
ABSTRACT
In contrast to central nervous system neurons, dorsal root ganglia (DRG) neurons can switch to a regenerative state after peripheral axotomy. In a screen for chromatin regulators of the regenerative responses in this conditioning lesion paradigm, we identified Tet methylcytosine dioxygenase 3 (Tet3) as upregulated in DRG neurons, along with increased 5-hydroxymethylcytosine (5hmC). We generated genome-wide 5hmC maps in adult DRG, which revealed that peripheral and central axotomy (leading to no regenerative effect) triggered differential 5hmC changes that are associated with distinct signaling pathways. 5hmC was altered in a large set of regeneration-associated genes (RAGs), including well-known RAGs, such as Atf3, Bdnf, and Smad1, that regulate axon growth potential of DRG neurons, thus supporting its role for RAG regulation. Our analyses also predicted HIF-1, STAT, and IRF as potential transcription factors that may collaborate with Tet3 for 5hmC modifications. Intriguingly, central axotomy resulted in widespread 5hmC modifications that had little overlap with those of peripheral axotomy, thus potentially constituting a roadblock for regeneration. Our study revealed 5hmC dynamics as a previously unrecognized epigenetic mechanism underlying the divergent responses after axonal injury.
Introduction
DNA methylation regulates diverse sets of genes that control cellular identity and differentiation state.Citation1 Initially thought to be a stable and heritable modification, DNA methylation is in fact a highly dynamic process, serving as a basic epigenetic mechanism to regulate neurodevelopmentCitation2 and neural plasticity in learning and memory.Citation3 DNA methylation is also frequently altered in CNS disorders.Citation4,5 The identification of enzymes responsible for DNA (de)methylation further highlights its dynamic nature. The addition of methyl group to cytosine (5mC) is catalyzed by DNA methyltransferases (Dnmts). DNA demethylation, on the other hand, occurs through an intermediate base, 5-hydroxymethylcytosine (5hmC), and is catalyzed by the Tet methylcytosine dioxygenase family (Tet1–3).Citation6
Besides serving as an intermediate in DNA demethylation, 5hmC may have regulatory roles in its own right.Citation7 The 5hmC modification is relatively abundant in the central nervous system (CNS), —more than 10-fold higher than in embryonic stem (ES) cells—, and accounts for ∼40% of modified cytosines in CNSCitation8–10 Comprehensive genome-wide mapping of 5hmC has been performed in ES cells,Citation11 and in CNS tissues at different developmental stagesCitation7,12 or under pathological conditions, such as cocaine abuse.Citation13 Collectively, they form the foundation to understand the influences of this base modification in neurodevelopment and CNS disorders. In contrast, virtually nothing is known about the 5hmC dynamics in nerve injury and axon regeneration.Citation14,15 Hence, insights into the 5hmC state would provide a new perspective for understanding epigenetic regulation of regenerative injury responses.
Compared to their embryonic counterparts, adult CNS neurons exhibit a diminished capacity for axon regrowth.Citation16,17 This is partly attributed to epigenetic repression of pro-growth genes or stable expression of growth-inhibiting genes after completion of axonal wiring.Citation18 To assess the role of 5hmC in the regenerative responses after axon injury, we took advantage of the well-established conditioning lesion paradigm in sensory neurons in dorsal root ganglia (DRG). DRG neurons share unique features of both CNS and peripheral nervous system (PNS) neurons, as they project both a central and a peripheral axon branch. Whereas central axotomy leaves DRG neurons in a state refractory to regeneration, peripheral axotomy switches DRG neurons into a regenerative state that promotes regrowth of both peripheral and central axon branches.Citation19,20 The conditioning lesion effect is transcription dependent, highlighting the importance of gene regulatory mechanisms for axon regeneration.Citation21
Earlier effort has been focused on identifying regeneration-associated genes (RAGs),Citation22-25 yet little is known of the transcriptional mechanisms underlying the induction and sustained expression of the pro-growth genes. Dynamic changes of chromatin landscapes are proposed to set the stage for coordinated regulation for entire classes of RAGs.Citation18 Previously, our laboratory has identified histone acetylation as a critical epigenetic mechanism in the conditioning lesion paradigm.Citation26 We showed that increased histone acetylation promoted access of the pro-regenerative transcription factor Smad1 to target genes. Increasing histone acetylation levels by specific HDAC inhibitors resulted in orchestrated transcriptional changes of a large repertoire of RAGs, as well as enhanced axon growth potential and sensory axon regeneration in a spinal cord injury (SCI) model.Citation26 However, HDAC inhibitors induced only a fraction of the RAGs tested, indicating engagement of additional regulatory mechanisms for the regenerative responses after the conditioning lesion.
To identify novel chromatin regulators of regenerative injury responses, we screened for epigenetic factors that are differentially regulated in adult DRG after peripheral axotomy as compared with naive state with no injury. We identified Tet3 as specifically upregulated in conditioned DRG, along with elevated 5hmC levels. To understand the influence of 5hmC reconfigurations in the regenerative responses, we generated comprehensive epigenomic maps of 5hmC distributions and dynamics under different regenerative states of adult DRG. While a large number of genomic loci displayed stable 5hmC configurations, axonal injury initiated widespread 5hmC alterations. A concept emerging from our analyses is that contrary to previous notion, central axotomy of DRG neurons is not a static event, but instead a dynamic process that results in genome-wide 5hmC reconfigurations with little overlap with peripheral axotomy. Hence, central axotomy appears to engage divergent 5hmC-regulated signaling pathways, which may constitute a roadblock for regeneration. Furthermore, we demonstrated that conditioning lesion resulted in unique acquisition or deletion of 5hmC modifications in about half of RAGs, many of which play important roles in neurite outgrowth. Our study also predicted a set of transcription factor families, including hypoxia-inducible factor 1 (HIF-1), signal transducer and activator of transcription (STAT), and interferon regulatory factor (IRF), that may assist Tet3 for 5hmC modifications. Together, our results suggest a novel role for 5hmC epigenetics in regulating regenerative injury responses.
Results
Tet3 is upregulated after conditioning lesion
In order to identify novel epigenetic regulators during axon regeneration, we screened by qRT-PCR array transcriptional changes of over 80 chromatin regulators in adult DRG 24 h after sciatic nerve transection (peripheral lesion, PL) as compared with contralateral uninjured control DRG (ctrl). We identified Tet3 as the most upregulated gene after the conditioning lesion (). We then compared transcriptional changes of Tet family members (Tet1–3) after PL or T8 dorsal column transection (central lesion, CL). Tet3 was the only member of the Tet family that was upregulated at 24 h in DRG after PL relative to CL (). Time-course analysis revealed that Tet3 was significantly induced in PL-DRG at 12 h post-lesion, remained elevated at 24 h, but started to decline after 3 d ().
Figure 1. Induction of Tet3 in conditioned DRG (A) Screen for transcriptional changes of epigenetic regulators in conditioned DRG vs. contralateral naive DRG by qRT-PCR array at 24 h after lesion. Tet3 showed highest induction among genes tested (blue box). (B) qRT-PCR showed specific induction of Tet3, but not other Tet family members in conditioned DRG with peripheral lesion (PL) as compared with DRG with central lesion (CL) at 24 h after injury. qRT-PCR of conditioned DRG also showed upregulation of Gadd45a, but not of other enzymes involved in DNA (de)methylation. Results were normalized to housekeeping gene Rpl13a. (C) Time-course analysis of Tet3 induction by qRT-PCR comparing conditioned PL-DRG to contralateral control DRG. (D) Diagram of DNA (de)methylation pathways. (E) Immunohistochemistry showed upregulation of TET3 (green, arrows) in the nuclei of conditioned PL-DRG neurons (NeuN, red) at 24 h after peripheral lesion as compared with control with no injury. DAPI (blue) was used for nuclear counterstaining. (F-G) Immunocytochemistry of cultured DRG neurons showed levels of TET3 (F) and 5hmC (G) in nuclei of DRG neurons at 2h, 24 h and 96 h in vitro. Scale bars: 50 μm (E), 25 μm (F-G). Representative images from three independent sets of experiments are shown.
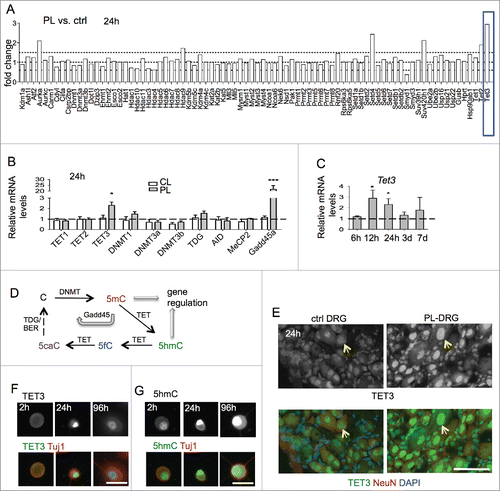
As TET enzymatic activity plays a key role in DNA hydroxymethylation/demethylation (), we performed additional qRT-PCR analysis on genes involved in regulation of DNA methylation. We detected a marked induction of growth arrest and DNA damage-inducible gene 45 α (Gadd45a) in conditioned PL-DRG at 24 h as compared with CL-DRG, but no significant changes in other genes tested (). Consistently, Gadd45a, a regulator of active DNA demethylation ,27 has been shown previously to be a highly upregulated RAG in the conditioning lesion paradigm.Citation28 Tet3 upregulation was confirmed by immunohistochemistry (IHC) in conditioned DRG, with predominantly nuclear localization in sensory neurons ().
Cultured DRG neurons undergo a process that is analogous to the in vivo conditioning lesion, with similar regenerative responses, as the cell dissociation step severs both axon branches.Citation29–31 We therefore conducted immunocytochemistry (ICC) on cultured DRG neurons, which allows more detailed cellular analysis. We found that Tet3 protein levels were low in freshly dissociated DRG neurons, increased in nuclei at 24 h, and waned by 96 h after plating, mirroring the in vivo results (). Tet3 induction hence occurs early after axonal injury, supporting the importance of early modulation of chromatin states to facilitate RAG induction.
Upregulated levels of Tet3 have been shown to lead to accumulation of 5hmC,Citation32–34 we therefore examined 5hmC levels by ICC, which confirmed elevated 5hmC immunosignals in the nuclei of cultured DRG neurons (). Specifically, freshly dissociated DRG neurons showed a moderate baseline level of 5hmC at 2 h after plating, but a marked increase at 24 h in culture, consistent with initiation of an in vitro conditioning program. Notably, the 5hmC increases were sustained at 96 h in vitro, even though Tet3 upregulation had waned (). These data confirmed a correlation between Tet3 induction and 5hmC accumulations in conditioned DRG neurons and supported the model that while Tet3 expression changes may be relatively transient, 5hmC may serve as a lasting epigenetic mark.Citation7,35
Dynamic genomic 5hmC patterns in adult DRG in different regenerative states
To identify genome-wide 5hmC reconfigurations in adult DRG under different regenerative states, we collected lumbar 4–6 DRGs at 24 h after either PL or CL in independent triplicate experiments. Contralateral, uninjured DRGs from mice subjected to PL were used as controls. Extracted chromatin was subjected to 5hmC capture using the β-glucosyltransferase-based covalent method (Hydroxymethyl Collector),Citation9 followed by genome-wide short-read sequencing. Around 60 million uniquely mapped, non-duplicated reads per sample were obtained (Table S2). As input controls, DNA samples without 5hmC enrichment were sequenced for each condition.
We first assessed global distributions of 5hmC signals using input-normalized 10 kb binned read count data. Principal component analysis revealed that the triplicates of each DRG condition clustered together (), indicating both reproducibility of each replicate and highly similar patterns of genomic 5hmC configurations within each DRG condition, but divergent 5hmC patterns among the three DRG conditions.
Figure 2. Genomic mapping revealed distinct 5 hmC distributions in adult DRG under different injury conditions (A) Principal component analysis (PCA) of input-normalized 5hmC binned data (10 kb bins) showed clustering of three independent biological replicates for each DRG injury condition. (B) Hierarchically-clustered heatmap of input-normalized 5hmC signals in genome-wide binned data (10 kb bins, filtered for RPKM>1) in adult DRGs under different regenerative conditions. Blue box highlights cluster of bins with conditioning lesion-specific 5hmC gain, and orange box highlights bins with conditioning-lesion-specific 5hmC loss. (C) Global visualization of 5hmC signals in and around gene bodies by ngs.plot. Read densities at all RefSeq transcripts were summed for each DRG injury condition and distributed from 3 kb upstream of transcription start site (TSS) to 3 kb downstream of transcription end site (TES). (D) Bar graph showing numbers of all 5hmC peaks in different DRG conditions as determined by MACS (top), and numbers of gene-associated 5hmC peaks (from TSS-3kb to TES+1kb; bottom). (E) Distributions of 5hmC MACS peaks per chromosome in different DRG conditions, as compared with values expected according to chromosome size or number of encoded genes. See also Table S2.
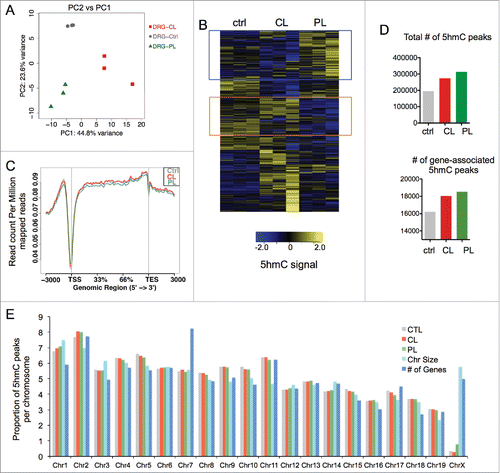
When the binned 5hmC data were compared and visualized as a hierarchically-clustered heatmap, we could distinguish genomic loci with differential 5hmC configurations among the control, CL, and PL DRG conditions (). Notably, one cluster displayed 5hmC gains in PL relative to ctrl or CL-DRG, while another cluster showed 5hmC depletions in PL-DRG. It was also evident that a substantial number of loci displayed 5hmC alterations in CL relative to ctrl or PL-DRG (). This suggests that even though CL induced neither Tet3 upregulation nor enhanced regenerative potential, it nevertheless resulted in widespread 5hmC reconfigurations throughout the genome. Together, our data lend evidence for dynamic 5hmC modifications in adult DRG induced after either peripheral or central axotomy.
Earlier studies revealed a predominant association of 5hmC with gene bodies,Citation11 we therefore generated global visualization of 5hmC read densities in and near gene bodies, which showed that the distributions of 5hmC densities were not random; instead, the highest 5hmC densities were located in gene bodies and in regions ∼1 kb upstream of transcription start site (TSS) (). In contrast, there was a gradual trailing along 3 kb downstream of transcription end site (TES). Moreover, 5hmC densities were markedly lower at the TSS sites, similar to that in earlier 5hmC mappings in CNS tissues.Citation12 Of note, the overall genic 5hmC densities patterns were comparable among the three DRG conditions ().
Peak-calling analysis revealed that the genome-wide 5hmC peak number was highest in PL-DRG (314,203) as compared with CL-DRG (274,544) or ctrl-DRG (195,662) (). PL-DRG also had the highest number of gene-associated 5hmC peaks (18,520) as compared with CL-DRG (18,032) or ctrl-DRG, which had far less (16,206) ().
Chromosomal distribution analysis showed a relatively even distribution of 5hmC peaks with a noticeable depletion on the X chromosome (). Substantial depletion of 5hmC on chromosome X has also been observed in CNS tissues,Citation12 supporting conserved 5hmC-associated regulatory mechanisms on chromosome X in both CNS and PNS. In comparison, the relative distributions of 5hmC peaks on autosomes were largely proportional to chromosome size and gene numbers encoded in each chromosome in all three DRG conditions with a few exceptions such as chromosome 1 and 7 (). Together, our results demonstrate that genomic features of 5hmC distributions in PNS are similar to those reported in CNS.Citation12 Our data also highlight specific loci with 5hmC changes induced by the conditioning lesion, thus raising the tantalizing possibility of a link between 5hmC modifications and the regenerative responses/RAG regulation in conditioned DRG.
Central and peripheral axotomy lead to distinct differentially hydroxymethylated regions
To comprehensively localize regeneration-associated differentially 5-hydroxymethylated regions (DhMRs) on a genome-wide scale, we first conducted pairwise comparisons of 5hmC signals between different DRG states using the diffReps program.Citation36 We identified 12,244 DhMRs in PL-vs-ctrl comparison, and 22,944 DhMRs in PL-vs-CL comparison (nominal P-value < 10−4; Supplemental Data Sets 1 and 3). There was a relatively even split of 5hmC gain and loss for PL-vs-ctrl DhMRs, and a bias toward 5hmC loss in PL-vs-CL DhMRs (3:1 ratio, ). There was nearly three times the number of CL-vs-ctrl DhMRs (35,242), with a strong bias toward 5hmC gain (5:1 ratio; Supplemental Data Set 2). The fact that more DhMRs display 5hmC gain in CL than in PL may suggest that increased levels of Tet3 in PL-DRG could contribute to full cytosine demethylation. Of note, the number of DhMRs may also be influenced by technical variations among the triplicates.
Figure 3. Distinct 5hmC reconfigurations after peripheral and central lesion of DRG (A) Bar graph showing the number of differentially hydroxymethylated regions (DhMRs) among pairwise comparisons between different DRG injury conditions. The numbers of DhMRs with 5hmC gain or loss are listed on the right. (B) Pie charts showing association of DhMRs with genomic features. (C) Pie charts showing association of DhMRs with CpG islands, shores, shelves, and open sea. (D) Bar graph showing the number of genes containing DhMRs for each comparison. The proportion of genes containing single or multiple DhMRs are indicated by colored boxes. (E) Venn diagram revealing small overlap between the two sets of DhMRs induced by CL or PL relative to control (left), and between the two sets of DhMRs that are associated with gene bodies (right). (F) Alignment of genome-wide DhMRs according to the extent of 5hmC gain or loss highlighting little overlap between different sets of pairwise comparisons. (G) Hierarchical clustering of enriched pathways associated with either CL-vs-ctrl or PL-vs-ctrl DhMRs by Ingenuity Comparative Analysis shows the top differential categories for Diseases and Functions (left) and Canonical Pathways (right).
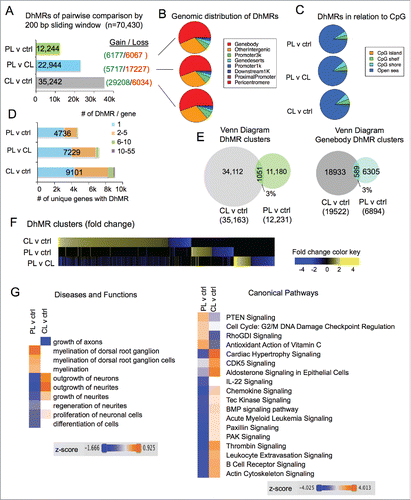
Genomic distribution analyses showed that ∼55% of DhMRs were in gene bodies, ∼5–7% at 1–3 kb promoter regions of TSS, ∼2% within 1 kb promoter regions of TSS, and ∼30% in intergenic regions (). In relation to CpG islands, the overwhelming majority of DhMRs (∼90%) was in “open sea,” while ∼5% were detected at CpG shelves, ∼6% at CpG shores, and less than 0.5% on CpG islands ().
We next determined the number of genes containing the DhMRs. There were nearly twice as many genes containing CL-vs-ctrl DhMRs (9101) than PL-vs-ctrl DhMRs (4736), while 7229 genes contained PL-vs-CL DhMRs (). Notably, a significant number of genes contained multiple DhMRs, ranging from 2 to as many as 55 ().
To determine whether PL and CL resulted in similar or distinct 5hmC modifications, we intersected the two sets of DhMRs (PL-vs-ctrl and CL-vs-ctrl), and surprisingly found only ∼3% overlapping DhMR clusters (1051). Similarly, there were only 3% overlapping gene body-associated DhMRs between the two sets (). Of note, on rare occasions, neighboring DhMRs in one set may share the same overlapping DhMR from the other set, in which cases, they were consolidated into one DhMR cluster, thus rendering a slightly decreased overall number of DhMR clusters than that of DhMRs (compare ). To further compare genomic distribution of DhMRs, we aligned all the 70,430 DhMRs, sorted according to the extent of 5hmC gain or loss for each pair-wise comparison, and it was evident that PL and CL resulted in markedly different 5hmC modification patterns with little overlap between the two ().
For functional annotation of DhMR-associated genes, we performed comparative Ingenuity Pathway Analysis (IPA) of gene sets associated with PL-vs-ctrl or CL-vs-ctrl DhMRs. Remarkably, the most divergent Diseases and Functions categories include growth of axons, outgrowth of neurites, and regeneration of neurites according to their activation z-score (). Likewise, the top differential DhMR-associated Canonical Pathways included many of the signaling pathways known to regulate axonal growth and regeneration, such as PTENCitation37 and BMP signaling,Citation38 as well as pathways important for cytoskeleton dynamics, such as RhoGDI and actin cytoskeleton signaling (). Of note, the directionality of the z-score is dictated by the direction of 5hmC changes in DhMRs, and thus does not necessarily indicate increased or decreased functional activation or signaling strength. Together, our results indicate that PL and CL elicit remarkably different genomic patterns of 5hmC modifications, which exert divergent influences on signaling pathways important for neurite outgrowth. These results also provide a fresh perspective on the underlying epigenetic mechanism of the conditioning effect induced by PL in contrast to a repressed regenerative state induced by CL.
5hmC dynamics in RAG regulation after conditioning lesion
Previously, we have performed Affymetrix array studies to identify differentially expressed genes in adult DRGs after conditioning lesion compared with central axotomy.Citation31 This comparison has the benefit of excluding genes triggered by injury alone. A total of 365 RAGs were identified in PL-vs-CL comparison after 24 h post-injury, with 231 genes being upregulated and 134 downregulated (). To explore the relationship of 5hmC alternations and transcriptional regulation, we intersected this set of RAGs with the PL-vs-CL DhMRs. We found that among the 365 RAGs, 161 genes (44%) contained DhMRs, among which 99 were upregulated and 62 downregulated RAGs (). Hence, nearly half of the PL-induced RAGs displayed 5hmC alterations, suggesting an active role of 5hmC-associated mechanisms in the transcriptional regulation of RAGs.
Figure 4. 5hmC dynamics and RAG regulation in conditioned DRG (A) Table listing the number of differentially regulated regeneration-associated genes (RAGs) in adult DRG at 24 h after PL compare with CL, and the number of RAGs that contained PL-vs-CL DhMRs. (B) Pie charts showing distribution of DhMRs associated with RAGs regarding genomic features (left) and CpG densities (right). (C) Diagram showing distribution of DhMRs associated with RAGs according to directionality of transcriptional and 5hmC changes after PL compare with CL at 24 h post-injury. DE: differentially expression genes. DR: DhMR. (D) Tables listing the top up- (left) or downregulated RAGs (right) in PL-DRG as compared with CL-DRG and the characteristics of the associated DhMRs. (E-F) Examples of genomic views of 5hmC changes of the three different DRG conditions in two highly regulated RAGs (E), and genomic views of PL-vs-CL DhMRs in the indicated RAGs (F), with green boxes indicating gain in 5hmC, and red boxes loss in 5hmC. (G) Pie chart showing the proportion of RAGs containing one or multiple DhMRs. See also Fig. S1.
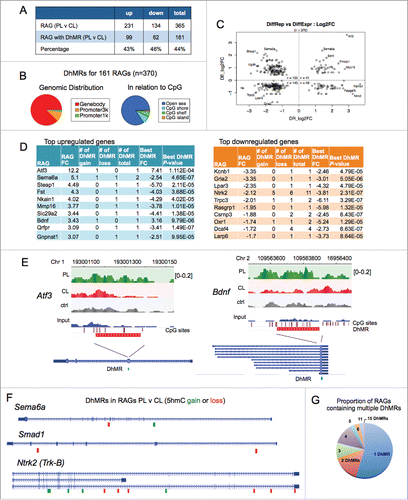
The genomic distribution analysis of the 370 DhMRs associated with the 161 RAGs showed that they were mostly located in gene bodies (88%), with only a small fraction located at promoter regions (10% at 1–3 kb of TSS and 2% within 1 kb of TSS) (). Even for these actively regulated genes in the conditioning lesion paradigm, there was no apparent association of DhMRs with CpG islands in that 5hmC changes occurred mostly in “open sea” (). The 161 RAGs were distributed relatively evenly throughout the genome with noticeable paucity on chromosome X (Fig. S1).
We next analyzed correlation of transcriptional changes of these 161 RAGs with 5hmC alterations (). Activating transcription factor 3 (Atf3) was the top upregulated RAG that also displayed largest 5hmC gain (). Atf3 is a well-known RAG that enhances the intrinsic growth state of DRG neurons.Citation39 It contained one PL-vs-CL DhMR with 5hmC gain in exon 2 (). Brain-derived neurotrophic factor (Bdnf), an important neurotrophin regulating axon growth,Citation40 is another top ranked upregulated RAG with concomitant 5hmC gain in the last exon (). Sema6a, ranked as the second upregulated RAG, is a transmembrane axon guidance molecule with axon repulsion activity through Plexin-A2.Citation41,42 It encompasses two PL-vs-CL DhMRs located in introns, with one showing 5hmC gain and one 5hmC loss (). Smad1, a regeneration-associated transcription factor that has been extensively characterized in our earlier studies, is upregulated specifically after PL, and its activation enhances axon growth potential of DRG neurons.Citation31,38 It contained two DhMRs in introns, both showing 5hmC loss (). Among the most downregulated RAGs, many contained multiple DhMRs (). For instance, Ntrk2 (also known as TrkB, a BDNF receptor) contained 5 DhMRs with 5hmC gain and 6 DhMRs with 5hmC loss, all of which are located in introns except for one located in the last exon ().
We next tested the model that gene upregulation may be correlated with 5hmC gain, and, conversely, gene downregulation with 5hmC loss. However, when the 370 RAG-associated DhMRs were classified into four groups according to the directionality of 5hmC and transcriptional changes, there was no clear correlation, with RAGs scattered in all four quadrants (). This may be attributed to the fact that close to half of the RAGs contained multiple DhMRs ( and ), often with divergent 5hmC changes in a single gene. We then selected one DhMR with the largest absolute fold-change to represent that gene, but still found no obvious correlation as suggested by the model: only around half of the RAGs showed concordant 5hmC changes in relation to the direction of transcriptional changes (Fig. S1B). We next focused on promoter region DhMRs, which represent only 12% of RAG-associated DhMRs. Even when separating promoter from gene body DhMRs, there was still no clear evidence that the directionality of 5hmC changes was correlated with the direction of transcriptional changes (Fig. S1C). We thus conclude that complex regulatory mechanisms must be at play to convey the 5hmC modifications to genomic output. It is noteworthy that the functional annotation of multiple loci within one gene displaying 5hmC modifications remains unclear at this point.
Unique 5hmC modifications specifically regulated in the conditioning lesion paradigm
Next, we reasoned that comparisons of 5hmC distributions across the three DRG conditions would allow us to identify DhMRs that are unique to the conditioning lesion or commonly shared by both PL and CL, the latter of which reflects injury-induced 5hmC changes. A Venn diagram of overlapping DhMRs from the three pair-wise comparisons delineated seven distinct DhMR groups ( and S2–3). Group a, or PL-unique DhMRs, consisted of 1,036 DhMR clusters that exhibited altered 5hmC signals specifically in PL relative to both ctrl and CL-DRG (, Supplemental Data Set 4). Among them, 552 loci showed 5hmC gain and 471 showed 5hmC loss (). Hence, the three way comparisons allowed us to significantly narrow down the DhMRs to the ones uniquely associated with the regenerative state triggered by PL.
Figure 5. Conditioning lesion-specific DhMRs (A) Venn diagram of three sets of DhMRs from pairwise comparisons identifying DhMR clusters that were unique to PL or CL, as well as injury-associated DhMRs that were shared by both. (B) PL-specific (group a) DhMRs separated into the group with specific 5hmC gain (left) and the group with specific 5hmC loss (right). Normalized 5hmC signals (reads per million divided by bin size) were plotted continuously upstream, within, and downstream of each PL-specific DhMRs and averaged across these regions. Total number of DhMRs is indicated at the top. (C) Pie chart showing the association of group a DhMRs with genomic features (left). The numbers of genes associated with group a DhMRs (5hmC gain or loss) are shown on the right. (D) Volcano plot of PL-specific DhMRs according to 5hmC fold changes and P-values. The location of Bdnf is highlighted in red. (E) Top ranked group a DhMRs with the largest fold change (FC) of either 5hmC gain or loss. (F) Genomic views of top ranked PL-specific DhMRs, exhibiting unique gain (P2rx3 and Defb28) or loss (Ap1g1) of 5hmC after PL compared with CL or control. (G) Ingenuity annotations of gene functions of group a DhMR-associated genes. (H) Genomic views of Stat3 and Camk2d showing locations of DhMRs. See also Figs. S2-S5.
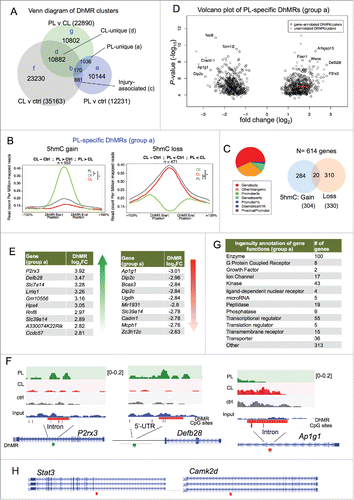
Around half of the group a PL-specific DhMRs were located in gene bodies (). Among a total of 614 genes containing group a DhMRs, 284 showed 5hmC gain and 310 genes showed 5hmC loss in PL, while 20 genes showed both 5hmC gain and loss (). Volcano plot identified genes with the most dynamically changed DhMRs (). Genome browser views of the top ranked genes, e.g., P2rx3 or Defb28, clearly demonstrated intragenic 5hmC gain at introns or 5’-UTR, respectively (). Ap1g1, on the other hand, showed markedly decreased 5hmC signals in an intron (). Annotation of biological function of group a DhMR-associated genes by IPA identified enzymes (100 genes), transcriptional regulators (55), kinases (43), as well as transporters (36), G protein coupled receptors (8) and microRNA (5) (). Notably, Stat3, a transcription factor shown to promote axon growth in DRG neurons,Citation43 displayed PL-specific 5hmC loss; calcium/calmodulin-dependent protein kinase II delta (Camk2d), a central regulator of plasticity,Citation44 also showed PL-specific 5hmC loss ().
Interestingly, the number of loci displaying CL-specific 5hmC alterations (group d) outnumbered group a DhMRs by 10-fold (10,882 DhMRs; Fig. S2A-C; Supplemental Data Set 7). This further supports an active chromatin remodeling process initiated by CL. Notably, an overwhelming majority of group d DhMRs (10,396) showed 5hmC gain (Fig. S2B).
Group c consisted of 881 axotomy-associated DhMR clusters that were shared by both PL and CL relative to ctrl (Fig. S2D, Supplemental data set 6). Among them, 540 loci showed 5hmC gain and 309 loci showed 5hmC loss after axotomy (Fig. S2D-G). Group b had 170 DhMR clusters that were overlapped by all three pair-wise comparisons (Fig. S3A; Supplemental Data Set 5). Among them, 69 DhMR showed 5hmC gain in both PL- and CL-relative to control DRGs, but the extent of 5hmC gain was more pronounced in CL than PL (Fig. S3A). Sixty-two DhMR showed divergent 5hmC changes, with 5hmC gain in CL and loss in PL compared with control (Fig. S3A). A complete list of the other scenarios of 5hmC changes is shown in Fig. S4. Group e-g DhMRs included a large number of loci displaying 5hmC changes present in only one pairwise comparison, without overlap with other pairwise comparisons (Figs. S3B-D, Supplemental Data Sets 8–10). As stated earlier, intersecting pair-wise comparisons of 5hmC maps allowed us to narrow down to specific DhMRs reflecting regeneration- or injury-associated 5hmC signatures. For instance, PL-vs-ctrl comparison yielded 12,244 DhMRs, the majority of which belong to group e DhMRs (10,144); and only 1036 belong to group a, PL-specific DhMRs.
We further analyzed the 161 DhMR-containing RAGs induced by PL and asked whether they contain group a, PL-specific DhMRs. We found that 18 of them exhibited PL-specific 5hmC changes (Fig. S5A), including growth factor signaling genes, such as Bdnf and Ntrk2 (TrkB), as well as cytoskeletal regulators such as Nedd4l, RhoC and Arhgap24 (Fig. S5B).
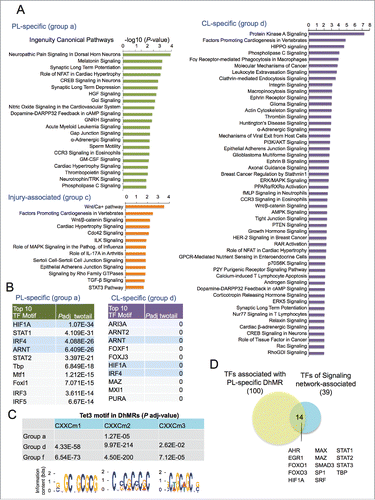
Lastly, we applied Ingenuity Pathway Analysis (IPA) for enrichment of Canonical Pathways of DhMR-associated genes. In genes associated with group a DhMRs, enriched pathways included neuropathic pain signaling in dorsal horn neurons, synaptic long-term potentiation or depression (). Other top-ranked pathways included NFAT, CREB, and Neurotrophin/TRK signaling, all of which have been linked to axonal growth.Citation45,46 (). In comparison, the top enriched pathways for group d genes included Protein Kinase A, Hippo, Integrin, and Ephrin receptor signaling (). It also included PI3K/AKT signaling, axon guidance signaling, ERK/MAPK signaling and PTEN signaling. In contrast, injury-associated, group c genes were enriched for Wnt, Cdc42, TGF-β, and STAT3 pathways (). Together, these data implicate distinct signaling pathways as regulated by 5hmC-associated mechanisms in different axonal injury settings.
Figure 6. Canonical pathways and predicted TFs associated with DhMRs (A) Top ranked Canonical Pathways of gene sets that are associated with different groups of DhMRs, as determined by Ingenuity Pathway Analysis (IPA). (B) Top ranked over-represented TF motifs identified by MEME-AME (with adjusted P-value <0.001 as cutoff) in group a and group d DhMRs (from all genomic regions, not restricted to only promoter regions). Shared motifs are highlighted in blue. (C) Table listing the adjusted P-values showing significant enrichment of known Tet3 DNA binding motifs (shown at the bottom) in all the three groups of DhMRs. (D) Venn diagram showing overlap of TFs whose binding motifs were enriched in group a, PL-specific DhMRs and TFs identified in a study of axon regeneration associated signaling networks.Citation25 See also Fig. S6.
Enriched transcription factor binding motifs in DhMRs
To uncover potential transcription regulators of 5hmC dynamics after axonal injury, we performed transcription factor motif (TF motif) enrichment analysis among the DhMRs in group a and group d (using DhMRs of all regions, and not limited to promoter regions). One hundred overrepresented TF motifs were identified in group a DhMRs (adjusted P-value < 0.001), including Smad1, a known pro-growth transcription factorCitation26,47 For group d DhMRs, 361 TF motifs were overrepresented. Comparing the top 10 most significantly enriched TF motifs, we found that seven TF motifs were unique to each group, notably, IRF and STAT motifs were highly overrepresented in group a DhMRs, while forkhead box (FOX) motifs were enriched in group d DhMRs (). Three TF motifs were shared, i.e., hypoxia-inducible factor 1-α (HIF1A), aryl hydrocarbon receptor nuclear translocator (ARNT), and interferon regulatory factor 4 (IRF4) (). Interestingly, HIF-1 is a heterodimeric TF consisting of HIF1A, the α subunit, and ARNT, the β subunit. The fact that both α and β subunits of HIF-1 were identified as top ranked enriched TF motifs in our unbiased approach supports its central role in regulating 5hmC dynamics after axonal injury.
The CXXC DNA-binding domain of TET3 has three recognized binding motifs.Citation34 We found that CXXC motif 2 was overrepresented in Group a DhMRs (adjusted P-value < 1.3 × 10−4), while all the three CXXC motifs were enriched in group d DhMRs (). We also analyzed group f DhMRs, which excluded PL-associated DhMRs, and found that all three Tet3 CXXC motifs were also enriched (). These results suggest that even though Tet3 is upregulated only in PL-DRG, the baseline Tet3 activity may contribute to 5hmC reconfigurations in all three DRG conditions.
Next, we investigated whether TFs identified through our unbiased motif analysis of DhMRs correlated with signaling network-associated transcription factors that had been described to regulate RAGs after conditioning lesion.Citation25 We found that among the 39 signaling network-associated transcription factors, 14 overlapped with the ones identified in our motif search, including HIF1A, STAT family, FOXO ().
Finally, we conducted Ingenuity upstream regulator analyses of the genes associated with PL- or CL-specific DhMRs. Among the top 10 most significant predicted upstream regulators, four (FOS, ERG, TGFB1, and ESR1) were shared by PL and CL, but the others did not overlap (Fig. S6), suggesting engagement of both shared and distinct regulators by PL and CL to regulate DhMR-associated genes.
Discussion
Cytosine hydroxymethylation is an epigenetic mark that modifies DNA-protein interactions, thereby influencing transcriptional output and cellular state. To set up a platform to understand the function of 5hmC dynamics in axon regeneration, we constructed detailed epigenomic maps of 5hmC modifications in adult DRG under three different regenerative conditions: peripheral lesion, which switches DRG neurons into a growth state; central lesion, which leaves DRG neurons in a state refractory to regeneration; and naive state with no injury. Functional annotations and comparative analyses of 5hmC maps provide a new perspective of a previously unrecognized epigenetic mechanism underlying the divergent regenerative responses after axonal injury.
Common genomic features of 5hmC distributions in PNS and CNS
The enhanced regenerative potential of PNS neurons after conditioning lesion provides an entry point to understand epigenetic mechanisms underlying the regenerative injury responses and coordinated regulation of RAGs that are absent in CNS neurons. Previous mapping studies have revealed distinct genomic distributions of 5hmC in ES cells and brain tissues.Citation9,11,12 To our knowledge, the current study represents the first genome-wide mapping of 5hmC in the PNS tissue. We revealed common genomic features of 5hmC configurations in CNS and PNS. For instance, there is a striking depletion of 5hmC marks on chromosome X in adult DRG, similar to that in CNS tissues, such as hippocampus and cerebellum.Citation12 This suggests engagement of similar 5hmC regulatory mechanisms in both CNS and PNS. Another conserved feature of 5hmC distributions between CNS and PNS is a marked low density of 5hmC near TSS sites and enriched 5hmC at gene bodies.Citation7,12 It is noteworthy that in ES cells, 5hmC has been found to accumulate at certain TSS sites, where it is associated with repressor complexes.Citation11,48
Previous studies on CNS tissues indicated that 5hmC signals remain relatively stable in certain genomic loci, but exhibit localized changes in others.Citation12 Our results similarly showed relative stability of 5hmC signals at many loci in adult DRG under injury conditions, echoing the findings in CNS studies. However, dynamically regulated loci are also abundant, with some exhibiting 5hmC modifications specific to peripheral lesion, others to central lesion or both. The intersection of three pairwise comparisons allowed us to narrow down the list of genomic loci that show PL-specific or CL-specific 5hmC modifications, as well as injury-associated 5hmC changes, thus providing additional understanding of this base modification in different injury settings.
5hmC-mediated regulation of RAGs after conditioning lesion
Overexpression of a single RAG, such as ATF3 or STAT3, is usually not sufficient to activate the axon regeneration program.Citation39,49 Instead, coordinated activation of many RAGs involved in multiple pathways is likely needed. Epigenetic changes hold potential for regulating entire classes of growth supporting or growth inhibiting genes. We found that nearly half of the RAGs induced by PL display 5hmC modifications at 24 h after lesion, including well-know RAGs, such as Atf3 and Bdnf, thus supporting an active role for 5hmC-associated mechanism in their regulation. Our findings on 5hmC-associated regulation of Bdnf in conditioned DRG also extends earlier reports on an active role of DNA methylation for activity-dependent gene regulation of Bdnf during neurodevelopmentCitation50 and learning and memory.Citation51
The mechanism by which 5hmC modifications regulate transcription remains to be determined. Oxidation of 5mC reduces the affinity of 5mC-specific binding proteins, including HDAC-associated repressive complexes, thereby switching transcriptional repression to transcriptional activation or a permissive state.Citation52 In this regard, 5hmC may function primarily to offset the repressive effect of 5mC by simply reducing 5mC levels. On the other hand, 5hmC may interact independently with specific DNA binding proteins to regulate transcription. It has been reported that methyl-CpG binding protein, MeCP2, can bind to 5hmC in gene bodies.Citation53 Of note, we find concordant changes in the direction of 5hmC modification and transcriptional alteration only in a subset of RAGs. Hence, for other RAGs, 5hmC may serve as an intermediate for cytosine demethylation. Our analyses of separating out gene body and promoter DhMRs also did not find evidence that the directionality of 5hmC changes predicts the direction of transcription changes. Another confounding factor is that around half of DhMR-associated RAGs contain multiple DhMRs, often displaying both 5hmC gains and losses. The proportion of the RAGs containing multiple DhMRs appeared similar to that of all DhMR-associated genes, which argues against a model whereby multiple DhMRs in a gene may denote more active regulation. The functional significance of multiple DhMRs in one gene requires future clarification and complex regulatory mechanisms may be at play to convey the 5hmC configurations into genomic output.
Our data did not distinguish 5hmC at CpG vs. non-CpG cytosine methylation sites (CpH; H stands for A, C, or T). DNA methylation at CpH sites has been identified for nearly 25% of all methylated cytosines in mouse ES cells, and is enriched in gene bodies and implicated in regulation of developmentally activated genes.Citation11,54 Higher resolution maps will be needed to resolve 5hmC modification on a single base level. A previous DNA methylation microarray analysis found only a modest number of genes differentially methylated after PL or CL, but none of the RAGs examined displayed significant alterations in DNA methylation at promoters or CpG islands.Citation15 In fact, most gene promoters were not significantly methylated or showed any changes in either axon injury condition. Of note, this microarray analysis focused on gene promoters and CpG islands, thus could not directly assess quantitative methylation levels at other genomic loci. Nevertheless, it echoes our findings that the overwhelming majority of 5hmC modifications, hence DNA methylation reconfigurations, occur at gene bodies and at “open sea,” and not on gene promoters or CpG islands.
The 5hmC modification by itself is insufficient to predict transcriptional changes, since only a subset of genes containing PL-vs-CL DhMRs showed significant transcriptional changes in our Affymetrix array screen for RAGs. It suggests that a changing chromatin landscape merely sets the stage for coordinated gene regulation. Our current study focuses on one time point, i.e., 24 h after axotomy, and it is likely that widespread 5hmC changes at 24 h may lead to sustained transcriptional changes of more genes at later time points. Additionally, Affymetrix array studies might underestimate transcriptional changes as compared with next-generation sequencing, a task worthy of future endeavor. Future studies conducted at later time points will address the timeframe under which specific 5hmC changes are maintained after axonal injury. The refinement of genome-wide distributions of 5hmC modifications in specific cell types will also shed further light on the influences of 5hmC in regulating cell type specific injury responses in adult DRG.
Central axotomy may switch DRG neurons to a repressed growth state
In the conditioning lesion paradigm, following axotomy of the peripheral branch of DRG neurons, retrograde transport of injury signals to the cell body of the neuron results in induction of RAGs.Citation55 Peculiarly, injury to the central branch of DRG axons does not lead to such activation of RAGs, thereby resulting in regenerative failure after spinal cord injury.Citation56 Our study revealed distinct 5hmC dynamics as a potential epigenetic mechanism underlying major differences in intrinsic transcriptional networks and, correspondingly, distinct regenerative states of DRG induced by PL or CL.
Long thought to be a static event, our study nonetheless highlights that the central axotomy results in widespread 5hmC modifications, despite the absence of Tet3 upregulation. Clearly, baseline enzymatic activities of TET3 or other TETs may induce and modify 5hmC throughout the genome in CL-DRG. In fact, we detected nearly 3-fold the numbers of DhMRs triggered by central axotomy as compare with peripheral axotomy, with little overlap between the two. Thus, the difference in global levels of 5hmC is not as relevant as the distinct genomic patterns of 5hmC modifications in response to peripheral vs. central axotomy. Aside from changes of expression level, Tet3 activity and its recruitment to specific genomic loci may be also otherwise regulated, affecting target gene changes in axon regeneration. Strikingly, IPA comparison analysis showed divergent Diseases and Functions and engagement of differential Canonical Pathways of the gene sets associated with PL- and CL-induced DhMRs. An intriguing perspective raised by our study is that CL may initiate a detrimental injury response that actively sets DRG neurons to a growth-repressed state. Hence, regenerative strategy would need not only to mimic favorable changes of the injury responses incurred by the PL, but also to mitigate those unfavorable changes incurred by the CL.
The underlying mechanisms by which PL and CL lead to distinct 5hmC modifications remain unclear, although our IPA analysis suggests that different axotomy paradigms may engage distinct upstream regulators to collaborate with TET3 or other Tets for converting 5mC to 5hmC, or further oxidization toward unmethylated cytosine. Indeed, motif enrichment analyses revealed enriched TF binding motifs at the DhMRs, with some specific for PL or CL, and others shared by both. For instance, IRF or STAT family TFs may participate in PL-specific 5hmC modifications, while FOX family TFs may contribute to CL-specific 5hmC changes. Our earlier studies demonstrated a collaborative mechanism of Smad1 and HDAC1 in RAG regulation,Citation26 hence different regeneration-associated TFs may interact with different epigenetic factors for gene regulation in the regenerative responses.
One notable shared TF motif among DhMRs is HIF-1, with both α and β units ranked as the top overrepresented TF motifs in our unbiased approach, suggesting a central role of HIF-1 in regulating injury responses after axotomy. A recent study demonstrated a role of HIF-1 in regulating multiple injury-induced genes in DRG neurons and a contribution to the conditioning lesion effect.Citation57 Our study thus extends further in suggesting 5hmC dynamics as a potential mechanism by which HIF-1 regulates transcriptional changes after axotomy. It is worth discussing, however, that our studies found enrichment of HIF-1 motifs in both PL- and CL-specific DhMRs, thus additional co-regulators must be at play to impart the specificity of HIF-1 target genes after PL as compared with CL.
A recent computational analysis identified 39 hub TFs that provide cross talk between 400 redundant axonal signaling networks in response to axonal injury in DRGs.Citation25 Encouragingly, there was a substantial overlap between our list of TFs implicated in 5hmC modifications and the 39 hub TFs implicated in DRG injury response, including HIF1A and STAT. Furthermore, recent multi-level bioinformatics analyses have revealed a specific regeneration-associated gene network that is coordinately regulated by RAGs, signaling pathways, and TFs, many of which also showed overlap with our 5hmC regulated Canonical Pathways and upstream TFs such as EGR1, STAT, and SMAD.Citation58 We also revealed potential 5hmC-associated mechanisms in regulating the hub TFs, such as ATF3 and STAT3, both of which contain PL-induced DhMRs. Hence, our data constructed new connections between signaling pathways, epigenetic regulator Tet, and transcription co-regulators, such as HIF-1, STAT, and IRF, for coordinated gene regulation in the regenerative responses after axonal injury.
In summary, genome-wide 5hmC mapping points to major influences of 5hmC in axon injury responses and regenerative potential through coordinated regulation of entire classes of genes in collaboration with specific transcription factors. The 5hmC reference epigenome in adult DRG provides a framework for future exploration of 5hmC-mediated mechanisms in the regenerative responses and RAG regulation. Altogether, our data suggest that changing the transcriptional state of adult neurons through epigenetic factors is a worthy strategy for enhancing regenerative potential.
Materials and methods
Animal Procedures
All surgeries were performed on 6- to 8-week-old CD1 female mice (Charles River Laboratories) in accordance with the guidelines and protocols approved by the Institutional Animal Care and Use Committee at Icahn School of Medicine at Mount Sinai. For dorsal column lesion, a T8 laminectomy was performed and ascending sensory fibers were transected using iris micro scissors (Fine Science Tools) to a depth of 0.8 mm. For the peripheral conditioning lesion, the right sciatic nerve was transected at mid-thigh level. Ipsilateral lumbar 4, 5, and 6 (L4–L6) DRGs and contralateral naive L4–L6 DRGs with no injury were collected. Of note, sciatic nerve lesion affects largely DRG neurons in lumbar L4–6 (all subpopulations), whereas T8 dorsal column lesion mainly affects medium/large-sized DRG neurons with low-threshold mechanoreceptors (Aβ fibers).
Immunohistochemistry
Mice were perfused intracardially with 4% paraformaldehyde (PFA) in 0.1 M phosphate buffer, and L4, L5, and L6 DRGs were dissected. DRGs were post-fixed in 4% PFA for 5 min, cryoprotected in 30% sucrose, embedded in OCT, and cryosectioned at 10 μm thickness. Sections were permeabilized with 0.1% Triton X-100 (Sigma), treated with blocking buffer (0.1 M phosphate buffer, 5% normal goat serum), and incubated overnight at 4°C with antibodies rabbit anti-TET3 (EMD Millipore, 1:500), rabbit anti-5hmC (Active Motif, 1:500), or mouse anti-β-III-Tubulin (Sigma, 1:1,000). After incubation with fluorescent secondary antibodies (Invitrogen), the sections were counterstained with DAPI (1:1,000; Invitrogen) and mounted with Fluoromount (EMS). Images were captured with an Axio Imager.A2 (Zeiss) microscope equipped with an AxioCam MRc camera.
Genomic DNA isolation
L4, L5, and L6 DRGs were dissected from animals subjected to dorsal column lesion or peripheral lesion 24 h prior. Contralateral DRGs from animals subjected to the peripheral lesion were collected as uninjured controls. DRGs from three separate animals were pooled for each biological replicate. Tissue was digested overnight at 65°C in TNES with 0.5 mg/ml Proteinase K. RNA was removed by digestion with RNase A (Thermo Scientific) for 1 h at 37°C. After RNase treatment, DNA was precipitated using high salt and ethanol.
5hmC capture and library construction
DNA was sheared into fragments of an average size of 250 bp using the Biorupter Twin (Diagenode) device, using the “low” setting and 30 cycles of 30 s “on,” followed by 30 s “off.” Fragment size was confirmed by gel electrophoresis, and DNA concentrations were confirmed before enrichment. Enrichment for 5hmC was performed with the Hydroxymethyl Collecter-Seq Kit (Active Motif) according to manufacturer's instruction. Briefly, 4.3 μg of sheared genomic DNA was used for each sample. The DNA was treated with a β-glucosyltransferase enzyme to chemically modify 5hmC nucleotides with a sugar moiety, which was then conjugated to biotin. The biotinylated 5hmC containing DNA fragments were captured with streptavidin conjugated to magnetic beads, washed, and eluted as 5hmC-enriched DNA. The 5hmC enriched DNA samples, plus one un-enriched sample from each experimental condition, designated “input,” were prepared for Illumina sequencing using the NEBNext ChIP-seq kit (New England Biolabs). Briefly, 10 ng DNA of each 5hmC enriched or input sample were subjected to end repair, A-tailing, adaptor ligation, size selection, and PCR enrichment according to manufacturer's directions. DNA concentrations and library fragment sizes were assessed by Qubit and Bioanalyzer. Libraries were submitted to the New York Genome Center for Illumina 50 bp paired-end sequencing on an Illumina HiSeq 2,500 device, with 6 barcoded samples in each lane.
qRT-PCR
RNAs from freshly collected DRGs or cultured DRG neurons were isolated using RNeasy Micro kit (QIAGEN). A total of 500 ng of total RNA was used for reverse transcription reaction (SuperScript III RT, Invitrogen). qRT-PCR was performed using SYBR Green PCR master mix (QuantaBiosciences) in ABI PCR System. The melting curve of each sample was measured to ensure the specificity of the products. Data were normalized to the housekeeping gene RPL13a and analyzed using the ΔΔCt method. Primers used in quantitative PCR are listed in Table S1. At least three independent experiments were performed for each condition.
Bioinformatics analyses
The 5hmC sequencing data was first controlled for quality using metrics generated by FastQC (v0.11.2).Citation59 Raw sequencing reads were aligned to the mouse mm9 genome using default settings of Bowtie (v2.2.0).Citation60 Only uniquely mapped reads were retained and the alignments were subsequently filtered using the SAMtools package (v0.1.19)Citation61 to remove duplicate reads. For the analysis of global distributions of 5hmC signals, the mouse genome was divided into 10 kb bins and the number of aligned reads for each bin were counted. Bins with RPKM > 1 for at least one sample were kept and the rest discarded. Read counts were variance stabilized, then normalized by input to yield an enrichment value. Finally, bins with enrichment > 1 for at least one sample were retained for use in the analysis, resulting in ∼20,000 bins. A hierarchical-clustered heatmap was generated using the default R heatmap.2 function, which defaults to clustering Euclidean distance using the “complete” tree-building method.
Peak-calling was performed using MACS (v2.1.1)Citation62 with the “--broad -q 0.01” setting. Differential analysis between samples was performed using diffReps (v1.55.4)Citation36 with settings of 200 bp window size, 20 bp moving step size, and a nominal P-value of 0.0001 to generate candidate regions. Annotation of called peaks and differential regions to their genomic features (promoters, gene bodies, intergenic, etc.) was performed using the Region Analysis program of diffReps (v1.55.4),Citation36 and read alignment profile plots were generated using ngs.plot (v2.47).Citation63
The transcription factor motif enrichment analysis was performed using the AME program of the MEME Suite (v4.11.1)Citation64 with “--method ranksum --scoring avg --rsmethod quick” settings.
Accession numbers
The 5hmC-enriched DNA sequencing data reported in this study has been deposited at the NCBI Gene Expression Omnibus (GEO) data archive and can be accessed with accession code GSE85972.
Statistical analysis
Prism GraphPad software was used for Student's t test analysis of qRT-PCR data.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Author contributions
E. L and L. S. performed all bioinformatics analyses, data interpretation, and graph preparation. A. K.-C. performed experiments and participated in all aspects of the study and data quantification. R. F. participated in experimental design and data interpretation. H.Z. designed the study, analyzed the data, and wrote the paper. All authors contributed to preparation of the manuscript.
Supplementary_data.zip
Download Zip (21.7 MB)Acknowledgments
We thank all members of Zou laboratory for discussions. This work was supported by NIH/NINDS (R01 NS073596), New York State Spinal Cord Injury Research program (DOH01-C30603GG-3450000, DOH01-C30832GG-3450000), and Irma T. Hirschl / Monique Weill-Caulier Scholar Award to H.Z.
Related Research Data
References
- Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet 2003; 33(Suppl):245-54; PMID:12610534; http://dx.doi.org/10.1038/ng1089
- Lister R, Mukamel EA, Nery JR, Urich M, Puddifoot CA, Johnson ND, Lucero J, Huang Y, Dwork AJ, Schultz MD, et al. Global epigenomic reconfiguration during mammalian brain development. Science 2013; 341:1237905; PMID:23828890; http://dx.doi.org/10.1126/science.1237905
- Day JJ, Sweatt JD. DNA methylation and memory formation. Nat Neurosci 2010; 13:1319-23; PMID:20975755; http://dx.doi.org/10.1038/nn.2666
- Gräff J, Kim D, Dobbin MM, Tsai LH. Epigenetic regulation of gene expression in physiological and pathological brain processes. Physiol Rev 2011; 91:603-49; PMID:21527733; http://dx.doi.org/10.1152/physrev.00012.2010
- Kinde B, Gabel HW, Gilbert CS, Griffith EC, Greenberg ME. Reading the unique DNA methylation landscape of the brain: Non-CpG methylation, hydroxymethylation, and MeCP2. Proc Natl Acad Sci U S A 2015; 112:6800-6; PMID:25739960; http://dx.doi.org/10.1073/pnas.1411269112
- Wu SC, Zhang Y. Active DNA demethylation: many roads lead to Rome. Nat Rev Mol Cell Biol 2010; 11:607-20; PMID:20683471; http://dx.doi.org/10.1038/nrm2950
- Hahn MA, Qiu R, Wu X, Li AX, Zhang H, Wang J, Jui J, Jin SG, Jiang Y, Pfeifer GP, et al. Dynamics of 5-hydroxymethylcytosine and chromatin marks in Mammalian neurogenesis. Cell Rep 2013; 3:291-300; PMID:23403289; http://dx.doi.org/10.1016/j.celrep.2013.01.011
- Globisch D, Munzel M, Muller M, Michalakis S, Wagner M, Koch S, Bruckl T, Biel M, Carell T. Tissue distribution of 5-hydroxymethylcytosine and search for active demethylation intermediates. PloS One 2010; 5:e15367; PMID:21203455; http://dx.doi.org/10.1371/journal.pone.0015367
- Song CX, Szulwach KE, Fu Y, Dai Q, Yi C, Li X, Li Y, Chen CH, Zhang W, Jian X, et al. Selective chemical labeling reveals the genome-wide distribution of 5-hydroxymethylcytosine. Nat Biotechnol 2011; 29:68-72; PMID:21151123; http://dx.doi.org/10.1038/nbt.1732
- Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, Agarwal S, Iyer LM, Liu DR, Aravind L, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 2009; 324:930-5; PMID:19372391; http://dx.doi.org/10.1126/science.1170116
- Ficz G, Branco MR, Seisenberger S, Santos F, Krueger F, Hore TA, Marques CJ, Andrews S, Reik W. Dynamic regulation of 5-hydroxymethylcytosine in mouse ES cells and during differentiation. Nature 2011; 473:398-402; PMID:21460836; http://dx.doi.org/10.1038/nature10008
- Szulwach KE, Li X, Li Y, Song CX, Wu H, Dai Q, Irier H, Upadhyay AK, Gearing M, Levey AI, et al. 5-hmC-mediated epigenetic dynamics during postnatal neurodevelopment and aging. Nat Neurosci 2011; 14:1607-16; PMID:22037496; http://dx.doi.org/10.1038/nn.2959
- Feng J, Shao N, Szulwach KE, Vialou V, Huynh J, Zhong C, Le T, Ferguson D, Cahill ME, Li Y, et al. Role of Tet1 and 5-hydroxymethylcytosine in cocaine action. Nat Neurosci 2015; 18:536-44; PMID:25774451; http://dx.doi.org/10.1038/nn.3976
- Iskandar BJ, Rizk E, Meier B, Hariharan N, Bottiglieri T, Finnell RH, Jarrard DF, Banerjee RV, Skene JHP, Nelson A, et al. Folate regulation of axonal regeneration in the rodent central nervous system through DNA methylation. J Clin Invest 2010; 120:1603-16; PMID:20424322; http://dx.doi.org/10.1172/JCI40000
- Puttagunta R, Tedeschi A, Soria MG, Hervera A, Lindner R, Rathore KI, Gaub P, Joshi Y, Nguyen T, Schmandke A, et al. PCAF-dependent epigenetic changes promote axonal regeneration in the central nervous system. Nat Commun 2014; 5:3527; PMID:24686445; http://dx.doi.org/10.1038/ncomms4527
- Goldberg JL. How does an axon grow? Genes Dev 2003; 17:941-58; PMID:12704078; http://dx.doi.org/10.1101/gad.1062303
- Zhou FQ, Snider WD. Intracellular control of developmental and regenerative axon growth. Philos Trans R Soc Lond B Biol Sci 2006; 361:1575-92; PMID:16939976; http://dx.doi.org/10.1098/rstb.2006.1882
- Wong JK, Zou H. Reshaping the chromatin landscape after spinal cord injury. Front Biol (Beijing) 2014; 9:356-66; PMID:25554728; http://dx.doi.org/10.1007/s11515-014-1329-8
- Neumann S, Woolf CJ. Regeneration of dorsal column fibers into and beyond the lesion site following adult spinal cord injury. Neuron 1999; 23:83-91; PMID:10402195; http://dx.doi.org/10.1016/S0896-6273(00)80755-2
- Richardson PM, Issa VM. Peripheral injury enhances central regeneration of primary sensory neurones. Nature 1984; 309:791-3; PMID:6204205; http://dx.doi.org/10.1038/309791a0
- Smith DS, Pate Skene JH. A transcription-dependent switch controls competence of adult neurons for distinct modes of axon growth. J Neurosci 1997; 17:646-58; PMID:8987787
- Blesch A, Lu P, Tsukada S, Alto LT, Roet K, Coppola G, Geschwind D, Tuszynski MH. Conditioning lesions before or after spinal cord injury recruit broad genetic mechanisms that sustain axonal regeneration: superiority to camp-mediated effects. Exp Neurol 2012; 235:162-73; PMID:22227059; http://dx.doi.org/10.1016/j.expneurol.2011.12.037
- Bonilla IE, Tanabe K, Strittmatter SM. Small proline-rich repeat protein 1A is expressed by axotomized neurons and promotes axonal outgrowth. J Neurosci 2002; 22:1303-15; PMID:11850458
- Costigan M, Befort K, Karchewski L, Griffin RS, D'Urso D, Allchorne A, Sitarski J, Mannion JW, Pratt RE, Woolf CJ. Replicate high-density rat genome oligonucleotide microarrays reveal hundreds of regulated genes in the dorsal root ganglion after peripheral nerve injury. BMC Neurosci 2002; 3:16; PMID:12401135; http://dx.doi.org/10.1186/1471-2202-3-16
- Michaelevski I, Segal-Ruder Y, Rozenbaum M, Medzihradszky KF, Shalem O, Coppola G, Horn-Saban S, Ben-Yaakov K, Dagan SY, Rishal I, et al. Signaling to transcription networks in the neuronal retrograde injury response. Sci Signal 2010; 3:ra53; PMID:20628157; http://dx.doi.org/10.1126/scisignal.2000952
- Finelli MJ, Wong JK, Zou H. Epigenetic regulation of sensory axon regeneration after spinal cord injury. J Neurosci 2013; 33:19664-76; PMID:24336730; http://dx.doi.org/10.1523/JNEUROSCI.0589-13.2013
- Ma DK, Guo JU, Ming GL, Song H. DNA excision repair proteins and Gadd45 as molecular players for active DNA demethylation. Cell Cycle 2009; 8:1526-31; PMID:19377292; http://dx.doi.org/10.4161/cc.8.10.8500
- Befort K, Karchewski L, Lanoue C, Woolf CJ. Selective up-regulation of the growth arrest DNA damage-inducible gene Gadd45 alpha in sensory and motor neurons after peripheral nerve injury. Eur J Neurosci 2003; 18:911-22; PMID:12925017; http://dx.doi.org/10.1046/j.1460-9568.2003.02827.x
- Doron-Mandel E, Fainzilber M, Terenzio M. Growth control mechanisms in neuronal regeneration. FEBS Lett 2015; 589:1669-77; PMID:25937120; http://dx.doi.org/10.1016/j.febslet.2015.04.046
- Frey E, Valakh V, Karney-Grobe S, Shi Y, Milbrandt J, DiAntonio A. An in vitro assay to study induction of the regenerative state in sensory neurons. Exp Neurol 2015; 263:350-63; PMID:25447942; http://dx.doi.org/10.1016/j.expneurol.2014.10.012
- Zou H, Ho C, Wong K, Tessier-Lavigne M. Axotomy-induced Smad1 activation promotes axonal growth in adult sensory neurons. J Neurosci 2009; 29:7116-23; PMID:19494134; http://dx.doi.org/10.1523/JNEUROSCI.5397-08.2009
- Gu TP, Guo F, Yang H, Wu HP, Xu GF, Liu W, Xie ZG, Shi L, He X, Jin SG, et al. The role of Tet3 DNA dioxygenase in epigenetic reprogramming by oocytes. Nature 2011; 477:606-10; PMID:21892189; http://dx.doi.org/10.1038/nature10443
- Li X, Wei W, Zhao QY, Widagdo J, Baker-Andresen D, Flavell CR, D'Alessio A, Zhang Y, Bredy TW. Neocortical Tet3-mediated accumulation of 5-hydroxymethylcytosine promotes rapid behavioral adaptation. Proc Natl Acad Sci U S A 2014; 111:7120-5; PMID:24757058; http://dx.doi.org/10.1073/pnas.1318906111
- Xu Y, Xu C, Kato A, Tempel W, Abreu JG, Bian C, Hu Y, Hu D, Zhao B, Cerovina T, et al. Tet3 CXXC domain and dioxygenase activity cooperatively regulate key genes for Xenopus eye and neural development. Cell 2012; 151:1200-13; PMID:23217707; http://dx.doi.org/10.1016/j.cell.2012.11.014
- Santos F, Peat J, Burgess H, Rada C, Reik W, Dean W. Active demethylation in mouse zygotes involves cytosine deamination and base excision repair. Epigenetics Chromatin 2013; 6:39; PMID:24279473; http://dx.doi.org/10.1186/1756-8935-6-39
- Shen L, Shao NY, Liu X, Maze I, Feng J, Nestler EJ. diffReps: detecting differential chromatin modification sites from ChIP-seq data with biological replicates. PloS One 2013; 8:e65598; PMID:23762400; http://dx.doi.org/10.1371/journal.pone.0065598
- Liu K, Lu Y, Lee JK, Samara R, Willenberg R, Sears-Kraxberger I, Tedeschi A, Park KK, Jin D, Cai B, et al. PTEN deletion enhances the regenerative ability of adult corticospinal neurons. Nat Neurosci 2010; 13:1075-81; PMID:20694004; http://dx.doi.org/10.1038/nn.2603
- Parikh P, Hao Y, Hosseinkhani M, Patil SB, Huntley GW, Tessier-Lavigne M, Zou H. Regeneration of axons in injured spinal cord by activation of bone morphogenetic protein/Smad1 signaling pathway in adult neurons. Proc Natl Acad Sci 2011; 108:E99–E107; PMID:21518886; http://dx.doi.org/10.1073/pnas.1100426108
- Seijffers R, Mills CD, Woolf CJ. ATF3 increases the intrinsic growth state of DRG neurons to enhance peripheral nerve regeneration. J Neurosci 2007; 27:7911-20; PMID:17652582; http://dx.doi.org/10.1523/JNEUROSCI.5313-06.2007
- Cohen-Cory S, Kidane AH, Shirkey NJ, Marshak S. Brain-derived neurotrophic factor and the development of structural neuronal connectivity. Dev Neurobiol 2010; 70:271-88; PMID:20186709; http://dx.doi.org/10.1002/dneu.20774
- Shim SO, Cafferty WB, Schmidt EC, Kim BG, Fujisawa H, Strittmatter SM. PlexinA2 limits recovery from corticospinal axotomy by mediating oligodendrocyte-derived Sema6A growth inhibition. Mol Cell Neurosci 2012; 50:193-200; PMID:22564823; http://dx.doi.org/10.1016/j.mcn.2012.04.007
- Suto F, Tsuboi M, Kamiya H, Mizuno H, Kiyama Y, Komai S, Shimizu M, Sanbo M, Yagi T, Hiromi Y, et al. Interactions between plexin-A2, plexin-A4, and semaphorin 6A control lamina-restricted projection of hippocampal mossy fibers. Neuron 2007; 53:535-47; PMID:17296555; http://dx.doi.org/10.1016/j.neuron.2007.01.028
- Miao T, Wu D, Zhang Y, Bo X, Subang MC, Wang P, Richardson PM. Suppressor of cytokine signaling-3 suppresses the ability of activated signal transducer and activator of transcription-3 to stimulate neurite growth in rat primary sensory neurons. J Neurosci 2006; 26:9512-9; PMID:16971535; http://dx.doi.org/10.1523/JNEUROSCI.2160-06.2006
- Colbran RJ, Brown AM. Calcium/calmodulin-dependent protein kinase II and synaptic plasticity. Curr Opin Neurobiol 2004; 14:318-27; PMID:15194112; http://dx.doi.org/10.1016/j.conb.2004.05.008
- Graef IA, Wang F, Charron F, Chen L, Neilson J, Tessier-Lavigne M, Crabtree GR. Neurotrophins and netrins require calcineurin/NFAT signaling to stimulate outgrowth of embryonic axons. Cell 2003; 113:657-70; PMID:12787506; http://dx.doi.org/10.1016/S0092-8674(03)00390-8
- Ma TC, Barco A, Ratan RR, Willis DE. cAMP-responsive element-binding protein (CREB) and cAMP co-regulate activator protein 1 (AP1)-dependent regeneration-associated gene expression and neurite growth. J Biol Chem 2014; 289:32914-25; PMID:25296755; http://dx.doi.org/10.1074/jbc.M114.582460
- Finelli MJ, Murphy KJ, Chen L, Zou H. Differential phosphorylation of Smad1 integrates BMP and neurotrophin pathways through Erk/Dusp in axon development. Cell Rep 2013; 3:1592-606; PMID:23665221; http://dx.doi.org/10.1016/j.celrep.2013.04.011
- Wu H, D'Alessio AC, Ito S, Wang Z, Cui K, Zhao K, Sun YE, Zhang Y. Genome-wide analysis of 5-hydroxymethylcytosine distribution reveals its dual function in transcriptional regulation in mouse embryonic stem cells. Genes Dev 2011; 25:679-84; PMID:21460036; http://dx.doi.org/10.1101/gad.2036011
- Bareyre FM, Garzorz N, Lang C, Misgeld T, Buning H, Kerschensteiner M. In vivo imaging reveals a phase-specific role of STAT3 during central and peripheral nervous system axon regeneration. Proc Natl Acad Sci U S A 2011; 108:6282-7; PMID:21447717; http://dx.doi.org/10.1073/pnas.1015239108
- Martinowich K, Hattori D, Wu H, Fouse S, He F, Hu Y, Fan G, Sun YE. DNA methylation-related chromatin remodeling in activity-dependent BDNF gene regulation. Science 2003; 302:890-3; PMID:14593184; http://dx.doi.org/10.1126/science.1090842
- Yu NK, Baek SH, Kaang BK. DNA methylation-mediated control of learning and memory. Mol Brain 2011; 4:5; PMID:21247469; http://dx.doi.org/10.1186/1756-6606-4-5
- Nan X, Ng HH, Johnson CA, Laherty CD, Turner BM, Eisenman RN, Bird A. Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature 1998; 393:386-9; PMID:9620804; http://dx.doi.org/10.1038/30764
- Mellen M, Ayata P, Dewell S, Kriaucionis S, Heintz N. MeCP2 binds to 5hmC enriched within active genes and accessible chromatin in the nervous system. Cell 2012; 151:1417-30; PMID:23260135; http://dx.doi.org/10.1016/j.cell.2012.11.022
- Lister R, Pelizzola M, Dowen RH, Hawkins RD, Hon G, Tonti-Filippini J, Nery JR, Lee L, Ye Z, Ngo QM, et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 2009; 462:315-22; PMID:19829295; http://dx.doi.org/10.1038/nature08514
- Cho Y, Sloutsky R, Naegle KM, Cavalli V. Injury-induced HDAC5 nuclear export is essential for axon regeneration. Cell 2013; 155:894-908; PMID:24209626; http://dx.doi.org/10.1016/j.cell.2013.10.004
- Afshari FT, Kappagantula S, Fawcett JW. Extrinsic and intrinsic factors controlling axonal regeneration after spinal cord injury. Expert Rev Mol Med 2009; 11:e37; PMID:19968910; http://dx.doi.org/10.1017/S1462399409001288
- Cho Y, Shin JE, Ewan EE, Oh YM, Pita-Thomas W, Cavalli V. Activating Injury-Responsive Genes with Hypoxia Enhances Axon Regeneration through Neuronal HIF-1alpha. Neuron 2015; 88:720-34; PMID:26526390; http://dx.doi.org/10.1016/j.neuron.2015.09.050
- Chandran V, Coppola G, Nawabi H, Omura T, Versano R, Huebner EA, Zhang A, Costigan M, Yekkirala A, Barrett L, et al. A Systems-Level Analysis of the Peripheral Nerve Intrinsic Axonal Growth Program. Neuron 2016; 89:956-70; PMID:26898779; http://dx.doi.org/10.1016/j.neuron.2016.01.034
- Andrews S. FastQC: a quality control tool for high throughput sequence data. Available online at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc
- Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 2009; 10:R25; PMID:19261174; http://dx.doi.org/10.1186/gb-2009-10-3-r25
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, Genome Project Data Processing S. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009; 25:2078-9; PMID:19505943; http://dx.doi.org/10.1093/bioinformatics/btp352
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol 2008; 9:R137; PMID:18798982; http://dx.doi.org/10.1186/gb-2008-9-9-r137
- Shen L, Shao N, Liu X, Nestler E. ngs.plot: Quick mining and visualization of next-generation sequencing data by integrating genomic databases. BMC Genomics 2014; 15:284; PMID:24735413; http://dx.doi.org/10.1186/1471-2164-15-284
- McLeay RC, Bailey TL. Motif Enrichment Analysis: a unified framework and an evaluation on ChIP data. BMC Bioinformatics 2010; 11:165; PMID:20356413; http://dx.doi.org/10.1186/1471-2105-11-165