
ABSTRACT
Preterm birth (PTB) affects one in six Black babies in the United States. Epigenetics is believed to play a role in PTB; however, only a limited number of epigenetic studies of PTB have been reported, most of which have focused on cord blood DNA methylation (DNAm) and/or were conducted in white populations. Here we conducted, by far, the largest epigenome-wide DNAm analysis in 300 Black women who delivered early spontaneous preterm (sPTB, n = 150) or full-term babies (n = 150) and replicated the findings in an independent set of Black mother-newborn pairs from the Boston Birth Cohort. DNAm in maternal blood and/or cord blood was measured using the Illumina HumanMethylation450 BeadChip. We identified 45 DNAm loci in maternal blood associated with early sPTB, with a false discovery rate (FDR) <5%. Replication analyses confirmed sPTB associations for cg03915055 and cg06804705, located in the promoter regions of the CYTIP and LINC00114 genes, respectively. Both loci had comparable associations with early sPTB and early medically-indicated PTB, but attenuated associations with late sPTB. These associations could not be explained by cell composition, gestational complications, and/or nearby maternal genetic variants. Analyses in the newborns of the 110 Black women showed that cord blood methylation levels at both loci had no associations with PTB. The findings from this study underscore the role of maternal DNAm in PTB risk, and provide a set of maternal loci that may serve as biomarkers for PTB. Longitudinal studies are needed to clarify temporal relationships between maternal DNAm and PTB risk.
Introduction
Despite more than a half century of intensive research and intervention efforts, preterm birth (PTB, birth before 37 weeks of gestation) remains the single most important maternal and child health challenge in the US, accounting for ∼35% of infant deaths and contributing to disability among survivors [Citation1]. Of particular concern is the US Black population, who continues to suffer from the highest prevalence of PTB (∼17%) in the world. The most challenging and major obstacle to preventing and treating PTB has been our incomplete understanding of its etiology and biological mechanisms.
With recent advances in high-throughput genomic technologies, researchers are now turning to epigenetics as a way of understanding the mechanistic pathways underlying the development of complex traits and diseases. DNA methylation (DNAm), one of the most-studied epigenetic mechanisms, is a heritable but also reversible addition of a methyl or hydroxymethyl group to the 5-carbon position of cytosine in a cytosine-phosphate-guanine (CpG) context. DNAm could regulate high-order DNA structure and affect gene expression via transcriptional regulation of genes and microRNA [Citation2], control of alternative promoter usage [Citation3] and/or alternative splicing [Citation4]. Because DNAm variations can reflect both genetic and environmental exposures, there is the potential to identify novel disease-associated genes and pathways that might not be discovered through genetic or environmental epidemiologic studies alone. There is also increasing interest in methylation profiling to locate disease-related epigenetic regions using peripheral blood, which could be used as potent disease biomarkers [Citation5].
So far, while several genome-wide epigenetic studies have been reported for PTB [Citation6–12], most of these have been focused on cord blood [Citation6,Citation8–11]. Cord blood has a unique cell type, nucleated red blood cells, which have an extreme methylation profile. Recently, an algorithm for estimating cell proportions in cord blood using newly developed cord reference panels has been published [Citation13], which allows investigators to account for the heterogeneity in DNAm resulting from differences in the proportion of this cell type. There are both maternal and fetal contributions to PTB, and it is likely that the impacts of maternal and fetal DNAm on PTB may differ from one another [Citation12]. However, to date, there have been only two epigenome-wide association studies to examine maternal DNAm for PTB [Citation7,Citation12]. Parets et al. [Citation7], in a small study composed of 40 Black women, identified no associations after controlling for multiple testing, although this finding may have been due to limited statistical power in such a small study. The study by Burris et al., showed that mothers in the highest quartile of 1st trimester LINE-1 DNAm had longer gestations and lower odds of PTB than those in the lowest quartile [Citation12]. Although this study had a decent sample size (n = 914), it focused on global methylation rather than site-specific methylation.
The aim of this study was to investigate the epigenome-wide DNAm associations with spontaneous PTB (sPTB) in Black women from the Boston Birth Cohort (BBC) using a two-stage approach. In the discovery stage, we analyzed 150 mothers with early sPTBs and 150 mothers with full-term births (TBs). In the replication stage, we replicated the findings from the discovery sample in an independent set of Black mother-newborn pairs from the same cohort, using both maternal and cord blood samples. We further explored whether the early sPTB associated loci showed comparable associations with other PTB subtypes, i.e., late sPTB and early medically-indicated PTB (mPTB). By incorporating existing genome-wide genotyping data, we then investigated whether the identified associations were under genetic control.
Results
Population characteristics
At the discovery stage, there were 300 Black mothers enrolled from the BBC. After data cleaning (see Methods), the discovery sample included 146 Black mothers with early sPTBs (cases) and 144 Black mothers with TBs (controls). Cases and controls were comparable on age at delivery, pre-pregnancy body mass index (BMI), smoking during pregnancy, alcohol drinking during pregnancy, psychosocial stress during pregnancy, parity and illicit drug use (all P > 0.05, ). As expected, gestational complications, such as hypertensive disorders, diabetes/gestational diabetes (DM/GDM) and intrauterine inflammation, were more prevalent in cases than in controls (all P < 0.05, ).
Table 1. Population characteristics of the discovery and the replication samples from the BBC.
CpG-specific DNAm associations at the discovery stage
We identified 45 autosomal sites (39 sites within gene regions and 6 intergenic) with significantly altered methylation levels in mothers with early sPTBs compared to mothers with TBs, at a false discovery rate (FDR) <5% (, and Supplemental Table S1). There were nearly equal proportions of hypo- (n = 19) and hyper- methylated (n = 26) sites, with the magnitude of DNAm change between cases and controls ranging from 0.3% to 4.4% ( and Supplemental Table S1). The most significant differentially methylated site for early sPTB was cg03915055 at chromosome 2q24.1, which is located in the promoter region of the cytohesin 1 interacting protein (CYTIP) gene. The methylation level at cg03915055 was about 4.1% lower in mothers with early sPTBs than in mothers with TBs (P = 1.1 × 10−13, ). No CpG sites on the X chromosome were found to be differentially methylated in mothers with early sPTBs compared to mothers with TBs ().
Figure 1. Manhattan plot for the genome-wide DNA methylation associations with early spontaneous preterm birth in 290 Black women from the Boston Birth Cohort. The dashed line represents false discovery rate = 5%.
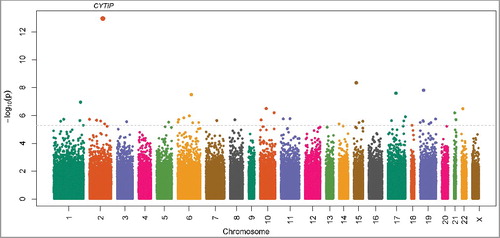
Table 2. The top DNA methylation loci associated with early spontaneous PTB in the discovery sample and their associations with PTB subtypes in the replication sample.
The associations between the identified 45 differentially methylated sites and early sPTB were largely unchanged when gestational complications including hypertensive disorders, GM/DGM, and/or intrauterine inflammation were adjusted in the regression model (Supplemental Figure S1).
Replication analyses and further characterization in other PTB subtypes
The identified 45 differentially methylated sites in the discovery sample were then taken forward for replication in the maternal sample from 110 Black mother-newborn pairs. There were 109 samples from Black mothers (54 with TBs, 7 with early sPTBs, 34 with late sPTBs and 14 with early medically-indicated PTBs (mPTB)) that passed the quality control steps, and their population characteristics are presented in . We found that three out of the 45 sites (cg03915055, cg06804705 and cg10126715) were hypomethylated in 7 mothers with early sPTBs compared to 54 mothers with TBs at P < 0.05 (, and Supplemental Table S1). Both cg03915055 and cg06804705 were among the top three CpG sites associated with early sPTB in the discovery sample (>3% methylation difference, ) and showed similar association directions and effect sizes with early sPTB in the replication sample (P < 0.05); thus, we focused our subsequent analyses on these two sites.
Figure 2. The distribution of DNA methylation levels at the two validated sites in both the discovery and replication Black maternal samples from the Boston Birth Cohort, stratified by term vs. preterm status of the neonates. TB: term birth; sPTB: spontaneous PTB; mPTB: medically-indicated PTB. P values shown in the figure were calculated using the regression models to indicate a significant DNAm difference between mothers in each PTB subgroup and mothers with TBs.
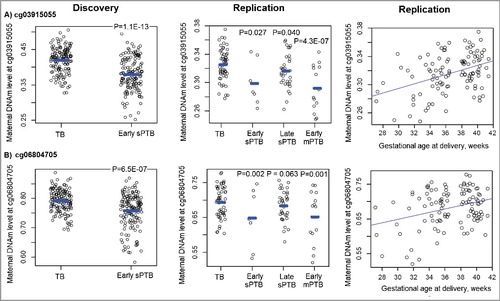
When other PTB subtypes were analyzed, we found that DNAm levels at both cg03915055 and cg06804705 were lower in the 34 mothers with late sPTBs than in the 54 mothers with TBs, although the effect sizes were smaller than those for early sPTB (). Interestingly, significant associations were also observed between these two sites and early mPTB ( and , all P ≤ 0.001), with the association directions and effect sizes similar to what we observed for early sPTB (). When gestational age (GA) was analyzed as the outcome, both sites showed dose-responsive and positive correlations with GA in these 109 maternal samples (, P = 3.8 × 10−6 for cg03915055 and P = 2.2 × 10−5 for cg06804705).
Cord blood DNAm levels at the two validated sites and their associations with PTB
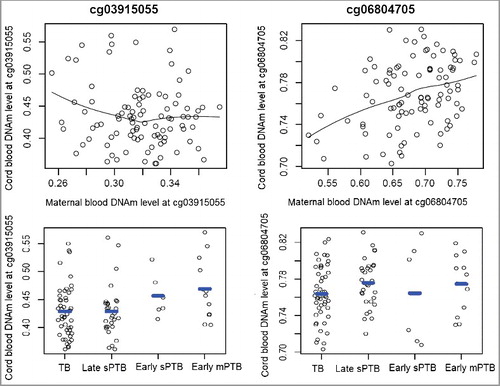
At the replication stage, DNAm levels in cord blood from the 110 mother-newborn pairs were also measured and 96 cord blood samples passed the quality control steps. Analyses of the 96 mother-newborn pairs showed that there was a moderate and positive mother-newborn DNAm correlation at cg06804705 (r = 0.32, P = 0.001), but no mother-newborn correlation was found at cg03915055 (r = −0.04, P = 0.67, ). We found no significant associations between cord blood DNAm levels at these two sites and PTB outcomes (all P > 0.05, ).
Figure 3. The maternal-cord blood DNA methylation correlation at cg03915055 and cg06804705, and cord blood DNA methylation difference at these two sites between term and PTB neonates. TB: term birth; sPTB: spontaneous PTB; mPTB: medically-induced PTB.
DNAm levels at the two replicated sites and maternal factors
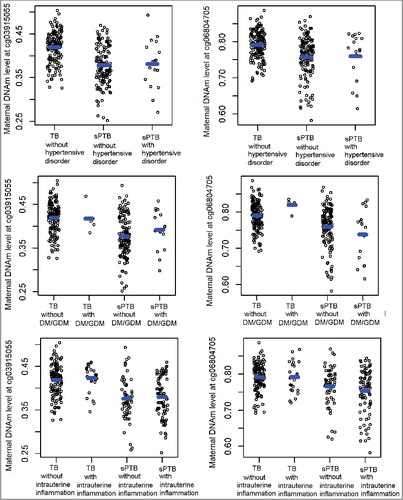
We further investigated the potential that the identified sPTB-associated DNAm changes at cg03915055 and cg06804705 were driven by gestational complications in the discovery sample. As shown in Supplemental Table S2, only intrauterine inflammation was significantly associated with methylation levels at both sites. However, when we stratified samples by sPTB status, the associations between intrauterine inflammation and DNAm disappeared, suggesting that intrauterine inflammation is not a confounder or mediator for the DNAm-sPTB associations (). Similarly, DNAm levels at both sites were comparable between sPTB mothers with and without DM/GDM, between TB mothers with and without DM/GDM, as well as between sPTB mothers with and with hypertensive disorders, all suggesting that the identified sPTB-DNAm associations were not influenced by these gestational complications (). Likewise, we found no associations for other maternal factors or estimated blood cell compositions with DNAm levels at the two sites (Supplemental Table S2).
Figure 4. Distributions of DNA methylation at cg03915055 and cg06804705 in mothers from the discovery sample, stratified by TB/PTB status combined with the existence of pregnancy complications. TB: term birth; sPTB: spontaneous PTB; DM/GDM: diabetes and gestational diabetes.
DNAm levels at the two replicated sites are not associated with local sequence variation
After combining the available genome-wide genotyping data in the discovery sample, there were 453 single nucleotide polymorphisms (SNPs) and 1032 SNPs with a minor allele frequency ≥ 0.01, respectively, located within 500 kb of cg03915055 and cg06804705. We found modest SNP associations for the two replicated methylation loci, with the most significant associations occurring between cg03915055 and rs113310199 (P = 0.003) and between cg06804705 and rs466939 (P = 0.002). Both associations, however, were no longer significant after correcting for multiple testing (FDR > 0.30). None of these SNPs were significantly associated with risk of early sPTB (data not shown).
Discussion
In the largest epigenome-wide study of PTB in high-risk US Black women to date, we identified and validated significant associations between maternal DNAm at two sites and early sPTB, which cannot be explained by maternal genetic variants, maternal cell type proportion, gestational complications or other maternal factors measured in this study. These two identified differentially methylated sites, located in the promoter region of the CYTIP and LINC00114 genes, respectively, were both hypomethylated in whole blood from mothers with early sPTBs compared to mothers with TBs, while no such associations were observed in cord blood, suggesting that the maternal and fetal epigenome may contribute to sPTB in different ways. Taken together, our findings strengthen our understanding of the role of maternal DNAm in sPTB risk, and provide a set of maternal loci that may act as biomarkers for sPTB.
The top differentially methylated site, cg03915055, is located in the promoter region of the CYTIP gene. A previous study showed that the DNAm level of this site was inversely correlated with CYTIP gene expression in breast tissue [Citation14]. CYTIP (also known as CYBR) transcription can be up-regulated by cytokines such as interleukin (IL)-2 and IL-12 in cultured lymphocytes [Citation15]. It may play a role in leukocyte trafficking (especially in response to proinflammatory cytokines under stressful conditions) and in T-cell receptor mediated signaling [Citation16,Citation17]. The study by O'Brien et al. demonstrated a 10-fold upregulation of CYTIP expression in the human myometrium during labor [Citation18]. Of interest, myometrial smooth muscle cells constitute the contractile machinery of the uterus and may offer a therapeutic target for the prevention of premature myometrial contractions [Citation19]. Although the exact function of CYTIP in the myometrium remains to be elucidated, the current available evidence suggests the possible involvement of CYTIP in the signal transduction mechanisms associated with labor.
Another sPTB-associated methylation site identified in this study (cg06804705) is annotated to LINC00114, a long intergenic non-protein coding RNA (lincRNA). LincRNA is involved in many biological processes, including transcriptional control, epigenetic modification and post-transcriptional control of mRNA [Citation20], and may play a role in cell differentiation, immune response and human diseases [Citation21,Citation22], Recent studies have demonstrated that long non-protein coding RNA was differentially expressed in human placentas derived from women with premature preterm rupture of membrane (PPROM), PTBs, premature rupture of the membranes (PROM) and TBs [Citation23]. In addition, differentially expressed long non-protein coding RNA, as identified from the human placenta samples of women with sPTBs and PPROM, may regulate their associated mRNA through differential mechanisms and connect the ubiquitin-proteasome system with infection-inflammation pathways [Citation24]. With this evidence, we suspect that the associations between DNAm alterations at cg06804705 and sPTB may occur partly via infection-inflammation pathways, which, however, need further validation.
Our data also demonstrated that DNAm levels at both loci (cg03915055 and cg06804705) have comparable associations with both early sPTB and early mPTB, but attenuated associations with late sPTB, suggesting that the two sites may represent the markers for the degree of prematurity. Should our findings be confirmed by future studies, they may stimulate future mechanistic studies to identify unique and common factors/pathways underlying various subtypes of PTB.
The fetal membranes are known to express one maternal allele and one paternal allele for each genetic variant. Previous genetic studies have demonstrated that the risk of recurrence of preterm delivery was transferred through mothers, not through fathers, and that fetal genotype is of relatively little importance in understanding genetic patterns of preterm delivery [Citation25]. DNAm is tissue-specific. Although it has been proposed that both the maternal and fetal epigenome contribute to PTB, such studies are very limited. In comparison, our findings clearly demonstrated that the associations of the promoter methylation of the CYTIP and LINC00114 genes with sPTB were observed in maternal blood but not in cord blood, suggesting that the impact of the maternal epigenome and fetal epigenome on the length of gestation may, at least in part, differ from one another.
Due to the cross-sectional nature of this study, we cannot directly assign a temporal or cause-effect relationship for the observed associations. Combining genetic and epigenetic data in typical Mendelian randomization analyses has been suggested to identify causal methylation changes due to genetic variation. However, we failed to identify any nearby SNP that was significantly associated with the methylation level at the two identified loci and/or with PTB, which may indicate that the two loci do not lie in the casual pathway downstream of the nearby SNPs. Although we did not find any maternal non-genetic factors that may lead to the identified sPTB-associated DNAm changes in this study, we still cannot exclude the possibility that such DNAm changes are driven by other unmeasured maternal factors (such as nutrition, air pollutants or metal exposures) and/or gene-environment interactions. Further studies are needed to explore these possibilities.
Although the underlying reasons for the observed methylation associations could not be determined in this study, we believe that our findings are still encouraging in regards to the epigenetic epidemiology of PTB; in this light, we offer two alternative implications: First, our results may reflect involvement of DNAm in the etiology of PTB. There are a number of mechanisms associated with PTB, which could be associated with epigenetic changes themselves. This explanation is also consistent with the potential functions of the two annotated genes, as we demonstrated above. Second, it is also possible that the identified sPTB-associated loci undergo DNAm changes in late pregnancy, which are functionally related to the adverse health consequences of mothers who experience PTB. If so, our findings may lead to new epidemiologic research towards understanding the mechanisms responsible for negative health outcomes in mothers with PTBs and to lessening such negative consequences. Our findings also suggest intriguing future studies to explore whether these identified sPTB-associated DNAm changes are detectable as early as during pregnancy, and whether they can be used as early predictive biomarkers for sPTB, for which neither causality nor functional knowledge is necessary.
There were several limitations to this study. First, as mentioned above, because this was a cross-sectional study we could not assign a cause-effect relationship to the observed associations, and we cannot exclude the possibility of reverse causation. Second, DNAm was measured in maternal peripheral blood leukocytes, which may or may not reflect the methylation of the maternal tissues (i.e., the myometrium) responsible for the conditions leading to PTB. Third, DNAm levels of the identified sites were not validated using other techniques such as pyrosequencing and no functional study was conducted. However, previous studies have demonstrated that the Infinium HumanMethylation450 BeadChip array used in this study correlates well with direct pyrosequencing [Citation26,Citation27]. Fourth, questions remain regarding how to interpret small DNAm differences, mostly those <5%. It is possible that the small changes that we identified in whole blood may be reflective of larger changes in a specific cell type [Citation28]. Previous studies have also shown that the small changes in methylation can have a strong functional effect on transcriptional activity [Citation29]. We confirmed the magnitude and direction of effects for several sPTB-related DNAm changes in our small underpowered replication sample but future studies in large cohorts are needed to replicate these findings with strong statistical support. Finally, DNAm is one of several epigenetic mechanisms that work in conjunction with one another to regulate gene expression, and, as such, further work should be done to understand how histone modifications or other mechanisms may affect the risk of PTB.
In summary, we demonstrated novel associations of DNAm levels in the promoter regions of the CYTIP and LINC00114 genes with sPTB in maternal whole blood but not in cord blood. Future studies using maternal samples collected prepregnancy and/or during early pregnancy are needed to distinguish maternal methylation changes that lie in the causal pathway from those that are the consequence of PTB. Such findings will greatly contribute to our understanding of PTB etiology and to the identification of early predictive markers and new drug targets for the prevention and treatment of PTB.
Methods
The Boston Birth Cohort (BBC) and study population
Both the discovery and the replication sample of this study were enrolled from the BBC, which was initiated in 1998 with a rolling enrollment in Boston, MA, as detailed elsewhere [Citation30]. Briefly, the BBC enrolled mothers who delivered singleton live preterm or low birthweight (<2500 g) infants, and mothers who delivered term and normal birth-weight infants. Pregnancies that were a result of in vitro fertilization, multiple gestations (e.g., twins, triplets), fetal chromosomal abnormalities or major birth defects were excluded. Each enrolled participant, after giving written informed consent, completed a questionnaire interview to assess maternal characteristics, lifestyle and diet information. A maternal blood sample was obtained within 24–72 hours after delivery and a cord blood sample was obtained at delivery, and both were stored in −80°C freezers. The study protocol was approved by the Institutional Review Boards (IRB) of Boston University Medical Center, the Ann & Robert H. Lurie Children's Hospital of Chicago, and the Johns Hopkins Bloomberg School of Public Health.
The discovery sample included 150 Black women who delivered early sPTB neonates (23–336/7 weeks of gestation, Cases) and 150 Black women who delivered TB neonates (39–42 weeks of gestation, Controls) from the BBC. Cases and controls were frequency matched on maternal age (+/− 5 years), parity, year of delivery and sex of the neonates. In the replication stage, 110 Black maternal-newborn pairs were enrolled, including 7 pairs with early sPTBs (<34 weeks of gestation), 34 pairs with late sPTBs (34–36.5 weeks of gestation), 14 pairs with early medically indicated PTBs (mPTB, <34 weeks of gestation) and 55 pairs with TBs (>37 weeks of gestation), all independent of the discovery sample. Both maternal whole blood and matched cord blood from these 110 maternal-newborn pairs were collected for DNAm measurements.
Definition of phenotypes of interest and covariates
We defined sPTB as a birth occurring secondary to documented active preterm labor (uterine contractions with cervical effacement and dilation at <37 weeks), or premature rupture of membranes at <37 weeks without uterine contractions, or both. Early sPTB was defined as sPTB at <34 weeks of gestation and late sPTB was defined as sPTB at 34–366/7 weeks of gestation. Early mPTB was defined as a birth delivered by medical induction or Cesarean section at <34 weeks without uterine contractions or rupture of membranes.
Maternal variables, such as smoking during pregnancy, maternal psychosocial stress during pregnancy and maternal illicit drug use, were collected based on a standard maternal questionnaire interview, as we reported previously [Citation31]. Maternal complications during pregnancy, including GDM and hypertensive disorders, were collected via maternal electronic medical record abstraction. Intrauterine inflammation of maternal origin was defined based on clinical signs of chorioamnionitis (intrapartum fever >38°C) and/or histologic chorioamnionitis [Citation32].
Genome-wide genotyping data from the discovery sample
All of the discovery samples were genome-wide genotyped at the Center for Inherited Disease Research (CIDR), using the Illumina HumanOmni2.5-4v1 array. Genotyping data cleaning was performed according to the protocol described by Laurie et al. [Citation33]. Principal component analyses (PCA) were conducted using Eigenstrat [Citation34], with all European, American, African, and Asian individuals in the 1000 Genomes Project as the reference, and the first three principle components were applied to capture genetic ancestry for each sample in the study.
Methylation measurements and quality control steps in the discovery sample
Genomic DNA was isolated from EDTA-treated peripheral white blood cells. Case and control DNA samples, all diluted to 50–100 ng/uL and evenly distributed across each plate to minimize batch effects, were shipped to the Center for Genetic Medicine, Northwestern University Feinberg School of Medicine for methylation profiling. Briefly, 0.5 μg of genomic DNA was bisulfite-converted using the EZ-96 DNA Methylation™ Gold Kit, and DNAm levels at 485,512 loci were measured using the Infinium HumanMethylation450 BeadChip, according to the manufacturer's instructions. For each sample, a raw intensity file (.idat) was processed and several quality control steps were performed using the R/Bioconductor package ‘minfi’ [Citation35], as we reported previously [Citation36]. We removed 3 samples with DNA quality issues; 3 of non-African ancestry based on PCA using genome-wide genotyping data; 1 that was biologically related to another enrolled woman; and 3 that were identified as outliers with a median log2 intensity value <10.5 in DNAm measurement. At the locus level, we removed 574 loci that had a detection P-value (a measure of probe performance) >0.01 in more than 10% of the samples; 51,665 loci that had an annotated SNP (minor allele frequency >0.01) at the measured and neighboring locus (+/− 1 bp depending on probe strand orientation) and/or were previously reported to be cross-reactive [Citation37]; and 416 Y-chromosome loci. These filters resulted in high quality DNAm data for 432,857 sites (including 10,107 X-chromosome sites) among 290 samples for subsequent analyses.
Using the ‘minfi’ package [Citation35], a stratified quantile normalization procedure was applied to the raw data, and normalized beta values (β), ranging from 0–1 for 0% to 100% methylated, were obtained for the 290 maternal samples. To account for potential batch effects, beta values and the corresponding M values (logit-transformed beta values) were ComBat-transformed [Citation38] using the ‘sva’ package [Citation39] with the array number as the surrogate for the batches. ComBat-transformed M values, reported as being superior to beta values for identification of differential methylation [Citation40], were utilized for downstream statistical analyses. ComBat-transformed beta values were used for plotting purposes since they are more intuitive.
Methylation measurements in the replication sample
The same DNAm measurement and quality control steps that were used in the discovery sample, as mentioned above, were applied to the replication sample. To reduce bias, each mother-newborn pair sample was distributed in the plate so that they were all measured on the same array. After quality control steps, one maternal and 14 cord blood samples were removed from further analyses.
Cell proportion estimation
Using the estimateCellCounts function in the ‘minfi’ package [Citation35], the distribution of each cell type, including CD4+, CD8+ T cells, B cells, monocytes, granulocytes, natural killer cells and nucleated red blood cells (specific to cord blood), was inferred for each maternal and cord blood sample, respectively, based on external adult and cord blood reference DNAm signatures of the constituent cell type from Illumina HumanMethylation450 BeadChips [Citation13,Citation41,Citation42].
Identification of differentially methylated sites and replication
To identify differentially methylated sites associated with early sPTB in the discovery sample, we used the ‘limma’ package [Citation43] to fit a linear regression model with the ComBat-transformed M value at each site as the outcome and sPTB status (1 = early sPTB cases, 0 = TB controls) as the independent variable, adjusting for potential confounders including maternal age, maternal smoking, genetic ancestry and cell composition. The false discovery rate (FDR) was estimated to correct for multiple testing.
The identified methylation sites were then validated for their associations with early sPTB in the maternal replication sample (comprised of 7 mothers with early sPTBs and 54 mothers with TBs) using similar linear regression models as were applied in the discovery sample. For the replicated methylation sites, we then tested whether they were differentially methylated in mothers with late sPTB (n = 34) and in mothers with early mPTBs (n = 14) compared to mothers with TBs (n = 54), using linear regression models. These sites were further assessed for mother-newborn correlation on DNAm levels by performing Spearman correlation analysis in 96 mother-newborn pairs. We also tested whether these validated sites were differentially methylated in cord blood between PTB cases and TB controls using linear regression models.
Identification of SNPs and/or environmental factors that affect DNAm levels at replicated methylation sites
We searched further for associations between methylation levels at the validated methylation sites and the common genetic variants (with a minor allele frequency >2%) within 500 kb of these probes, under an additive effects model adjusting for cell composition, maternal smoking status and genetic ancestry. This analysis was done in the discovery sample due to a lack of genotyping data in the replication samples. We further performed several additional analyses in the discovery sample to explore whether the methylation levels at the validated methylation sites were influenced by measured covariates and/or gestational complications using linear regression models. All of these analyses were performed using R version 3.3.1.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
KEPI_A_1287654_s02.docx
Download MS Word (90 KB)Acknowledgements
We thank all of the study participants for their support and help with the study. We are also grateful for the dedication and hard work of the field team at the Department of Pediatrics, Boston University School of Medicine, and for the help and support of the obstetric nursing staff at Boston Medical Center.
Additional information
Funding
Related Research Data
References
- Callaghan WM, MacDorman MF, Rasmussen SA, et al. The contribution of preterm birth to infant mortality rates in the United States. Pediatrics. 2006;118:1566–1573. doi:10.1542/peds.2006-0860. PMID:17015548
- Lopez-Serra P, Esteller M. DNA methylation-associated silencing of tumor-suppressor microRNAs in cancer. Oncogene. 2012;31:1609–1622. doi:10.1038/onc.2011.354. PMID:21860412
- Laurent L, Wong E, Li G, et al. Dynamic changes in the human methylome during differentiation. Genome Res. 2010;20:320–331. doi:10.1101/gr.101907.109. PMID:20133333
- Maunakea AK, Nagarajan RP, Bilenky M, et al. Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature. 2010;466:253–257. doi:10.1038/nature09165. PMID:20613842
- Heyn H, Esteller M. DNA methylation profiling in the clinic: applications and challenges. Nat Rev Gene. 2012;13:679–692. doi:10.1038/nrg3270. PMID:22945394
- Parets SE, Conneely KN, Kilaru V, et al. Fetal DNA methylation associates with early spontaneous preterm birth and gestational age. PloS One. 2013;8:e67489. doi:10.1371/journal.pone.0067489. PMID:23826308
- Parets SE, Conneely KN, Kilaru V, et al. DNA methylation provides insight into intergenerational risk for preterm birth in African Americans. Epigenetics. 2015;10:784–792. doi:10.1080/15592294.2015.1062964. PMID:26090903
- Fernando F, Keijser R, Henneman P, et al. The idiopathic preterm delivery methylation profile in umbilical cord blood DNA. BMC Genomics. 2015;16:736. doi:10.1186/s12864-015-1915-4. PMID:26419829
- Lee H, Jaffe AE, Feinberg JI, et al. DNA methylation shows genome-wide association of NFIX, RAPGEF2 and MSRB3 with gestational age at birth. Int J Epidemiol. 2012;41:188–199. doi:10.1093/ije/dyr237. PMID:22422452
- Schroeder JW, Conneely KN, Cubells JC, et al. Neonatal DNA methylation patterns associate with gestational age. Epigenetics. 2011;6:1498–1504. doi:10.4161/epi.6.12.18296. PMID:22139580
- Cruickshank MN, Oshlack A, Theda C, et al. Analysis of epigenetic changes in survivors of preterm birth reveals the effect of gestational age and evidence for a long term legacy. Genome Med. 2013;5:96. doi:10.1186/gm500. PMID:24134860
- Burris HH, Rifas-Shiman SL, Baccarelli A, et al. Associations of LINE-1 DNA Methylation with preterm birth in a prospective cohort study. J Dev Orig Health Dis. 2012;3:173–181. doi:10.1017/S2040174412000104. PMID:22720130
- Bakulski KM, Feinberg JI, Andrews SV, et al. DNA methylation of cord blood cell types: Applications for mixed cell birth studies. Epigenetics. 2016;11:354–362. doi:10.1080/15592294.2016.1161875. PMID:27019159
- Fleischer T, Frigessi A, Johnson KC, et al. Genome-wide DNA methylation profiles in progression to in situ and invasive carcinoma of the breast with impact on gene transcription and prognosis. Genome Biol. 2014;15:435. doi:10.1186/s13059-014-0435-x, PMID:25146004
- Tang P, Cheng TP, Agnello D, et al. Cybr, a cytokine-inducible protein that binds cytohesin-1 and regulates its activity. Proc Nat Acad Sci USA. 2002;99:2625–2629. doi:10.1073/pnas.052712999. PMID:11867758
- Chen Q, Coffey A, Bourgoin SG, et al. Cytohesin binder and regulator augments T cell receptor-induced nuclear factor of activated T Cells.AP-1 activation through regulation of the JNK pathway. J Biol Chem. 2006;281:19985–19994. doi:10.1074/jbc.M601629200. PMID:16702224
- Coppola V, Barrick CA, Bobisse S, et al. The scaffold protein Cybr is required for cytokine-modulated trafficking of leukocytes in vivo. Mol Cell Biol. 2006;26:5249–5258. doi:10.1128/MCB.02473-05. PMID:16809763
- O'Brien M, Morrison JJ, Smith TJ. Upregulation of PSCDBP, TLR2, TWIST1, FLJ35382, EDNRB, and RGS12 gene expression in human myometrium at labor. Reproductive Sci. 2008;15:382–393. doi:10.1177/1933719108316179. PMID:18497345
- Mosher AA, Rainey KJ, Bolstad SS, et al. Development and validation of primary human myometrial cell culture models to study pregnancy and labour. BMC Pregnancy Childbirth. 2013;13(Suppl 1):S7. doi:10.1186/1471-2393-13-S1-S7, PMID:23445904
- Esteller M. Non-coding RNAs in human disease. Nat Rev Genet. 2011;12:861–874. doi:10.1038/nrg3074. PMID:22094949
- Wilusz JE, Sunwoo H, Spector DL. Long noncoding RNAs: functional surprises from the RNA world. Genes Dev. 2009;23:1494–1504. doi:10.1101/gad.1800909. PMID:19571179
- Okazaki Y, Furuno M, Kasukawa T, et al. Analysis of the mouse transcriptome based on functional annotation of 60,770 full-length cDNAs. Nature. 2002;420:563–573. doi:10.1038/nature01266. PMID:12466851
- Luo X, Shi Q, Gu Y, et al. LncRNA pathway involved in premature preterm rupture of membrane (PPROM): an epigenomic approach to study the pathogenesis of reproductive disorders. PloS One. 2013;8:e79897. doi:10.1371/journal.pone.0079897. PMID:24312190
- Luo X, Pan J, Wang L, et al. Epigenetic regulation of lncRNA connects ubiquitin-proteasome system with infection-inflammation in preterm births and preterm premature rupture of membranes. BMC Pregnancy Childbirth. 2015;15:35. doi:10.1186/s12884-015-0460-0. PMID:25884766
- Boyd HA, Poulsen G, Wohlfahrt J, et al. Maternal contributions to preterm delivery. Am J Epidemiol. 2009;170:1358–1364. doi:10.1093/aje/kwp324. PMID:19854807
- Roessler J, Ammerpohl O, Gutwein J, et al. Quantitative cross-validation and content analysis of the 450k DNA methylation array from Illumina, Inc. BMC Res Notes. 2012;5:210. doi:10.1186/1756-0500-5-210. PMID:22546179
- Nestor CE, Barrenas F, Wang H, et al. DNA methylation changes separate allergic patients from healthy controls and may reflect altered CD4+ T-cell population structure. PLoS Genet. 2014;10:e1004059. doi:10.1371/journal.pgen.1004059. PMID:24391521
- Liang L, Willis-Owen SA, Laprise C, et al. An epigenome-wide association study of total serum immunoglobulin E concentration. Nature. 2015;520:670–674. doi:10.1038/nature14125. PMID:25707804
- Murphy SK, Adigun A, Huang Z, et al. Gender-specific methylation differences in relation to prenatal exposure to cigarette smoke. Gene. 2012;494:36–43. doi:10.1016/j.gene.2011.11.062. PMID:22202639
- Wang X, Zuckerman B, Pearson C, et al. Maternal cigarette smoking, metabolic gene polymorphism, and infant birth weight. Jama. 2002;287:195–202. doi:10.1001/jama.287.2.195. PMID:11779261
- Wang G, Divall S, Radovick S, et al. Preterm birth and random plasma insulin levels at birth and in early childhood. JAMA. 2014;311:587–596. doi:10.1001/jama.2014.1. PMID:24519298
- Gupta M, Mestan KK, Martin CR, et al. Impact of clinical and histologic correlates of maternal and fetal inflammatory response on gestational age in preterm births. J Matern Fetal Neonatal Med. 2007;20:39–46. doi:10.1080/14767050601156861. PMID:17437198
- Laurie CC, Doheny KF, Mirel DB, et al. Quality control and quality assurance in genotypic data for genome-wide association studies. Genet Epidemiol. 2010;34:591–602. doi:10.1002/gepi.20516. PMID:20718045
- Price AL, Patterson NJ, Plenge RM, et al. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006;38:904–909. doi:10.1038/ng1847. PMID:16862161
- Aryee MJ, Jaffe AE, Corrada-Bravo H, et al. Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics. 2014;30:1363–1369. doi:10.1093/bioinformatics/btu049. PMID:24478339
- Hong X, Hao K, Ladd-Acosta C, et al. Genome-wide association study identifies peanut allergy-specific loci and evidence of epigenetic mediation in US children. Nat Comm. 2015;6:6304. doi:10.1038/ncomms7304. PMID:25710614
- Chen YA, Lemire M, Choufani S, et al. Discovery of cross-reactive probes and polymorphic CpGs in the Illumina Infinium HumanMethylation450 microarray. Epigenetics. 2013;8:203–209. doi:10.4161/epi.23470. PMID:23314698
- Johnson WE, Li C, Rabinovic A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics. 2007;8:118–127. doi:10.1093/biostatistics/kxj037. PMID:16632515
- Leek JT, Johnson WE, Parker HS, et al. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics. 2012;28:882–883. doi:10.1093/bioinformatics/bts034. PMID:22257669
- Du P, Zhang X, Huang CC, et al. Comparison of Beta-value and M-value methods for quantifying methylation levels by microarray analysis. BMC Bioinformatics. 2010;11:587. doi:10.1186/1471-2105-11-587. PMID:21118553
- Jaffe AE, Irizarry RA. Accounting for cellular heterogeneity is critical in epigenome-wide association studies. Genome Biol. 2014;15:R31. doi:10.1186/gb-2014-15-2-r31. PMID:24495553
- Houseman EA, Accomando WP, Koestler DC, et al. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinformatics. 2012;13:86. doi:10.1186/1471-2105-13-86. PMID:22568884
- Smyth GK. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol. 2004;3:1–25, ISSN (Online) 1544–6115. doi:10.2202/1544-6115.1027. PMID:16646809