencing primer. After the pyrosequencing reaction, the percentage of methylation for each CpG was analyzed using the PyroMark™ Q24 software. Methylated and unmethylated EpiTect Control DNA (QIAGEN, Sweden) were run in each pyrosequencing reaction. Methylation values for the 5 CpGs were subjected to epigenetic classification using the support vector machine (SVM) model as previously described by Queiros et al.
Statistical analysis
Pearson X2 was applied to investigate the association between the epigenetic classification and other prognostic markers. The X2 test was used to assess whether tumor cell purity had an impact on epigenetic classification. We compared the distribution of the 3 epigenetic subgroups in cases with tumor content below or above the median value (90%) and found no differences (P = 0.45). The Kruskal Wallis test was employed to assess differences in IGHV identity between the epigenetic subgroups. The Genesis software was used to perform hierarchal clustering analysis and to generate heatmaps.Citation30 Kaplan-Meier analysis was performed to construct survival curves for TTFT and OS. TTFT was calculated from the date of diagnosis until the starting date of initial treatment or last follow-up. OS was calculated from the date of diagnosis until date of death (all deaths) or last follow-up. The log-rank test was used to evaluate differences between subgroups. A stepwise multivariate Cox regression analysis was applied to compare the prognostic significance of the epigenetic classification in relation to other prognostic markers. All statistical analyses were carried out using Statistica Software version 13 (Dell Inc., Tulsa, OK, USA).
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
This research project was supported by the Swedish Cancer Society, the Swedish Research Council, Uppsala University, Uppsala University Hospital, the Lion's Cancer Research Foundation (Uppsala), and Selander's Foundation, Uppsala. The authors gratefully thank Dr. JI Martín-Subero for helpful discussions.
Reference
- Strathdee G, Brown R. Aberrant DNA methylation in cancer: potential clinical interventions. Exp Rev Mol Med 2002; 4:1-17; PMID:14987388; http://dx.doi.org/10.1017/S1462399402004222
- Corcoran M, Parker A, Orchard J, Davis Z, Wirtz M, Schmitz OJ, Oscier D. ZAP-70 methylation status is associated with ZAP-70 expression status in chronic lymphocytic leukemia. Haematologica 2005; 90:1078-88; PMID:16079107
- Chantepie SP, Vaur D, Grunau C, Salaun V, Briand M, Parienti JJ, Heutte N, Cheze S, Roussel M, Gauduchon P, et al. ZAP-70 intron1 DNA methylation status: determination by pyrosequencing in B chronic lymphocytic leukemia. Leukemia Res 2010; 34:800-8; PMID:19944462; http://dx.doi.org/10.1016/j.leukres.2009.10.018
- Claus R, Lucas DM, Ruppert AS, Williams KE, Weng D, Patterson K, Zucknick M, Oakes CC, Rassenti LZ, Greaves AW, et al. Validation of ZAP-70 methylation and its relative significance in predicting outcome in chronic lymphocytic leukemia. Blood 2014; 124:42-8; PMID:24868078; http://dx.doi.org/10.1182/blood-2014-02-555722
- Claus R, Lucas DM, Stilgenbauer S, Ruppert AS, Yu L, Zucknick M, Mertens D, Buhler A, Oakes CC, Larson RA, et al. Quantitative DNA methylation analysis identifies a single CpG dinucleotide important for ZAP-70 expression and predictive of prognosis in chronic lymphocytic leukemia. J Clin Oncol 2012; 30:2483-91; PMID:22564988; http://dx.doi.org/10.1200/JCO.2011.39.3090
- Abreu C, Moreno P, Palacios F, Borge M, Morande P, Landoni AI, Gabus R, Dighiero G, Giordano M, Gamberale R, et al. Methylation status regulates lipoprotein lipase expression in chronic lymphocytic leukemia. Leukemia & lymphoma 2013; 54:1844-8; PMID:23614796; http://dx.doi.org/10.3109/10428194.2013.796057
- Moreno P, Abreu C, Borge M, Palacios F, Morande P, Pegazzano M, Bianchi S, Landoni AI, Agrelo R, Giordano M, et al. Lipoprotein lipase expression in unmutated CLL patients is the consequence of a demethylation process induced by the microenvironment. Leukemia 2013; 27:721-5; PMID:22828442; http://dx.doi.org/10.1038/leu.2012.212
- Strathdee G, Holyoake TL, Sim A, Parker A, Oscier DG, Melo JV, Meyer S, Eden T, Dickinson AM, Mountford JC, et al. Inactivation of HOXA genes by hypermethylation in myeloid and lymphoid malignancy is frequent and associated with poor prognosis. Clin Cancer Res 2007; 13:5048-55; PMID:17785556; http://dx.doi.org/10.1158/1078-0432.CCR-07-0919
- Strathdee G, Sim A, Parker A, Oscier D, Brown R. Promoter hypermethylation silences expression of the HoxA4 gene and correlates with IgVh mutational status in CLL. Leukemia 2006; 20:1326-9; PMID:16688227; http://dx.doi.org/10.1038/sj.leu.2404254
- Shinawi T, Hill V, Dagklis A, Baliakas P, Stamatopoulos K, Agathanggelou A, Stankovic T, Maher ER, Ghia P, Latif F. KIBRA gene methylation is associated with unfavorable biological prognostic parameters in chronic lymphocytic leukemia. Epigenetics 2012; 7:211-5; PMID:22430796; http://dx.doi.org/10.4161/epi.7.3.19222
- Hill VK, Dunwell TL, Catchpoole D, Krex D, Brini AT, Griffiths M, Craddock C, Maher ER, Latif F. Frequent epigenetic inactivation of KIBRA, an upstream member of the Salvador/Warts/Hippo (SWH) tumor suppressor network, is associated with specific genetic event in B-cell acute lymphocytic leukemia. Epigenetics 2011; 6:326-32; PMID:21173572; http://dx.doi.org/10.4161/epi.6.3.14404
- Raval A, Lucas DM, Matkovic JJ, Bennett KL, Liyanarachchi S, Young DC, Rassenti L, Kipps TJ, Grever MR, Byrd JC, et al. TWIST2 demonstrates differential methylation in immunoglobulin variable heavy chain mutated and unmutated chronic lymphocytic leukemia. J Clin Oncol 2005; 23:3877-85; PMID:15809452; http://dx.doi.org/10.1200/JCO.2005.02.196
- Irving L, Mainou-Fowler T, Parker A, Ibbotson RE, Oscier DG, Strathdee G. Methylation markers identify high risk patients in IGHV mutated chronic lymphocytic leukemia. Epigenetics 2011; 6:300-6; PMID:21051931; http://dx.doi.org/10.4161/epi.6.3.14038
- Kanduri M, Cahill N, Goransson H, Enstrom C, Ryan F, Isaksson A, Rosenquist R. Differential genome-wide array-based methylation profiles in prognostic subsets of chronic lymphocytic leukemia. Blood 2010; 115:296-305; PMID:19897574; http://dx.doi.org/10.1182/blood-2009-07-232868
- Kulis M, Heath S, Bibikova M, Queiros AC, Navarro A, Clot G, Martinez-Trillos A, Castellano G, Brun-Heath I, Pinyol M, et al. Epigenomic analysis detects widespread gene-body DNA hypomethylation in chronic lymphocytic leukemia. Nat Genet 2012; 44:1236-42; PMID:23064414; http://dx.doi.org/10.1038/ng.2443
- Cahill N, Bergh AC, Kanduri M, Goransson-Kultima H, Mansouri L, Isaksson A, Ryan F, Smedby KE, Juliusson G, Sundstrom C, et al. 450K-array analysis of chronic lymphocytic leukemia cells reveals global DNA methylation to be relatively stable over time and similar in resting and proliferative compartments. Leukemia 2013; 27:150-8; PMID:22922567; http://dx.doi.org/10.1038/leu.2012.245
- Queiros AC, Villamor N, Clot G, Martinez-Trillos A, Kulis M, Navarro A, Penas EM, Jayne S, Majid A, Richter J, et al. A B-cell epigenetic signature defines three biologic subgroups of chronic lymphocytic leukemia with clinical impact. Leukemia 2015; 29:598-605; PMID:25151957; http://dx.doi.org/10.1038/leu.2014.252
- Tobin G, Thunberg U, Johnson A, Eriksson I, Soderberg O, Karlsson K, Merup M, Juliusson G, Vilpo J, Enblad G, et al. Chronic lymphocytic leukemias utilizing the VH3-21 gene display highly restricted Vlambda2-14 gene use and homologous CDR3s: implicating recognition of a common antigen epitope. Blood 2003; 101:4952-7; PMID:12586612; http://dx.doi.org/10.1182/blood-2002-11-3485
- Thorselius M, Krober A, Murray F, Thunberg U, Tobin G, Buhler A, Kienle D, Albesiano E, Maffei R, Dao-Ung LP, et al. Strikingly homologous immunoglobulin gene rearrangements and poor outcome in VH3-21-using chronic lymphocytic leukemia patients independent of geographic origin and mutational status. Blood 2006; 107:2889-94; PMID:16317103; http://dx.doi.org/10.1182/blood-2005-06-2227
- Baliakas P, Agathangelidis A, Hadzidimitriou A, Sutton LA, Minga E, Tsanousa A, Scarfo L, Davis Z, Yan XJ, Shanafelt T, et al. Not all IGHV3-21 chronic lymphocytic leukemias are equal: prognostic considerations. Blood 2015; 125:856-9; PMID:25634617; http://dx.doi.org/10.1182/blood-2014-09-600874
- Puente XS, Bea S, Valdes-Mas R, Villamor N, Gutierrez-Abril J, Martin-Subero JI, Munar M, Rubio-Perez C, Jares P, Aymerich M, et al. Non-coding recurrent mutations in chronic lymphocytic leukaemia. Nature 2015; 526:519-24; PMID:26200345; http://dx.doi.org/10.1038/nature14666
- Strefford JC, Sutton LA, Baliakas P, Agathangelidis A, Malcikova J, Plevova K, Scarfo L, Davis Z, Stalika E, Cortese D, et al. Distinct patterns of novel gene mutations in poor-prognostic stereotyped subsets of chronic lymphocytic leukemia: the case of SF3B1 and subset #2. Leukemia 2013; 27:2196-9; PMID:23558524; http://dx.doi.org/10.1038/leu.2013.98
- Smedby KE, Hjalgrim H, Melbye M, Torrang A, Rostgaard K, Munksgaard L, Adami J, Hansen M, Porwit-MacDonald A, Jensen BA, et al. Ultraviolet radiation exposure and risk of malignant lymphomas. J Natl Cancer Inst 2005; 97:199-209; PMID:15687363; http://dx.doi.org/10.1093/jnci/dji022
- Hallek M, Cheson BD, Catovsky D, Caligaris-Cappio F, Dighiero G, Dohner H, Hillmen P, Keating MJ, Montserrat E, Rai KR, et al. Guidelines for the diagnosis and treatment of chronic lymphocytic leukemia: a report from the International Workshop on Chronic Lymphocytic Leukemia updating the National Cancer Institute-Working Group 1996 guidelines. Blood 2008; 111:5446-56; PMID:18216293; http://dx.doi.org/10.1182/blood-2007-06-093906
- Agathangelidis A, Darzentas N, Hadzidimitriou A, Brochet X, Murray F, Yan XJ, Davis Z, van Gastel-Mol EJ, Tresoldi C, Chu CC, et al. Stereotyped B-cell receptors in one-third of chronic lymphocytic leukemia: a molecular classification with implications for targeted therapies. Blood 2012; 119:4467-75; PMID:22415752; http://dx.doi.org/10.1182/blood-2011-11-393694
- Murray F, Darzentas N, Hadzidimitriou A, Tobin G, Boudjogra M, Scielzo C, Laoutaris N, Karlsson K, Baran-Marzsak F, Tsaftaris A, et al. Stereotyped patterns of somatic hypermutation in subsets of patients with chronic lymphocytic leukemia: implications for the role of antigen selection in leukemogenesis. Blood 2008; 111:1524-33; PMID:17959859; http://dx.doi.org/10.1182/blood-2007-07-099564
- Thunberg U, Johnson A, Roos G, Thorn I, Tobin G, Sallstrom J, Sundstrom C, Rosenquist R. CD38 expression is a poor predictor for VH gene mutational status and prognosis in chronic lymphocytic leukemia. Blood 2001; 97:1892-4; PMID:11263438; http://dx.doi.org/10.1182/blood.V97.6.1892
- Gunnarsson R, Staaf J, Jansson M, Ottesen AM, Goransson H, Liljedahl U, Ralfkiaer U, Mansouri M, Buhl AM, Smedby KE, et al. Screening for copy-number alterations and loss of heterozygosity in chronic lymphocytic leukemia–a comparative study of four differently designed, high resolution microarray platforms. Genes Chromosomes Cancer 2008; 47:697-711; PMID:18484635; http://dx.doi.org/10.1002/gcc.20575
- Cortese D, Sutton LA, Cahill N, Smedby KE, Geisler C, Gunnarsson R, Juliusson G, Mansouri L, Rosenquist R. On the way towards a ‘CLL prognostic index’: focus on TP53, BIRC3, SF3B1, NOTCH1 and MYD88 in a population-based cohort. Leukemia 2014; 28:710-3; PMID:24217197; http://dx.doi.org/10.1038/leu.2013.333
- Sturn A, Quackenbush J, Trajanoski Z. Genesis: cluster analysis of microarray data. Bioinformatics 2002; 18:207-8; PMID:11836235; http://dx.doi.org/10.1093/bioinformatics/18.1.207