
ABSTRACT
DNA methylation aberrancies are hallmarks of human cancers and are characterized by global DNA hypomethylation of repetitive elements and non-CpG rich regions concomitant with locus-specific DNA hypermethylation. DNA methylation changes may result in altered gene expression profiles, most notably the silencing of tumor suppressors, microRNAs, endogenous retorviruses and tumor antigens due to promoter DNA hypermethylation, as well as oncogene upregulation due to gene-body DNA hypermethylation. Here, we review DNA methylation aberrancies in human cancers, their use in cancer surveillance and the interplay between DNA methylation and histone modifications in gene regulation. We also summarize DNA methylation inhibitors and their therapeutic effects in cancer treatment. In this context, we describe the integration of DNA methylation inhibitors with conventional chemotherapies, DNA repair inhibitors and immune-based therapies, to bring the epigenome closer to its normal state and increase sensitivity to other therapeutic agents to improve patient outcome and survival.
Challenges to cancer therapy
According to the US National Cancer Institute (www.cancer.gov), over 100 forms of human cancer have been identified, spanning all tissues in the human body. Multiple tumor subtypes can develop from each tissue; however, the mechanisms of tumorigenesis are not identical between tumor types. This is compounded by the fact that even though human cancer is a result of genetic, epigenetic, and transcriptomic alterations, there is no uniform cancer genomic profile for each primary tumor tissue type or even between tumors within individual subgroups. This has necessitated the use of precision medicine and personalized approaches for targeting the specific genomic signatures for treatment of individual patients or patient subgroups.
The Cancer Genome Atlas (TCGA) took the early steps in addressing this conundrum by characterizing genetic, epigenetic, transcriptomic, and protein alterations in over 11,000 primary tumors spanning nearly 30 tumor types with the ultimate goal of identifying novel therapeutic targets (cancergenome.nih.gov). TCGA and other efforts (summarized in Citationref.1) have advanced our understanding of the genomic driver mutations, copy number alterations (CNAs), gene expression changes, and the contributions of DNA methylation aberrancies toward the cancer genome. TCGA has verified the majority of known driver somatic mutations in nearly 20 major forms of human cancers, and has characterized additional alterations and low frequency mutations thanks to advancements in next-generation sequencing and microarray technologies. However, these findings have only recently been described, and as a result, have not yet resulted in transforming cancer patient treatment.
This is especially relevant due to the generally low frequencies of genetic alterations in most types of cancers.Citation2,3 On the contrary, epigenetic alterations, most notably DNA methylation changes, are substantially more prevalent than genetic alterations in virtually every cancer type, further highlighting their importance as diagnostic tools and therapeutic targets.Citation4,5 Our understanding of cancer epigenetics, namely DNA methylation and chromatin modification changes, has been tremendously improved due to efforts from multi-institutional, international consortia including TCGA, the Encyclopedia of DNA Elements (ENCODE, www.encodeproject.org), the Roadmap Epigenetics Project (www.roadmapepigenomics.org), Therapeutically Applicable Research to Generate Effective Treatments (TARGET, https://ocg.cancer.gov/programs/target) for pediatric cancers, and others (reviewed in Citationref.1), as well as from technological advancements in sample throughput and the ability to profile increased genomic content. With these data, DNA methylation, histone modifications, and chromatin structure have become better understood, as are the mechanistic details of DNA methylation inhibition. This review focuses on DNA methylation changes in human cancers, their use as biomarkers of surveillance, attributes of DNA methylation inhibition, and therapeutic strategies using DNA methylation inhibitors with other treatment regimens.
DNA methylation
DNA methylation in mammalian organisms is largely restricted to the addition of a methyl group to the C-5 position of cytosine in a 5′-CG-3′ (CpG) sequence context, with non-CpG DNA methylation generally found in embryonic stem (ES) cell populations (reviewed in Citationref.6). Methylated CpGs are more prone to spontaneous deamination to thymine than the rate at which unmethylated CpGs deaminate to uracil; therefore, the CpG content in the human genome is only 20% of what is expected by sequence context alone (reviewed in Citationref.6). Indeed, the human genome is CpG depleted, and approximately 70% of all CpGs are methylated,Citation6,7 with exact levels dependent on the tissue type, mostly in transposable elements (LINE, SINE, and ERV) and intergenic regions of the human genome. However, there are regions of the genome, called CpG islands, that contain their expected CpG content, are unmethylated in normal somatic tissues, and are enriched (> 50%) in gene promoter regions.Citation6
Promoter CpG islands are generally not enriched in TATA boxes and other core promoter sequence motifs for focused transcription initiation, but rather show transcriptional permissivity in which transcription factor and RNA polymerase II binding to promoter CpG islands can result in switching between active and inactive transcriptional states (reviewed in Citationref.8). CpG islands do not have consensus sequences or fixed sizes, but are sources of transcription factor (TF) binding.Citation8 Several transcription factors, including Sp1, Nrf-1, E2F, and ETS, contain CpG dinucleotides in their recognition and binding sites.
Cancer-specific DNA methylation alterations
DNA methylation changes are hallmarks of every cancer type, and are highlighted by global DNA hypomethylation of repetitive elements and CpG-poor regions concomitant with gene-specific DNA hypermethylation.Citation9-11 DNA methylation alterations may result in gene expression changes, namely gene silencing due to CpG island (or CpG rich) promoter DNA hypermethylation and gene activation due to DNA hypomethylation of CpG-poor gene promoters. DNA hypomethylated genes can remain silenced by the presence of H3K27me3 repressive mark or the lack of required transcription factors. A recent study also demonstrated that DNA hypermethylation of gene bodies or transcribed regions is associated with oncogene overexpression, Citation12 showing that these gene expression consequences help propel tumorigenesis. In addition, the frequent occurrence of cancer-linked DNA hypermethylation and DNA hypomethylation is associated with tumor progression and tumor formation. Because of these attributes, DNA methylation alterations are thought to be early events in human tumorigenesis.
De Carvalho et al. performed genome-scale DNA methylation analyses of HCT116 colon cancer cells deficient for one or more DNA methyltransferases (DNMTs) to identify epigenetic driver genes that require DNA methylation for cancer cell survival. Citation13 These include, A disintegrin and metallopeptidase domain 2 (ADAM2), BCHE, CDO1, ESX1, interleukin-1 receptor-associated kinase 3 (IRAK3), P2RY14, and synaptonemal complex protein 3 (SYCP3), all of which are epigenetically silenced in human cancers and their DNA demethylation results in cell death. For example, IRAK3 negatively regulates the anti-apoptotic gene SURVIVIN and inhibits MAPK, NF-kB, and STAT3 signaling pathways, which are activated in numerous cancer types. In addition, DNA demethylation and re-expression of ADAM2 and SYCP3 can trigger apoptotic cell death.
In light of the epigenetic-based expression changes in human cancers, however, not all DNA methylation changes drive gene expression consequences. In fact, the majority of DNA methylation aberrancies are simply passenger events.Citation14-16 However, these markings do serve as biomarkers of disease, and have been used in translational studies with diagnostic and prognostic utility to correlate with patient survival, response to chemotherapy, and other clinical co-variates (reviewed in Citationref.17). In addition, cancer-specific methylated DNA biomarkers can be identified in circulating tumor cells (CTCs) and tumor-derived, cell-free DNA (cfDNA) in blood, urine, or other bodily fluids, as sensitive (early) detection protocols (reviewed in Citationrefs. 17,18).
CpG island methylator phenotypes in human cancers
DNA methylation alterations substantially outnumber somatic mutations in human cancers,Citation19 and individual tumor types can be stratified into subgroups based on DNA methylation profiles. In 1999, Toyota et al. first identified a unique subset of colorectal tumors positive for a CpG Island Methylator Phenotype (CIMP) that display extensive DNA hypermethylation at a unique set of CpG islands that remained unmethylated in other colorectal tumors and normal tissues.Citation20 Follow up experiments showed that CIMP tumors are preferentially located in the proximal (right) colon, are enriched in women of older age, and harbor the BRAF V600E (BRAFV600E) point mutation as well as MLH1 epigenetic silencing due to promoter DNA hypermethylation and microsatellite instability (MSI).Citation21 Colorectal CIMP is associated with poor patient outcome; however, this is not completely understood since outcome is also affected by BRAF and MSI status.Citation22-27 A comprehensive, pooled analysis of 33 published reports describing the correlation between CIMP status and patient outcome in 10,635 patients Citation28 showed that CIMP status is associated with shorter disease free survival (DFS) and overall survival (OS), irrespective of MSI status. In addition, colon cancer patients with microsatellite stable (MSS) disease were significantly associated with CIMP and reduced OS. CIMP status may confer a predictive DFS advantage for patients receiving the conventional 5-fluorouracil (5-FU) based adjuvant therapies.Citation28 These reports suggest that CIMP-specific DNA methylation may be used for predictive and prognostic biomarkers; however, the efficacy of DNA methylation inhibitors in improving CIMP patient survival and outcome remains to be determined.
CIMPs have been characterized for cancers derived from brain,Citation29 breast,Citation30,31 endometrium,Citation32,33 and stomach.Citation34-38 However, tumor types that do not contain CIMP-based subgroups also display DNA methylation alterations that correlate with tumor invasiveness and clinical outcome. For example, high frequencies of DNA methylation-derived gene expression alterations are present in renal cell carcinomas (RCCs).Citation16 Analysis of TCGA clear cell RCC (ccRCC) data indicated that DNA methylation alterations positively correlate with tumor aggressiveness,Citation39 and DNA methylation markers associated with ccRCC aggressiveness were characterized in a recent report.Citation40
DNA methyltransferases
Cytosine DNA methylation marks are placed by the enzymatic activities of DNMTs using S-adenosylmethionine (SAM) as a co-factor. As first modeled by Santi et al., DNMTs catalyze methyl-transfer by first forming a covalent intermediate of the DNMT active site amino acid cysteine residue bound to the C-6 position of the targeted cytosine base.Citation41 In this first step, the C-5/C-6 double bond becomes a single bond as the enzyme-DNA covalent intermediate is formed, providing the impetus for forming a bond between the C-5 position and the methyl group, thereby releasing SAM. The C-5/C-6 double bond reforms and the enzyme then releases for catalyzing additional methyl-transfer events.
DNMT1 is thought to be responsible for maintenance DNA methylation to copy DNA methylation patterns from parental DNA onto daughter DNA strands soon after cellular DNA replication (reviewed in Citationref.42). DNMT3A and DNMT3B are referred to as de novo DNMTs in that they create new DNA methylation marks at CpG sites that were originally unmethylated and play important roles during embryonic development and tumorigenesis.Citation42 DNMT2 is a tRNA-methyltransferase, while DNMT3L is a structural protein that is essential in coordinating DNMT3A and DNMT3B-associated DNA methylation during embryonic development.Citation42 DNMT3C was recently discovered by Barau et al.Citation43 and is essential for fertility via de novo DNA methylation of evolutionarily young retrotransposon promoters of the male germline. Experiments involving HCT116 colon cancer cells harboring hypomorphic knockdown of DNMT1 (DNMT1Δ2−5) and/or knockout of DNMT3B (DNMT3B−/−) showed that downregulating DNMT1 or DNMT3B alone did not substantially alter global DNA methylation levels; however DNMT1/DNMT3B double knockout (DKO, DNMT1−/− DNMT3B−/−) cells showed nearly complete (95%) DNA demethylation, suggesting that DNMT1 and DNMT3B work in concert to maintain DNA methylation marks.Citation44-46
Two DNMT3A isoforms have been identified,Citation47 described as DNMT3A and DNMT3A2. DNMT3A is expressed at low levels across all cell types and developmental stages and is localized to heterochromatin; however, DNMT3A2 is mainly expressed in ES cells and embryonic carcinomas and is thought to be the main DNMT3A isoform responsible for de novo DNA methylation in ES cells.Citation47 Moreover, DNMT3A and DNMT3A2 each connect to nucleosomes via interaction with DNMT3L.Citation48,49 With respect to human cancers, DNMT3A somatic mutations have been identified in acute myeloid leukemia (AML) and other blood-based malignancies.Citation50,51 TCGA identified DNMT3A mutations in over 50% of AML tumors, and showed correlation with NPM1 mutations. Interestingly, AML tumors with mutations in DNMT3A, NPM1, and FLT3 showed substantial DNA hypomethylation compared with apparently normal CD34+/CD34- white blood cells.Citation51
Over 30 DNMT3B splice-variant isoforms have been described, some possessing catalytic activity (DNMT3B1, DNMT3B2, DNMT3B6), while others are not catalytically active (DNMT3B3, DNMT3B4, DNMT3B5, DNMT3B7).Citation52-58 DNMT3B is important for gene-body DNA methylation in a manner irrespective of the isoforms' catalytic activity.Citation58 The catalytically inactive DNMT3B isoforms, most notably DNMT3B3, are expressed in human cancer cells and may serve as a beacon to recruit DNMT3A when DNMT3L is absent. DNMT3B may act as an accessory protein, much like DNMT3L, to recruit DNMT3A in establishing de novo DNA methylation patterns, especially in somatic cells that do not express DNMT3L.Citation58,59 DNMT3B is mainly involved in maintaining or restoring DNA methylation of CpGs located in gene bodies or transcribed regions by recognition of H3K36me3 marks.Citation12,58
DNA demethylation
Since the discovery and characterization of DNMTs, efforts to identify DNA demethylases, enzymes that remove DNA methylation marks, have been difficult and met with controversy. A mammalian DNA demethylase was reported in 1999 that was later identified as MBD2, Citation60,61 a member of the DNA methylation binding (MBD) protein family; Citation62,63 however, the DNA demethylase activity of MBD2 was not validated in other laboratories.Citation63 Several years later, enzymes belonging to the Ten Eleven Translocase (TET) family (TET1, TET2, TET3) were identified and were shown to function by converting 5-methylcytosine (5mC) to 5-hydroxymethylcytosine (5hmC) using ascorbic acid (vitamin C) as an enhancing cofactor.Citation64,65 TET1 was first described as leukemia-associated protein with a CXXC domain (LCX) by Ono et al., who sought to characterize novel fusion genes located on chromosome 10q22 that partner with MLL in AML patients with chromosome 10 and 11 translocations.Citation66 TET enzymes have homology to trypanosome base J binding proteins (JBP1 and JBP2) and contain 2-oxoglutatrate and iron-dependent dioxygenase activities similar those found in histone lysine demethylases.Citation64,67 The CXXC domain is also found in MBD proteins, implicating TET enzymes as possessing methylated DNA binding capabilities.
Subsequent studies showed TET enzymes further convert 5hmC to 5-formylcytosine (5fC) and 5-carboxylcytosine (5caC),Citation68,69 with both 5fC and 5caC recognized by the DNA glycosylase-mediated base excision repair pathway that ultimately results in the substitution of 5mC with an unmethylated cytosine residue. Unlike DNA methylation catalysis in which methyl marks are directly placed onto unmethylated cytosines, DNA demethylation can occur: 1) passively, by the inability of the DNA methyltransferase machinery to place methyl marks on nascent DNA strands; or 2) actively, by a multi-step process of TET-mediated oxidative conversion and removal by DNA repair mechanisms. The 5hmC marks are found at reduced levels (∼1%) compared with 5mC levels (4–5%) in the human genome; however, much like 5mC alterations, 5hmC alterations are present across several forms of human cancers.Citation70 Levels of 5mC and 5hmC and TET enzyme activities are paramount in understanding the functions and mechanisms of the cancer methylome. Interestingly, vitamin C plasma levels are generally low in cancer patients,Citation71 and the strategy of restoring vitamin C levels to physiologic levels may improve sensitivity and efficacy of TET-based DNA demethylation as an effective anti-cancer therapy.
IDH1 mutations drive CIMP in human gliomas
As mentioned above, CIMP subgroups exist in multiple human cancer types, in which subsets of tumors show cancer-specific DNA methylation at unique sets of CpG islands. A glioma-specific CIMP (G-CIMP) was reported by Noushmehr et al. in 2010, in which approximately 15% of the glioblastomas (grade IV gliomas) analyzed showed extensive DNA hypermethylation. G-CIMP patients show improved survival and younger age, as well as unique genomic profiles, including the presence of TP53 mutations and a lack of copy number alterations.Citation29 Among these, a heterozygous point mutation in isocitrate dehydrogenase 1 (IDH1), corresponding to the R132 amino acid (IDH1R132H), which was first identified by Parsons et al.Citation72 IDH1 functions as a dimer in the citric acid (TCA) cycle by converting isocitrate to α-ketoglutarate (α-KG).Citation73 IDH1R132H further converts α-KG to D-2-hydroxyglutarate (2-HG).Citation74 The latter is an inhibitor of TET activityCitation75 and subsequent TET-based DNA demethylation, which results in the hypermethylated DNA feature of these tumors. The introduction of IDH1R132H into cancer cells is sufficient to drive G-CIMP DNA hypermethylation, thus providing evidence of the driving effects of this mutation in cancer cells.Citation76,77
Extensive tumor genome characterization has also unveiled TET mutations in AML patients Citation51,78-83 and low frequency IDH mutations in colorectal cancers and cutaneous melanomas.Citation84,85 However, the presence of CIMPs in colon, breast, stomach and endometrial tumors does not correlate with any significant frequencies of IDH or TET mutations, suggesting that CIMP DNA methylation profiles may be generated by distinct mechanisms unique to each tumor type.
Chromatin structure and histone modifications
Chromatin modifications also shape the epigenome in regulating gene expression profiles in both normal and cancer cells. DNA of 146 bp in length is wrapped twice around histone octamers containing dimers of histones H2A, H2B, H3, and H4. Modifications of the N-terminal tail amino acids of Histones H3 and H4 are associated with euchromatic (open) and heterochromatic (closed) chromatin states (reviewed in Citation86). These are commonly referred to as writers (adding marks), erasers (removing markings), and readers (scanning and binding to specific chromatin markings for transcriptional activation or silencing).
The histone codeCitation87 describes how gene expression activity (off/on) is correlated with specific histone modifications of lysine and arginine amino acids of histone N-terminal tails. These modifications include methylation (me), acetylation (Ac), phosphorylation (P), ubiquitination (Ub), and others. Namely, histone H3 lysine acetylation at positions 9 (H3K9Ac) and 14 (H3K14Ac) is associated with open chromatin at promoter gene regions and active gene expression, as are histone H3 lysine 4 trimethylation (H3K4me3) markings at gene promoter regions that also block DNA methylation.Citation88,89 H3K4 monomethylation (HeK4me1) is a marking of enhancer regions of the genome, and H3K27 acetylation is a marking of active enhancer activity when accompanied by K3K4me1 marks.Citation89,90 H3K36me3 marks are found in gene-body regions of actively transcribed genes via the catalytic activity of the SETD2 histone methyltransferase.Citation89 SETD2 is thought to interact with DNMT3B in maintaining DNA methylation of most transcribed gene-body regions, thus providing additional evidence linking chromatin modifications (H3K36me3) and DNA methylation (5mC) marks in the human genome.Citation12,58
The repressive histone H3 lysine 27 trimethylation (H3K27me3) marks are placed by the EZH2 component of the Polycomb Repressive Complex 2 (PRC2),Citation86 while H3K9 methylation (H3K9me2 and H3K9me3) is linked to repression of repetitive elements and the pericentric chromosomal regions. Genomic regions marked by K3K9me and H3K27me may also harbor DNA methylation to provide an additional level of gene regulation;Citation91 however, H3K9me3 or H3K27me3 occupancy may be independent of DNA methylation within a specific genomic region. Gene regions with co-localization of H3K27me3 and H3K4me3 marks are referred to as bivalent chromatin, as they contain both active (H3K4me3) and repressive (H3K27me3) marks. These are of particular importance in ES cells, in which the presence of both marks at specific gene regions, mostly transcription factors and other developmental regulators, correlates with their being poised for potential gene activity when required during cellular differentiation and development.Citation92 H3K27me3 is retained upon differentiation to a state where gene silencing is required, while H3K4me3 is retained in differentiated cells in which gene activity is required.
Gene promoter regions marked by H3K27me3 occupancy are prone to cancer-specific DNA hypermethylation,Citation93-95 suggestive of a stem cell origin of human cancer. In this model, replacement of the permanently-marked repressive (H3K27me3) state from the flexible bivalent (H3K4me3 and H3K27me3) state after differentiation may predispose these sites for DNA hypermethylation due to epigenetic cross-talk between DNA methylation and chromatin machinery early during tumorigenesis. However, DNA methylation and H3K27me3 marks appear to be mutually exclusive in ES cells, and are not always coupled in human cancers. Experiments from Gal-Yam et al. Citation96 support 3 models of epigenetic switching in human cancers in which: 1) genes originally silenced by H3K27me3 marks acquire DNA hypermethylation but then lose the H3K27me3 mark in human cancers; 2) DNA hypermethylation occurs at genes not originally marked by H3K27me3 occupancy; and 3) H3K27me3 marks are added without DNA hypermethylation. Importantly, an appreciable number of demethylated promoter regions after treatment with DNA methylation inhibitors remain silenced by the retained presence or gain of H3K27me3 marks.Citation15,97 This is a critical aspect of epigenetic therapy, and suggests that treatment of cancer cells with DNA methylation inhibitors alone may not effectively activate genes that are epigenetically silenced in human cancers.
Somatic mutations of epigenetic modifier genes in human cancers
Both DNA methylation and chromatin modifications function in concert to regulate gene expression, with crosstalk between these 2 aspects of epigenetics as important components of the cancer epigenome. Indeed, the preponderance of mutations in epigenetic-modifying genes has been identified across multiple human cancer types suggests that cancer cells are epigenetically deregulated, leading to aberrant gene expression and subsequent signaling pathway alterations. Somatic mutations of genes involved in DNA methylation (DNMT3A, TET, IDH), histone methylation (MLL family, EZH2, SUZ12, SETD2), histone demethylation (KMD family), and chromatin remodeling (CHD family, ARID family, ATRX, SMARC family) have been described in multiple forms of human cancer (reviewed in Citationref.98), and are highlighted in .
Table 1. Epigenetic regulator genes altered in the major forms of human cancer. Genes are listed as their HUGO Gene Nomenclature IDs.
Methylated DNA binding proteins
Proteins with methylated CpG DNA binding specificity have been known for over 2 decades (reviewed in Citation99). Proteins with a methylated CpG binding domain (MBD) include MeCP2, and the MBD family (MBD1–6), and function by recognizing, reading and binding to methylated CpG marks, as well as binding to other proteins for transcriptional and chromatin structure regulation. For example, MBD2 is a member of the Mi-2/NuRD chromatin remodeling complex, and functions as a transcriptional repressor by interacting with methylated DNA and deacetylated histones. MBD4 recognizes methylated CpG marks; in addition, it recognizes and repairs mCpG/TpC mismatches due to 5mC deamination to thymine on one DNA strand. Other binding proteins include UHRF1 and Kaiso, among others.Citation99 UHRF1 binds to hemimethylated DNA in recruiting DNMT1 to replication forks for maintenance DNA methylation, and also binds to H3K9me3 and H3 Arginine 2 (H3Ar2) marks via its histone reader domain. Kaiso is a transcription factor that recruits histone deacetylases and the nuclear co-repressor (N-CoR complex), which contains SWI/SNF chromatin remodelers, to methylated DNA regions for gene repression.
microRNAs
microRNA (miRNA) expression changes are frequent events in human cancers. microRNA expression has profound effects on signaling pathways and cell growth, since miRNAs regulate the translation rate of the majority of coding genes in the genome, and can be categorized as having tumor suppressive or oncogenic capabilities (reviewed inCitation100). miR-15a and miR-16–1 were the first miRNAs to be associated with human cancer pathogenesis, as both are frequently deleted in chronic lymphocytic leukemia (CLL), and are therefore referred to as tumor suppressors (reviewed in Citationref.101). Subsequent reports identified miRNA amplifications and deletions in nearly every cancer type, with deletion of miRNAs that repress oncogenic signaling or activate tumor suppressors, as well as amplification of miRNAs that negatively regulate tumor suppressors or activate oncogenes.
Adding to the complexity of miRNA-based gene regulation, miRNA expression alterations as a result of aberrant DNA methylation have been described (reviewed in Citationrefs. 102,103), in which reduced miRNA expression results in increased tumor invasiveness and metastases and reduced growth control that can be reversed by treatment with DNA methylation inhibitors, implicating individually-characterized miRNAs as tumor suppressors. The first evidence described miR-127 DNA hypermethylation and silencing in primary human colon and prostate tumors.Citation104 miR-127 targets the proto-oncogene BCL6 by translational repression. BCL6 is an important oncogene as it suppresses p53 expression and is involved in DNA damage-based apoptotic programs in B cells. Treatment of tumor cells with DNA methylation inhibitors resulted in the re-activation of miR-127 and subsequent inhibition of BCL6, thus providing additional evidence of the utility of DNA methylation inhibition in reversing oncogenic signaling for cancer treatment. Recently, epigenetic silencing of miR-200c was found to be associated with lymph node metastases in breast cancer.Citation105 Likewise, DNA hypermethylation-based silencing of miR-490–3p in colorectal cancer is linked to cell proliferation, invasiveness, and the epithelial-mesenchymal transition (EMT).Citation106 Other examples of miRNA epigenetic silencing include miR-148a and miR-199a, which are silenced by DNA hypermethylation in nasopharyngeal carcinomasCitation107 and testicular germ cell tumors,Citation108 respectively.
Additional sets of miRNAs, termed epi-miRNAs, regulate epigenetic modifiers by specifically targeting DNMTs, histone deacetylases (HDACs), histone modifiers, repressors and other epigenetic regulators across a variety of tissue types, and, thereby have wide reaching effects on shaping the epigenomes in both normal and tumor cells.Citation103 Aberrant epi-miR expression is common in a variety of human cancers. For example, miR-101, which suppresses EZH2 translation, is downregulated in human cancers, resulting in increased EZH2 translation and aberrant placement of repressive H3K27me3 marks.Citation109 Another example is mir-29a/b/c, which directly inhibits DNMA3A and DNMT3B translation and/or transcription. miR-29a/b/c expression is inversely correlated with DNMT3A and DNMT3B mRNA expression in lung tumors.Citation110 Interestingly, lung cancer patients with low DNMT3A mRNA expression show increased survival compared with those with high DNMT3A mRNA expression, and are related to miR-29 expression in these tumors. These data suggest miR-29a/b/c function as tumor suppressors in restoring normal DNA methylation, and their silencing is associated with DNA hypermethylation.
Five-azacytidine-based inhibitors of DNA methylation
Currently, chemical inhibitors of several epigenetic modifiers have been developed (reviewed inCitation111) (). The first DNA methylation inhibitors, 5-azacytidine (5-Aza-CR) and 5-aza-2′-deoxycytidine (5-Aza-CdR), were synthesized over 50 y ago by Sorm, Piskala et al. (reviewed in Citationref.112) as a cytotoxic anticancer drug, much like 5-fluorouracil (5-FU), for the treatment of AML and acute lymphoblastic leukemia (ALL). Early trials evaluating the performance of high doses of 5-Aza nucleotides in leukemia patients resulted in only short-term effects accompanied by extensive cytotoxicity.Citation112
Work from Jones and TaylorCitation113 showed that in vitro application of 5-Aza-CR led to mouse embryo (10T1/2) differentiation into muscle cells due extensive genome-wide DNA hypomethylation and subsequent cellular reprogramming. 10T1/2 cell differentiation to myoblast-like cells is attributed to MyoD1 promoter DNA methylation changes.Citation114 Further research showed that upon delivery into the nucleus, Aza-substituted analogs are converted to Aza-triphosphate and incorporated into newly synthesized genomic DNA during DNA replication. Aza-incorporated DNA traps DNMTs to the site of purported DNA methylation, and effectively removes free DNMT from the nucleus, leading to passive DNA demethylation.Citation41 An additional model was described by Ghoshal et al.,Citation115 who showed that upon binding to aza-substituted cytosines, DNMT1 becomes poly-ubiquitinated and then degraded by the 26S proteasome, suggesting that multiple means of DNMT1 inactivation occur in response to 5-Aza treatment.
Both 5-Aza-CR and 5-Aza-CdR are effective DNA methylation inhibitors.Citation116 While 5-Aza-CR is activated by uridine-cytidine kinase and can be incorporated into both DNA and RNA, 5-Aza-CdR is activated by deoxycytidine kinase and is only incorporated into of newly synthesized DNA strands after replication. As a result, lower doses of 5-Aza-CdR are required for DNA demethylation, even though 5-Aza-CdR treatment results in high cellular toxicity and DNA damage. Aza-based drug treatments in cancer cell cultures and mouse models of cancer resulted in reduced numbers of viable tumor cells and tumor volumes. Both 5-Aza-CR and 5-Aza-CdR are FDA-approved for the treatment of MDS and leukemia patients under the trade names Vidaza and Dacogen/Decitabine, respectively.
Zebularine and SGI-110 show novel attributes as DNA methylation inhibitors
In addition to 5-Aza-CR and 5-Aza-CdR, other DNA methylation inhibitors, including zebularine and SGI-110, have been characterized. Zebularine, 1-(β-D-Ribofuranosyl)-2(1H)-pyrimidinone, was originally designed as a transition state inhibitor of cytidine deaminase, and has structural similarity to cytidine, but lacks the C-4 amino group present on cytidine nucleosides. Much like 5-azacytidine analogs, zebularine is also incorporated into nascent DNA strands and shows a similar mechanism of covalent DNMT trapping for passive DNA methylation inhibition,Citation117 although zebularine appears to preferentially inhibit DNMT1 over DNMT3A and DNMT3B.Citation118 Zebularine inhibits cell growth of a variety of cancer types, and re-activates genes silenced by DNA hypermethylation, including tumor suppressor and tumor antigen genes.Citation118-120 Moreover, zebularine improves radiosensitivity in both in vitro and in vivo settings.Citation121 Zebularine showed stability in aqueous solution and can be delivered orally in vivo, an important attribute for potential clinical utility. However, 1–2 orders of magnitude greater dosing is required for the same effect seen from 5-Aza-CdR treatments.Citation118-120 Additional pharmacokinetic studiesCitation122 concluded that zebularine has low bioavailability and frequent or continuous intravenous infusion is required for effective DNA demethylation.
Guadecitabine (SGI-110), is a modified version of 5-Aza-CdR, consisting of 5-Aza-CdR covalently bound to deoxyguanosine via phosphodiester linkage.Citation123,124 SGI-110 shows promising clinical utility, as it is less subject to deamination by cytidine deaminase, and therefore, displays improved stability and lower toxicity over 5-Aza-CdR alone.Citation123 SGI-110 is highly tolerated upon delivery, and is effective by both intraperitoneal and subcutaneous delivery methods in mice and patient derived xenograft (PDX) models of cancer.Citation123 Kuang et al. evaluated SGI-110 in hepatocellular carcinoma cell lines and PDX mouse models to guide future phase I/II clinical trials, either alone or in conjunction with oxaliplatin, Citation125 a platinum-based cytotoxic compound that inhibits DNA synthesis by forming cross-links with DNA. Low-dose SGI-110 treatment alone resulted in reduced cell proliferation, and pretreatment of HCC cells with low-dose SGI-110 resulted in significantly increased oxaliplatin sensitivity as well as inhibition of WNT/EGF/IGF signaling.
SGI-110 is currently being evaluated in 17 ongoing clinical trials across multiple cancer types, including AML, CMML, colorectal, germ cell, MDS, melanoma, lung and ovarian cancers, in which SGI-110 is administered either as a single agent or in combination with chemotherapeutic agents (www.clinicaltrials.gov). SGI1–110 clinical trials in liver and non-small cell lung cancer patients have recently completed. Issa and colleaguesCitation126 reported a completed phase I trial evaluating the efficacy of SGI-110 dose escalation and drug delivery frequencies in AML and MDS patients (either 5 consecutive days, once weekly for 4 weeks or twice per week for 4 weeks), with the goal of determining drug tolerance and the extent of DNA demethylation. SGI-110 was well tolerated, had a significantly longer half-life than 5-Aza-CdR and was most biologically effective when administered for 5 consecutive days. In agreement, global DNA demethylation, as measured by LINE-1 repetitive elements, was maximized at day 8 of the daily dosing schedule, and clinical response correlated with increased LINE-1 DNA demethylation. These experiments show promise for using SGI-110 in the clinic; however, combining SGI-110 with other chemotherapies has not yet been accomplished.
Non-nucleoside DNA methylation inhibitors
Non-nucleoside DNA methylation inhibitors have also been described, namely 5-fluoro-2′-deoxycytidine, (−)-epigallocatechin-3 gallate (EGCG), hydralazine, mitoxanthrone, N-acetylprocainamide, psammaplin A, procainamide and procaine (summarized in ref. Chuang, 2005 #788). Hydralazine is clinically administered for hypertension and was shown to reduce DNMT1 and DNMT3A expression. Procainamide has clinical utility in treating arrhythmia and was shown to inhibit DNMT activity. EGCG is a polyphenol in green tea that has shown DNA methylation inhibition and chemopreventive activities. However, comparative analyses showed that these non-nucleoside inhibitors are not as effective as 5-Aza-CdR in DNA demethylation and gene reactivation;Citation127 therefore, their clinical utility may be limited.
Mechanisms of 5-Aza-CdR-based DNA demethylation based on preclinical studies
Five-Aza-CdR induced DNA demethylation
Even though Aza-based DNA methylation inhibitors display anti-tumor attributes, they are not stable in aqueous solution, and a single high-dose treatment results only in transient effects of DNA demethylation and increased population doubling times, since DNA remethylation occurs within days after a single dose.Citation12,128 It is well known that Aza-nucleotide treatments of cancer cells result in the re-expression of genes silenced by promoter DNA hypermethylation (). Epigenetically-silenced genes have been well described in human cancers (reviewed in Citationref.129), and include CDKN2A (p16), MGMT, MLH1, SFRPs and BRCA1/2. Epigenetic silencing of these genes contributes to tumorigenesis by allowing the cell to escape cell cycle control [CDKN2A (p16)], DNA repair (BRCA1/2, MGMT, MLH1), and ligand-receptor based signaling (SFRPs). MGMT promoter DNA hypermethylation results in silencing of the gene and subsequently, the inability of the MGMT enzyme to methylate the O-6 position of guanine residues. MGMT DNA methylation is an important diagnostic in glioma patient care to determine if temozolomide can be administered. Temozolomide is an alkylating agent, and is effective in glioma treatment only when MGMT is silenced. As a result of MGMT silencing, DNA damage incurred by the drug cannot be repaired, and the cell enters into apoptotic programmed cell death.Citation130
Figure 1. DNA methylation and gene expression consequences after treatment with DNA methylation inhibitors. Circles represent CpG sites, with closed circles indicative of methylated CpGs and open circles representing unmethylated CpGs. Transcription start sites are indicated by the bent arrow. (A) Demethylation of CpG island promoter regions, resulting in gene activity. (B) Gene-body demethylation results in reduced expression. (C) Retained gene silencing by PRC2 occupancy after DNA methylation inhibitor treatment. HMA: Hypomethylating agent.
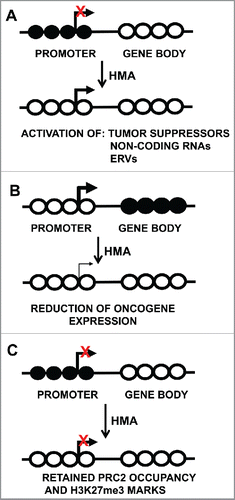
While identifying epigenetically silenced genes reactivated by 5-Aza-CR and 5-Aza-CdR is important for identifying potential epigenetic drivers, gene regions with both 5mC and H3K27me3 marks remain silenced after treatment with DNA methylation inhibitors due to retention of the H3K27me3 repressive mark Citation15,97 (). This is a critical issue for using DNA methylation inhibitors for therapeutic approaches, as treatment of cancer cells with DNA methylation inhibitors alone may not effectively activate genes that are epigenetically silenced in human cancers. Treatment schemes with DNA methylation and EZH2 inhibitors may help to effectively activate a larger set of epigenetically silenced genes, and ultimately bring the tumor cell epigenome closer to a normal epigenetic state.
Single low-dose, 24 h delivery of 5-Aza-CdR has more prolonged cell growth inhibition and reduced toxicity, although DNA remethylation still occurs. A report from Tsai a et al.Citation131 showed that human leukemia cells treated with low-dose (10–500 nM) 5-Aza-CdR displayed significantly reduced tumorigenicity, cellular toxicity, DNA damage and apoptosis. Low-dose 5-Aza-CdR was also effective in reducing tumorigenicity and tumor size in mouse xenograft models of breast cancer. Moreover, single low dose 5-Aza-CdR treatment resulted in loss of DNMT1 protein, together with sustained DNA demethylation of gene promoters and activation of epigenetically-silenced genes.Citation131 Pathway analyses of re-expressed genes showed increased cyclin dependent kinase (CDK) based cell cycle control and decreased AKT signaling.Citation131 The AKT pathway mediated epithelial to mesenchymal transition (EMT), which is associated with tumor progression and invasiveness. TGF-β signaling was also increased in the Aza-treated leukemia cells. In this pathway, TGF-β drives cell maturation and inhibits the renewal of progenitor cells. These features highlight the promise for DNA methylation inhibitors in clinical practice.
The reactivation of epigenetically-silenced genes after 5-Aza-CdR treatment has been well characterized. However, such treatment also reduces the overexpression of genes through DNA demethylation of gene bodies and transcribed regions, which are normally methylated in actively expressed genes Citation12 (). Downregulated genes include oncogenes and those involved in c-MYC regulated processes. This finding revealed an unexpected therapeutic advantage of 5-Aza-CdR and was the first evidence that DNMT inhibitors not only reactivate tumor suppressor genes, but also down-regulate overexpressed oncogenes in human cancer cells.
Gene-body DNA remethylation after 5-Aza-CdR treatment
While the mechanisms of DNA demethylation are becoming well understood, DNA remethylation kinetics is not universal for all genes across the genome. Indeed, Yang and colleaguesCitation12 showed that DNA demethylation occurs evenly across the human genome; however, DNA remethylation is differentiated by location and genomic context. DNA remethylation can be categorized by “Fast” or “Slow” kinetics after 5-Aza-CdR treatment of HCT116 colon cancer cells (). Among these, the Fast CpG sites quickly and completely remethylate to pre-treated levels, while the Slow CpGs remain demethylated for at least 60 d after 5-Aza-CdR treatment (). Interestingly, most of the Fast CpG sites were mostly located in non-CpG island gene-body regions and were positively correlated with gene expression. DNMT3B is required for remethylation of Fast sites after 5-Aza-CdR treatment, in which DNMT3B recognizes H3K36me3 marks as a beacon for DNA remethylation.
Figure 2. DNA remethylation kinetics after 5-aza-CdR treatment. Top: DNA remethylation of “Fast” (left) and “Slow” (right) CpG sites. Graphs are plotted as percent methylation (y-axis) vs. time after 5-Aza-CdR treatment (x-axis). Bottom: details of Fast and Slow rebounding genes.
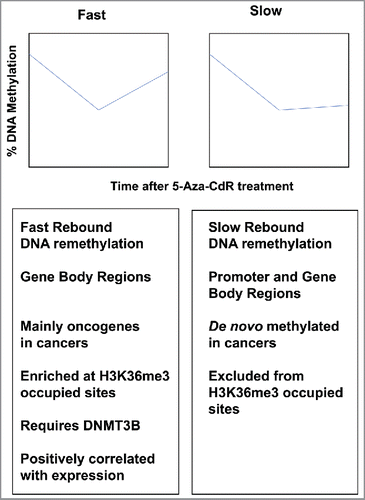
Since H3K36me3 marks are localized at gene bodies and are positively correlated with active gene expression, SETD2, which is responsible for H3K36me3 marks, is thought to interact with DNMT3B in this regard. Meta analyses showed that genes harboring Fast CpGs are overexpressed in human cancers and are enriched for pathways in which cell growth is stimulated. Similar numbers of Slow genes were located in gene promoter and gene-body regions, and most Slow CpG islands are cancer-associated targets of de novo DNA methylation. Slow loci located in gene bodies were not associated with H3K36me3 occupancy, but rather increased occupancy of H3K27me3 and H2A.Z marks, which are anti-correlated with DNA methylation. As a result, these chromatin modifications may block DNMT3B activity required for gene-body DNA remethylation. These findings highlight the importance of gene-body DNA methylation in gene regulation, as well as a novel mechanistic attribute of 5-Aza-CdR-based DNA demethylation.
Viral mimicry induced by 5-Aza-CdR treatment
The reactivation of epigenetically-silenced genes by 5-Azacytidine has been well described, however, additional mechanistic details were recently discovered. Low-dose 5-Aza-CdR treatment does not result in cellular toxicity but rather delayed DNA demethylation, suggesting that pathways unrelated to cytotoxicity may be activated. In general, endogenous retroviruses (ERVs) and other repetitive elements located in gene bodies are commonly silenced by DNA methylation in somatic cells, but can be reactivated after treatment with DNA methylation inhibitors.Citation132-134 Activated ERVs stimulate an immune response after 5-Aza-CdR treatment by allowing the expression of double stranded RNAs (dsRNAs), which are targeted by melanoma differentiation-associated protein 5 (MDA5). In turn, MDA5 becomes activated and recruits mitochondrial anti-viral signaling protein (MAVS), which induces interferon regulatory transcription factor 7 (IRF7), and subsequent antiviral response pathways. Importantly, these data show that activation of an interferon response is triggered by 5-Aza-CdR via ERV reactivation. Essentially, 5-Aza-CdR induces viral mimicry by deceiving the cancer cell to function in a viral-infected state. These paradigm-shifting findings not only support the efficacy of 5-Aza-CdR as a therapeutic tool, but also highlight the exploitation of dsRNA activation, ERV activation and MDA5 signaling as novel therapeutic targets and biomarkers for gauging treatment response.
Viral mimicry is also activated by vitamin C, a required co-factor for TET-based DNA demethylation. In vitro delivery of physiologic doses of vitamin C has a pronounced effect on DNA demethylation and gene reactivation when combined with low doses of 5-Aza-CdR, most notably, ERVs, dsRNAs and an interferon response.Citation135 Moreover, this is accompanied by increased apoptosis and inhibition of cancer cell proliferation. The synergistic boost in DNA demethylation is likely due to stimulation of both passive (Aza) and active (TET) DNA demethylation mechanisms. For clinical benefit, vitamin C is water soluble, inexpensive, abundant, orally available and easily administered.
Clinical efficacy of combining DNMT inhibitors with conventional chemotherapies based on preclinical testing
Preclinical models, including cell culture, 3-D organoids, patient derived xenografts (PDX) and genetic mouse models, provide time- and cost-effective means of determining potential clinical efficacy for the development of new treatments, schedules, and doses. These can also be used to determine whether the inclusion of DNA methylation inhibitors result in increased sensitivity to conventional cytotoxic chemotherapeutics or those used for specific pathway inhibition (). One example is the report from Ikehata et al.,Citation136 in which human colon cancer cell lines were treated with chemotherapies (5-fluorouracil, irinotecan, oxaliplatin), DNA methylation inhibitors (5-Aza-CR, 5-Aza-CdR, zebularine) and/or histone deacetylase inhibitors (Trichostatin A, SAHA, valproic acid) to determine if epigenetic therapies improve tumor toxicity. Indeed, the addition of DNA methylation inhibitors resulted in synergistic effects incurred by chemotherapy, and in particular, 5-Aza-CdR showed the most potent synergistic effect when administered with oxaliplatin.
Figure 3. Therapeutic approaches accompanying DNA methylation inhibition for cancer patient treatment. Cancer cells treated with DNA methylation inhibitors have activated tumor suppressors, ERVs and miRNAs, with reduced expression of oncogenes. Activated ERVs promote an immune response that can be further targeted by immune-based therapies. Activated genes may help promote sensitivity to conventional chemotherapies. DNA methylation inhibitors, most notably 5-Aza-CR and 5-Aza-CdR, cause DNA damage, thus sensitizing the treated cell to DNA repair inhibitors.
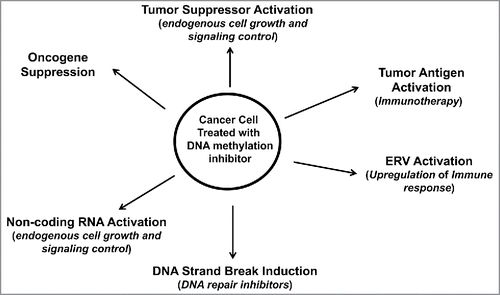
Combining DNA methylation inhibitors with immune-based therapies also shows clinical promise. Priming non-small cell lung cancer (NSCLC) cells with 5-Aza-CR (Vidaza) results in the activation of immune-related genes as well as those related to immune evasion.Citation137 Specifically, 5-Aza-CR upregulates PD-L1, a key mediator of immune tolerance, suggesting that epigenetic therapy combined with PD-L1 inhibitors to block immune checkpoints may be efficacious in driving immune activation and reducing immune inhibition.
DNA methylation inhibition may also have clinical efficacy when coupled with DNA repair inhibitors (). DNA methylation inhibitors cause DNA strand breaks, and cancer cell lines treated with 5-Aza-CdR showed prolonged γH2A.X expression, a marker of double-strand DNA breaks.Citation138 Therefore, DNMT inhibitors may radiosensitize cells for improved response to radiotherapies. The increase in DNA strand breaks after 5-Aza-CdR treatment of cancer cells is especially important in the clinical setting, as DNA repair inhibitors that inhibit poly-ADP ribose polymerase (PARP), a DNA repair protein that binds to double-strand breaks, are used in the clinic for cancer treatment. DNA mismatch repair systems are challenged in colorectal and endometrial cancers, suggesting that epigenetic therapy may be efficacious in these tumor types.
Findings from clinical trials
Clinical trials involving DNA methylation inhibitors, either implemented alone, in combination with conventional chemotherapies, have been mostly performed for MDS and AML patients (reviewed in Citation139,140). Indeed, a query of clinicaltrials.gov showed over 500 cancer-related clinical trials involving 5-Aza-CR or 5-Aza-CdR (query: “5-aza” and “cancer”). Approximately, 400 trials involved patients with MDS, leukemia [AML, chronic myeloid leukemia (CML), and chronic myelomonocytic leukemia (CMML)] and lymphoma, while most of the remaining 100 trials involve patients with solid tumors. A comparison of the clinical trials in patients with solid or liquid malignancies is shown in .
Figure 4. Clinical trials involving 5-Aza nucleotides in human cancers. Plot shows the number of trials completed, withdrawn, or in progress (y-axis) vs. individual tumor type (x-axis).
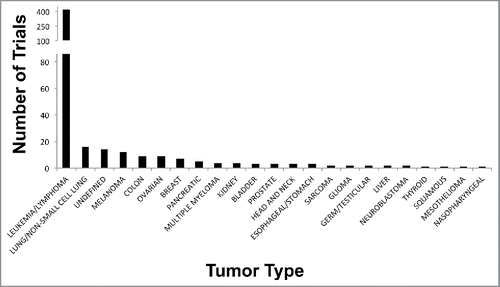
Five-Aza-CR and 5-Aza-CdR are FDA-approved for treatment of MDS and AML patients, and 5-Aza-CR is regarded as the standard of care for treating MDS patients who are not eligible for allogenic stem cell transplantation, the only known curative treatment of MDS. 5-Aza-CR treatment of MDS patients in the AML-001 trial resulted in a nearly 10-month increased OS and also increased the time of progression of MDS to AML. In agreement, AML patients enrolled in the same trial also showed similar increases in OS after receiving 5-Aza-CR as compared with conventional therapies.Citation140-143 In support of this, a large phase III trial evaluating 5-Aza-CR vs. conventional care in 488 newly diagnosed AML patients of advanced age and >30% bone marrow blastsCitation144 concluded that patients treated with 5-Aza-CR showed improved median overall survival, improved one-year survival rates after treatments and improved response metrics; however, remission rates were similar between both treatments.
Five-Aza-CR and 5-Aza-CdR are currently in clinical trials for patients with solid tumors, including lung, breast, ovarian and colon cancers, among others (). A phase I/II study of low-dose 5-Aza-CR and the HDAC inhibitor entinostat for metastatic non-small cell lung cancer (NSCLC) patients showed objective responses in a subset of patients.Citation145 DNA demethylation of APC, RASSF1A, CDH13, and CDKN2A (p16) in serial blood samples from NSCLC patients collected during treatment correlated with improved progression free survival (PFS) an OS.Citation145
Increased global DNA demethylation in treated tumor biopsies also correlated with longer survival in a similar phase I/II trial evaluating 5-azacitidine and entinostat in breast cancer patients.Citation146 While this drug combination was well tolerated in patients with HER2-negative tumors, it did not result in clinical activity in women with triple-negative breast cancer. Clinical response was evident in only a subset of women with hormone-resistant disease; however, ESR1 DNA hypomethylation and gene expression activation occurred in nearly one-half of the treated hormone-resistant patients, suggesting that these patients may benefit from both endocrine and epigenetic therapies.
DNA demethylating agents have also been used in trials for ovarian cancer patients. In general, ovarian cancer patients receive platinum-based therapies; however, resistance to platinum-based drugs is common. Fu et al.Citation147 reported on a phase I/II clinical trial of ovarian cancer patients with platinum-resistant or platinum refractory disease, in which patients were given sequential treatments of 5-Aza-CR followed by carboplatin. Pretreatment with 5-Aza-CR agent helped to overcome platinum resistance and inhibit the development of liver metastases in some patients, and demonstrates promise as a means of ovarian cancer treatment.
Colon cancer patients with metastatic disease have few treatment options. Agents targeting vascular endothelial growth factor receptor (VEGFR), epidermal growth factor (EGFR), and tyrosine kinase have improved outcomes for metastatic CRC patients. However, the survival benefit achieved with each novel therapeutic is modest, and only a fraction of patients demonstrate a measurable or durable response. Despite immense pre-clinical and clinical research efforts, only RAS mutation status exists to guide therapeutic decisions. KRAS mutations predict resistance to EGFR-based antibodies, including panitumumab and cetuximab, since KRAS is involved in signal transduction from ligand-bound EGFR from the cell membrane to the nucleus.Citation147
EGFR silencing may also be involved in treatment resistance for colorectal cancer patients. Scartozzi et al.Citation148 identified EGFR promoter DNA hypermethylation in 58% of primary colon tumors, and patients with EGFR promoter DNA methylation showed a 5 month shorter PFS and an 11.7-month shorter OS compared with those patients without EGFR promoter DNA methylation. In addition, a phase I/II trialCitation149 to assess the performance of 5-Aza-CdR and panitumumab in 20 metastatic colorectal cancer patients with wild-type KRAS tumors showed tolerance and activity to this drug combination. Partial responses were observed in 2/20 (10%) of patients and stable disease was observed in 10/20 (50%) of patients, suggesting that this drug combination may improve survival and quality of life in patients with metastatic colon cancer. Finally, a phase II study of 5-Aza-CR and entinostat in colon cancer patients,Citation150 was tolerated and a subset of patients showed DNA demethylation correlated with improved PFS.
Conclusions
Epigenetic therapies, namely DNA methylation and histone modifier inhibitors, show promise as effective anti-cancer agents. Aza-based DNA methylation inhibitors have been tested for nearly 50 years; however, new mechanistic details have recently emerged after combining preclinical drug treatments with genome-wide platforms for measuring DNA methylation changes. Treatment with DNA methylation inhibitors not only results in re-activation of tumor suppressor genes, but also repression of oncogenes and gene with demethylated gene-body/transcribed regions. In addition, the reactivation of endogenous retroviruses (ERVs) after 5-Aza-CdR results in viral mimicry and the initiation of an immune response that can be targeted with immune-based therapies. Combining DNA methylation inhibitors with conventional chemotherapies and those that specifically inhibit DNA repair show promise in pre-clinical models. Finally, integrating DNA methylation inhibitors with inhibitors of histone modifiers provides synergistic enhancement of DNA methylation inhibition, as does the addition of vitamin C, a cofactor of TET-based oxidative DNA demethylation. These combined methodologies () suggest that DNA methylation inhibitors serve as primers for activating epigenetically silenced genes that can be further targeted with complementary therapies. Evaluation of these treatment schemes in preclinical models and clinical trials will shed light on the efficacy for routine use in treating patients with a wide variety of human cancers.
Disclosure of potential conflicts of interest
Daniel J. Weisenberger is a consultant for Zymo Research Corporation.
Funding
This work was supported by the Vicky Joseph Cancer Research Lab (to G.L.), NIH 5R21 CA201865 (to G.L. and D.J.W.) and NIH/NCI P30 CA014089 (to D.J.W.).
Related Research Data
References
- Weisenberger DJ, Brown PJ. Network and consortia for epigenetic drug discovery. In: Egger G, Arimondo P, editors. Drug Discovery in Cancer Epigenetics. Waltham, MA USA: Academic Press; 2016. p 143-162
- Prasad V. Perspective: The precision-oncology illusion. Nature 2016; 537(7619):S63; PMID:27602743; https://doi.org/10.1038/537S63a
- Klauschen F, Andreeff M, Keilholz U, Dietel M, Stenzinger A. The combinatorial complexity of cancer precision medicine. Oncoscience 2014; 1(7):504-9; PMID:25594052; https://doi.org/10.18632/oncoscience.66
- Hoadley KA, Yau C, Wolf DM, Cherniack AD, Tamborero D, Ng S, Leiserson MD, Niu B, McLellan MD, Uzunangelov V, et al. Multiplatform analysis of 12 cancer types reveals molecular classification within and across tissues of origin. Cell 2014; 158(4):929-44; PMID:25109877; https://doi.org/10.1016/j.cell.2014.06.049
- Witte T, Plass C, Gerhauser C. Pan-cancer patterns of DNA methylation. Genome Med 2014; 6(8):66; PMID:25473433; https://doi.org/10.1186/s13073-014-0066-6
- Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet 2012; 13(7):484-92; PMID:22641018; https://doi.org/10.1038/nrg3230
- Robertson KD, Jones PA. DNA methylation: past, present and future directions. Carcinogenesis 2000; 21(3):461-7; PMID:10688866; https://doi.org/10.1093/carcin/21.3.461
- Deaton AM, Bird A. CpG islands and the regulation of transcription. Genes Dev 2011; 25(10):1010-22; PMID:21576262; https://doi.org/10.1101/gad.2037511
- Ehrlich M, Wang RY. 5-Methylcytosine in eukaryotic DNA. Science 1981; 212(4501):1350-7; PMID:6262918; https://doi.org/10.1126/science.6262918
- Feinberg AP, Vogelstein B. Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature 1983; 301:89-92; PMID:6185846; https://doi.org/10.1038/301089a0
- Gama-Sosa MA, Slagel VA, Trewyn RW, Oxenhandler R, Kuo KC, Gehrke CW, Ehrlich M. The 5-methylcytosine content of DNA from human tumors. Nucleic Acids Res 1983; 11:6883-6894; PMID:6314264; https://doi.org/10.1093/nar/11.19.6883
- Yang X, Han H, De Carvalho DD, Lay FD, Jones PA, Liang G. Gene body methylation can alter gene expression and is a therapeutic target in cancer. Cancer Cell 2014; 26(4):577-90; PMID:25263941; https://doi.org/10.1016/j.ccr.2014.07.028
- De Carvalho DD, Sharma S, You JS, Su SF, Taberlay PC, Kelly TK, Yang X, Liang G, Jones PA. DNA methylation screening identifies driver epigenetic events of cancer cell survival. Cancer Cell 2012; 21(5):655-67; PMID:22624715; https://doi.org/10.1016/j.ccr.2012.03.045
- Hinoue T, Weisenberger DJ, Lange CP, Shen H, Byun HM, Van Den Berg D, Malik S, Pan F, Noushmehr H, van Dijk CM, et al. Genome-scale analysis of aberrant DNA methylation in colorectal cancer. Genome Res 2012; 22(2):271-82; PMID:21659424; https://doi.org/10.1101/gr.117523.110
- Lay FD, Liu Y, Kelly TK, Witt H, Farnham PJ, Jones PA, Berman BP. The role of DNA methylation in directing the functional organization of the cancer epigenome. Genome Res 2015; 25(4):467-77; PMID:25747664; https://doi.org/10.1101/gr.183368.114
- Becket E, Chopra S, Duymich CE, Lin JJ, You JS, Pandiyan K, Nichols PW, Siegmund KD, Charlet J, Weisenberger DJ, et al. Identification of DNA methylation-independent epigenetic events underlying clear cell renal cell carcinoma. Cancer Res 2016; 76(7):1954-64; PMID:26759245; https://doi.org/10.1158/0008-5472.CAN-15-2622
- Laird PW. The power and the promise of DNA methylation markers. Nat Rev Cancer 2003; 3(4):253-66; PMID:12671664; https://doi.org/10.1038/nrc1045
- How Kit A, Nielsen HM, Tost J. DNA methylation based biomarkers: practical considerations and applications. Biochimie 2012; 94(11):2314-37; PMID:22847185; https://doi.org/10.1016/j.biochi.2012.07.014
- Schuebel KE, Chen W, Cope L, Glockner SC, Suzuki H, Yi JM, Chan TA, Van Neste L, Van Criekinge W, van den Bosch S, et al. Comparing the DNA hypermethylome with gene mutations in human colorectal cancer. PLoS Genet 2007; 3(9):1709-23; PMID:17892325; https://doi.org/10.1371/journal.pgen.0030157
- Toyota M, Ahuja N, Ohe-Toyota M, Herman JG, Baylin SB, Issa JP. CpG island methylator phenotype in colorectal cancer. Proc Natl Acad Sci U S A 1999; 96(15):8681-6; PMID:10411935; https://doi.org/10.1073/pnas.96.15.8681
- Weisenberger DJ, Siegmund KD, Campan M, Young J, Long TI, Faasse MA, Kang GH, Widschwendter M, Weener D, Buchanan D, et al. CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer. Nat Genet 2006; 38(7):787-93; PMID:16804544; https://doi.org/10.1038/ng1834
- Ogino S, Nosho K, Kirkner GJ, Kawasaki T, Meyerhardt JA, Loda M, Giovannucci EL, Fuchs CS. CpG island methylator phenotype, microsatellite instability, BRAF mutation and clinical outcome in colon cancer. Gut 2009; 58(1):90-6; PMID:18832519; https://doi.org/10.1136/gut.2008.155473
- Min BH, Bae JM, Lee EJ, Yu HS, Kim YH, Chang DK, Kim HC, Park CK, Lee SH, Kim KM, et al. The CpG island methylator phenotype may confer a survival benefit in patients with stage II or III colorectal carcinomas receiving fluoropyrimidine-based adjuvant chemotherapy. BMC Cancer 2011; 11:344; PMID:21827707; https://doi.org/10.1186/1471-2407-11-344
- Donada M, Bonin S, Barbazza R, Pettirosso D, Stanta G. Management of stage II colon cancer - the use of molecular biomarkers for adjuvant therapy decision. BMC Gastroenterol 2013; 13:36; PMID:23446022; https://doi.org/10.1186/1471-230X-13-36
- Shiovitz S, Bertagnolli MM, Renfro LA, Nam E, Foster NR, Dzieciatkowski S, Luo Y, Lao VV, Monnat RJ Jr, Emond MJ, et al. CpG island methylator phenotype is associated with response to adjuvant irinotecan-based therapy for stage III colon cancer. Gastroenterology 2014; 147(3):637-45; PMID:24859205; https://doi.org/10.1053/j.gastro.2014.05.009
- Phipps AI, Limburg PJ, Baron JA, Burnett-Hartman AN, Weisenberger DJ, Laird PW, Sinicrope FA, Rosty C, Buchanan DD, Potter JD, et al. Association between molecular subtypes of colorectal cancer and patient survival. Gastroenterology 2015; 148(1):77-87 e2; PMID:25280443; https://doi.org/10.1053/j.gastro.2014.09.038
- Kang KJ, Min BH, Ryu KJ, Kim KM, Chang DK, Kim JJ, Rhee JC, Kim YH. The role of the CpG island methylator phenotype on survival outcome in colon cancer. Gut Liver 2015; 9(2):202-7; PMID:25167802; https://doi.org/10.5009/gnl13352
- Juo YY, Johnston FM, Zhang DY, Juo HH, Wang H, Pappou EP, Yu T, Easwaran H, Baylin S, van Engeland M, et al. Prognostic value of CpG island methylator phenotype among colorectal cancer patients: a systematic review and meta-analysis. Ann Oncol 2014; 25(12):2314-27; PMID:24718889; https://doi.org/10.1093/annonc/mdu149
- Noushmehr H, Weisenberger DJ, Diefes K, Phillips HS, Pujara K, Berman BP, Pan F, Pelloski CE, Sulman EP, Bhat KP, et al. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell 2010; 17(5):510-22; PMID:20399149; https://doi.org/10.1016/j.ccr.2010.03.017
- Cancer Genome Atlas Research Network T. Comprehensive molecular portraits of human breast tumours. Nature 2012; 490(7418):61-70; PMID:23000897; https://doi.org/10.1038/nature11412
- Fang F, Turcan S, Rimner A, Kaufman A, Giri D, Morris LG, Shen R, Seshan V, Mo Q, Heguy A, et al. Breast cancer methylomes establish an epigenomic foundation for metastasis. Sci Transl Med 2011; 3(75):75ra25; PMID:21430268; https://doi.org/10.1126/scitranslmed.3001875
- Cancer Genome Atlas Research N, Kandoth C, Schultz N, Cherniack AD, Akbani R, Liu Y, Shen H, Robertson AG, Pashtan I, Shen R, et al. Integrated genomic characterization of endometrial carcinoma. Nature 2013; 497(7447):67-73; PMID:23636398; https://doi.org/10.1038/nature12113
- Zhang QY, Yi DQ, Zhou L, Zhang DH, Zhou TM. Status and significance of CpG island methylator phenotype in endometrial cancer. Gynecol Obstet Invest 2011; 72(3):183-91; PMID:21968189; https://doi.org/10.1159/000324496
- Toyota M, Ahuja N, Suzuki H, Itoh F, Ohe-Toyota M, Imai K, Baylin SB, Issa JP. Aberrant methylation in gastric cancer associated with the CpG island methylator phenotype. Cancer Res 1999; 59(21):5438-42; PMID:10554013
- An C, Choi IS, Yao JC, Worah S, Xie K, Mansfield PF, Ajani JA, Rashid A, Hamilton SR, Wu TT. Prognostic significance of CpG island methylator phenotype and microsatellite instability in gastric carcinoma. Clin Cancer Res 2005; 11(2 Pt 1):656-63; PMID:15701853
- Kusano M, Toyota M, Suzuki H, Akino K, Aoki F, Fujita M, Hosokawa M, Shinomura Y, Imai K, Tokino T. Genetic, epigenetic, and clinicopathologic features of gastric carcinomas with the CpG island methylator phenotype and an association with Epstein-Barr virus. Cancer 2006; 106(7):1467-79; PMID:16518809; https://doi.org/10.1002/cncr.21789
- Chang MS, Uozaki H, Chong JM, Ushiku T, Sakuma K, Ishikawa S, Hino R, Barua RR, Iwasaki Y, Arai K, et al. CpG island methylation status in gastric carcinoma with and without infection of Epstein-Barr virus. Clin Cancer Res 2006; 12(10):2995-3002; PMID:16707594; https://doi.org/10.1158/1078-0432.CCR-05-1601
- Cancer Genome Atlas Research N. Comprehensive molecular characterization of gastric adenocarcinoma. Nature 2014; 513(7517):202-9; PMID:25079317; https://doi.org/10.1038/nature13480
- Cancer Genome Atlas Research N. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature 2013; 499(7456):43-9; PMID:23792563; https://doi.org/10.1038/nature12222
- Wei JH, Haddad A, Wu KJ, Zhao HW, Kapur P, Zhang ZL, Zhao LY, Chen ZH, Zhou YY, Zhou JC, et al. A CpG-methylation-based assay to predict survival in clear cell renal cell carcinoma. Nat Commun 2015; 6:8699; PMID:26515236; https://doi.org/10.1038/ncomms9699
- Santi DV, Norment A, Garrett CE. Covalent bond formation between a DNA-cytosine methyltransferase and DNA containing 5-azacytosine. Proc Natl Acad Sci U S A 1984; 81(22):6993-7; PMID:6209710; https://doi.org/10.1073/pnas.81.22.6993
- Jones PA, Liang G. Rethinking how DNA methylation patterns are maintained. Nat Rev Genet 2009; 10(11):805-11; PMID:19789556; https://doi.org/10.1038/nrg2651
- Barau J, Teissandier A, Zamudio N, Roy S, Nalesso V, Herault Y, Guillou F, Bourc'his D. The DNA methyltransferase DNMT3C protects male germ cells from transposon activity. Science 2016; 354(6314):909-912; PMID:27856912; https://doi.org/10.1126/science.aah5143
- Rhee I, Bachman KE, Park BH, Jair KW, Yen RW, Schuebel KE, Cui H, Feinberg AP, Lengauer C, Kinzler KW, et al. DNMT1 and DNMT3b cooperate to silence genes in human cancer cells. Nature 2002; 416(6880):552-6; PMID:11932749; https://doi.org/10.1038/416552a
- Rhee I, Jair KW, Yen RW, Lengauer C, Herman JG, Kinzler KW, Vogelstein B, Baylin SB, Schuebel KE. CpG methylation is maintained in human cancer cells lacking DNMT1. Nature 2000; 404(6781):1003-7; PMID:10801130; https://doi.org/10.1038/35010000
- Egger G, Jeong S, Escobar SG, Cortez CC, Li TW, Saito Y, Yoo CB, Jones PA, Liang G. Identification of DNMT1 (DNA methyltransferase 1) hypomorphs in somatic knockouts suggests an essential role for DNMT1 in cell survival. Proc Natl Acad Sci U S A 2006; 103(38):14080-5; PMID:16963560; https://doi.org/10.1073/pnas.0604602103
- Chen T, Ueda Y, Xie S, Li E. A novel Dnmt3a isoform produced from an alternative promoter localizes to euchromatin and its expression correlates with active de novo methylation. J Biol Chem 2002; 277(41):38746-54; PMID:12138111; https://doi.org/10.1074/jbc.M205312200
- Nimura K, Ishida C, Koriyama H, Hata K, Yamanaka S, Li E, Ura K, Kaneda Y. Dnmt3a2 targets endogenous Dnmt3L to ES cell chromatin and induces regional DNA methylation. Genes Cells 2006; 11(10):1225-37; PMID:16999741; https://doi.org/10.1111/j.1365-2443.2006.01012.x
- Jeong S, Liang G, Sharma S, Lin JC, Choi SH, Han H, Yoo CB, Egger G, Yang AS, Jones PA. Selective anchoring of DNA methyltransferases 3A and 3B to nucleosomes containing methylated DNA. Mol Cell Biol 2009; 29(19):5366-76; PMID:19620278; https://doi.org/10.1128/MCB.00484-09
- Ley TJ, Ding L, Walter MJ, McLellan MD, Lamprecht T, Larson DE, Kandoth C, Payton JE, Baty J, Welch J, et al. DNMT3A mutations in acute myeloid leukemia. N Engl J Med 2010; 363(25):2424-33; PMID:21067377; https://doi.org/10.1056/NEJMoa1005143
- Cancer Genome Atlas Research Network T. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med 2013; 368(22):2059-2074; PMID:23634996; https://doi.org/10.1056/NEJMoa1301689
- Aoki A, Suetake I, Miyagawa J, Fujio T, Chijiwa T, Sasaki H, Tajima S. Enzymatic properties of de novo-type mouse DNA (cytosine-5) methyltransferases. Nucleic Acids Res 2001; 29(17):3506-12; PMID:11522819; https://doi.org/10.1093/nar/29.17.3506
- Saito Y, Kanai Y, Sakamoto M, Saito H, Ishii H, Hirohashi S. Overexpression of a splice variant of DNA methyltransferase 3b, DNMT3b4, associated with DNA hypomethylation on pericentromeric satellite regions during human hepatocarcinogenesis. Proc Natl Acad Sci U S A 2002; 99(15):10060-5; PMID:12110732; https://doi.org/10.1073/pnas.152121799
- Ostler KR, Davis EM, Payne SL, Gosalia BB, Exposito-Cespedes J, Le Beau MM, Godley LA. Cancer cells express aberrant DNMT3B transcripts encoding truncated proteins. Oncogene 2007; 26(38):5553-63; PMID:17353906; https://doi.org/10.1038/sj.onc.1210351
- Gopalakrishnan S, Van Emburgh BO, Shan J, Su Z, Fields CR, Vieweg J, Hamazaki T, Schwartz PH, Terada N, Robertson KD. A novel DNMT3B splice variant expressed in tumor and pluripotent cells modulates genomic DNA methylation patterns and displays altered DNA binding. Mol Cancer Res 2009; 7(10):1622-34; PMID:19825994; https://doi.org/10.1158/1541-7786.MCR-09-0018
- Gordon CA, Hartono SR, Chedin F. Inactive DNMT3B splice variants modulate de novo DNA methylation. PLoS One 2013; 8(7):e69486; PMID:23894490; https://doi.org/10.1371/journal.pone.0069486
- Shao G, Zhang R, Zhang S, Jiang S, Liu Y, Zhang W, Zhang Y, Li J, Gong K, Hu XR, et al. Splice variants DNMT3B4 and DNMT3B7 overexpression inhibit cell proliferation in 293A cell line. In Vitro Cell Dev Biol Anim 2013; 49(5):386-94; PMID:23636939; https://doi.org/10.1007/s11626-013-9619-z
- Duymich CE, Charlet J, Yang X, Jones PA, Liang G. DNMT3B isoforms without catalytic activity stimulate gene body methylation as accessory proteins in somatic cells. Nat Commun 2016; 7:11453; PMID:27121154; https://doi.org/10.1038/ncomms11453
- Sharma S, De Carvalho DD, Jeong S, Jones PA, Liang G. Nucleosomes containing methylated DNA stabilize DNA methyltransferases 3A/3B and ensure faithful epigenetic inheritance. PLoS Genet 2011; 7(2):e1001286; PMID:21304883; https://doi.org/10.1371/journal.pgen.1001286
- Bhattacharya SK, Ramchandani S, Cervoni N, Szyf M. A mammalian protein with specific demethylase activity for mCpG DNA. Nature 1999; 397(6720):579-83; PMID:10050851; https://doi.org/10.1038/17533
- Ramchandani S, Bhattacharya SK, Cervoni N, Szyf M. DNA methylation is a reversible biological signal. Proc Natl Acad Sci U S A 1999; 96(11):6107-12; PMID:10339549; https://doi.org/10.1073/pnas.96.11.6107
- Wolffe AP, Jones PL, Wade PA. DNA demethylation. Proc Natl Acad Sci U S A 1999; 96(11):5894-6; PMID:10339513; https://doi.org/10.1073/pnas.96.11.5894
- Bird A. DNA methylation patterns and epigenetic memory. Genes Dev 2002; 16(1):6-21; PMID:11782440; https://doi.org/10.1101/gad.947102
- Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, Agarwal S, Iyer LM, Liu DR, Aravind L, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 2009; 324(5929):930-5; PMID:19372391; https://doi.org/10.1126/science.1170116
- Blaschke K, Ebata KT, Karimi MM, Zepeda-Martinez JA, Goyal P, Mahapatra S, Tam A, Laird DJ, Hirst M, Rao A, et al. Vitamin C induces Tet-dependent DNA demethylation and a blastocyst-like state in ES cells. Nature 2013; 500(7461):222-6; PMID:23812591; https://doi.org/10.1038/nature12362
- Ono R, Taki T, Taketani T, Taniwaki M, Kobayashi H, Hayashi Y. LCX, leukemia-associated protein with a CXXC domain, is fused to MLL in acute myeloid leukemia with trilineage dysplasia having t(10;11)(q22;q23). Cancer Res 2002; 62(14):4075-80; PMID:12124344
- Iyer LM, Tahiliani M, Rao A, Aravind L. Prediction of novel families of enzymes involved in oxidative and other complex modifications of bases in nucleic acids. Cell Cycle 2009; 8(11):1698-710; PMID:19411852; https://doi.org/10.4161/cc.8.11.8580
- Ito S, Shen L, Dai Q, Wu SC, Collins LB, Swenberg JA, He C, Zhang Y. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science 2011; 333(6047):1300-3; PMID:21778364; https://doi.org/10.1126/science.1210597
- Booth MJ, Branco MR, Ficz G, Oxley D, Krueger F, Reik W, Balasubramanian S. Quantitative sequencing of 5-methylcytosine and 5-hydroxymethylcytosine at single-base resolution. Science 2012; 336(6083):934-7; PMID:22539555; https://doi.org/10.1126/science.1220671
- Pfeifer GP, Kadam S, Jin SG. 5-hydroxymethylcytosine and its potential roles in development and cancer. Epigenetics Chromatin 2013; 6(1):10; PMID:23634848; https://doi.org/10.1186/1756-8935-6-10
- Mayland CR, Bennett MI, Allan K. Vitamin C deficiency in cancer patients. Palliat Med 2005; 19(1):17-20; PMID:15690864; https://doi.org/10.1191/0269216305pm970oa
- Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Siu IM, Gallia GL, et al. An integrated genomic analysis of human glioblastoma multiforme. Science 2008; 321(5897):1807-12; PMID:18772396; https://doi.org/10.1126/science.1164382
- Cohen AL, Holmen SL, Colman H. IDH1 and IDH2 mutations in gliomas. Curr Neurol Neurosci Rep 2013; 13(5):345; PMID:23532369; https://doi.org/10.1007/s11910-013-0345-4
- Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, Fantin VR, Jang HG, Jin S, Keenan MC, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2010; 465(7300):966; PMID:20559394; https://doi.org/10.1038/nature09132
- Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SH, Ito S, Yang C, Wang P, Xiao MT, et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell 2011; 19(1):17-30; PMID:21251613; https://doi.org/10.1016/j.ccr.2010.12.014
- Duncan CG, Barwick BG, Jin G, Rago C, Kapoor-Vazirani P, Powell DR, Chi JT, Bigner DD, Vertino PM, Yan H. A heterozygous IDH1R132H/WT mutation induces genome-wide alterations in DNA methylation. Genome Res 2012; 22(12):2339-55; PMID:22899282; https://doi.org/10.1101/gr.132738.111
- Turcan S, Rohle D, Goenka A, Walsh LA, Fang F, Yilmaz E, Campos C, Fabius AW, Lu C, Ward PS, et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature 2012; 483(7390):479-83; PMID:22343889; https://doi.org/10.1038/nature10866
- Mardis ER, Ding L, Dooling DJ, Larson DE, McLellan MD, Chen K, Koboldt DC, Fulton RS, Delehaunty KD, McGrath SD, et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med 2009; 361(11):1058-66; PMID:19657110; https://doi.org/10.1056/NEJMoa0903840
- Ward PS, Patel J, Wise DR, Abdel-Wahab O, Bennett BD, Coller HA, Cross JR, Fantin VR, Hedvat CV, Perl AE, et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell 2010; 17(3):225-34; PMID:20171147; https://doi.org/10.1016/j.ccr.2010.01.020
- Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, Li Y, Bhagwat N, Vasanthakumar A, Fernandez HF, et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 2010; 18(6):553-67; PMID:21130701; https://doi.org/10.1016/j.ccr.2010.11.015
- Marcucci G, Maharry K, Wu YZ, Radmacher MD, Mrozek K, Margeson D, Holland KB, Whitman SP, Becker H, Schwind S, et al. IDH1 and IDH2 gene mutations identify novel molecular subsets within de novo cytogenetically normal acute myeloid leukemia: a Cancer and Leukemia Group B study. J Clin Oncol 2010; 28(14):2348-55; PMID:20368543; https://doi.org/10.1200/JCO.2009.27.3730
- Tefferi A, Lasho TL, Abdel-Wahab O, Guglielmelli P, Patel J, Caramazza D, Pieri L, Finke CM, Kilpivaara O, Wadleigh M, et al. IDH1 and IDH2 mutation studies in 1473 patients with chronic-, fibrotic- or blast-phase essential thrombocythemia, polycythemia vera or myelofibrosis. Leukemia 2010; 24(7):1302-9; https://doi.org/10.1038/leu.2010.113
- Wagner K, Damm F, Gohring G, Gorlich K, Heuser M, Schafer I, Ottmann O, Lubbert M, Heit W, Kanz L, et al. Impact of IDH1 R132 mutations and an IDH1 single nucleotide polymorphism in cytogenetically normal acute myeloid leukemia: SNP rs11554137 is an adverse prognostic factor. J Clin Oncol 2010; 28(14):2356-64; PMID:20368538; https://doi.org/10.1200/JCO.2009.27.6899
- Cancer Genome Atlas Research Network T. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012; 487(7407):330-7; PMID:22810696; https://doi.org/10.1038/nature11252
- Cancer Genome Atlas N. Genomic Classification of Cutaneous Melanoma. Cell 2015; 161(7):1681-96; PMID:26091043; https://doi.org/10.1016/j.cell.2015.05.044
- Margueron R, Reinberg D. Chromatin structure and the inheritance of epigenetic information. Nat Rev Genet 2010; 11(4):285-96; PMID:20300089; https://doi.org/10.1038/nrg2752
- Jenuwein T, Allis CD. Translating the histone code. Science 2001; 293(5532):1074-80; PMID:11498575; https://doi.org/10.1126/science.1063127
- Bannister AJ, Kouzarides T. Regulation of chromatin by histone modifications. Cell Res 2011; 21(3):381-95; PMID:21321607; https://doi.org/10.1038/cr.2011.22
- Hon GC, Hawkins RD, Ren B. Predictive chromatin signatures in the mammalian genome. Hum Mol Genet 2009; 18(R2):R195-201; PMID:19808796; https://doi.org/10.1093/hmg/ddp409
- Heinz S, Romanoski CE, Benner C, Glass CK. The selection and function of cell type-specific enhancers. Nat Rev Mol Cell Biol 2015; 16(3):144-54; PMID:25650801; https://doi.org/10.1038/nrm3949
- Cedar H, Bergman Y. Linking DNA methylation and histone modification: patterns and paradigms. Nat Rev Genet 2009; 10(5):295-304; PMID:19308066; https://doi.org/10.1038/nrg2540
- Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, Fry B, Meissner A, Wernig M, Plath K, et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 2006; 125(2):315-26; PMID:16630819; https://doi.org/10.1016/j.cell.2006.02.041
- Ohm JE, McGarvey KM, Yu X, Cheng L, Schuebel KE, Cope L, Mohammad HP, Chen W, Daniel VC, Yu W, et al. A stem cell-like chromatin pattern may predispose tumor suppressor genes to DNA hypermethylation and heritable silencing. Nat Genet 2007; 39(2):237-42; PMID:17211412; https://doi.org/10.1038/ng1972
- Schlesinger Y, Straussman R, Keshet I, Farkash S, Hecht M, Zimmerman J, Eden E, Yakhini Z, Ben-Shushan E, Reubinoff BE, et al. Polycomb-mediated methylation on Lys27 of histone H3 pre-marks genes for de novo methylation in cancer. Nat Genet 2007; 39(2):232-6; PMID:17200670; https://doi.org/10.1038/ng1950
- Widschwendter M, Fiegl H, Egle D, Mueller-Holzner E, Spizzo G, Marth C, Weisenberger DJ, Campan M, Young J, Jacobs I, et al. Epigenetic stem cell signature in cancer. Nat Genet 2007; 39(2):157-8; PMID:17200673; https://doi.org/10.1038/ng1941
- Gal-Yam EN, Egger G, Iniguez L, Holster H, Einarsson S, Zhang X, Lin JC, Liang G, Jones PA, Tanay A. Frequent switching of Polycomb repressive marks and DNA hypermethylation in the PC3 prostate cancer cell line. Proc Natl Acad Sci U S A 2008; 105(35):12979-84; PMID:18753622; https://doi.org/10.1073/pnas.0806437105
- Pandiyan K, You JS, Yang X, Dai C, Zhou XJ, Baylin SB, Jones PA, Liang G. Functional DNA demethylation is accompanied by chromatin accessibility. Nucleic Acids Res 2013; 41(7):3973-85; PMID:23408854; https://doi.org/10.1093/nar/gkt077
- Shen H, Laird PW. Interplay between the cancer genome and epigenome. Cell 2013; 153(1):38-55; PMID:23540689; https://doi.org/10.1016/j.cell.2013.03.008
- Shimbo T, Wade PA. Proteins that read DNA methylation. In: Jeltsch A, Jurkowska RA, editors. Advances in Experimental Medicine and Biology. DNA methyltransferases - Role and Function. Volume 945: Springer; 2016. p 303-20
- Adams BD, Kasinski AL, Slack FJ. Aberrant regulation and function of microRNAs in cancer. Curr Biol 2014; 24(16):R762-76; PMID:25137592; https://doi.org/10.1016/j.cub.2014.06.043
- Iorio MV, Croce CM. MicroRNA dysregulation in cancer: diagnostics, monitoring and therapeutics. A comprehensive review. EMBO Mol Med 2012; 4(3):143-59; PMID:22351564; https://doi.org/10.1002/emmm.201100209
- Lopez-Serra P, Esteller M. DNA methylation-associated silencing of tumor-suppressor microRNAs in cancer. Oncogene 2012; 31(13):1609-22; PMID:21860412; https://doi.org/10.1038/onc.2011.354
- Amodio N, Rossi M, Raimondi L, Pitari MR, Botta C, Tagliaferri P, Tassone P. miR-29s: a family of epi-miRNAs with therapeutic implications in hematologic malignancies. Oncotarget 2015; 6(15):12837-61; PMID:25968566; https://doi.org/10.18632/oncotarget.3805
- Saito Y, Liang G, Egger G, Friedman JM, Chuang JC, Coetzee GA, Jones PA. Specific activation of microRNA-127 with downregulation of the proto-oncogene BCL6 by chromatin-modifying drugs in human cancer cells. Cancer Cell 2006; 9(6):435-43; PMID:16766263; https://doi.org/10.1016/j.ccr.2006.04.020
- Damiano V, Brisotto G, Borgna S, di Gennaro A, Armellin M, Perin T, Guardascione M, Maestro R, Santarosa M. Epigenetic silencing of miR-200c in breast cancer is associated with aggressiveness and is modulated by ZEB1. Genes Chromosomes Cancer 2017; 56(2):147-158; PMID:27717206; https://doi.org/10.1002/gcc.22422
- Zheng K, Zhou X, Yu J, Li Q, Wang H, Li M, Shao Z, Zhang F, Luo Y, Shen Z, et al. Epigenetic silencing of miR-490-3p promotes development of an aggressive colorectal cancer phenotype through activation of the Wnt/beta-catenin signaling pathway. Cancer Lett 2016; 376(1):178-87; PMID:27037061; https://doi.org/10.1016/j.canlet.2016.03.024
- Li HP, Huang HY, Lai YR, Huang JX, Chang KP, Hsueh C, Chang YS. Silencing of miRNA-148a by hypermethylation activates the integrin-mediated signaling pathway in nasopharyngeal carcinoma. Oncotarget 2014; 5(17):7610-24; PMID:25277193; https://doi.org/10.18632/oncotarget.2282
- Chen BF, Suen YK, Gu S, Li L, Chan WY. A miR-199a/miR-214 self-regulatory network via PSMD10, TP53 and DNMT1 in testicular germ cell tumor. Sci Rep 2014; 4:6413; PMID:25231260; https://doi.org/10.1038/srep06413
- Friedman JM, Liang G, Liu CC, Wolff EM, Tsai YC, Ye W, Zhou X, Jones PA. The putative tumor suppressor microRNA-101 modulates the cancer epigenome by repressing the polycomb group protein EZH2. Cancer Res 2009; 69(6):2623-9; PMID:19258506; https://doi.org/10.1158/0008-5472.CAN-08-3114
- Fabbri M, Garzon R, Cimmino A, Liu Z, Zanesi N, Callegari E, Liu S, Alder H, Costinean S, Fernandez-Cymering C, et al. MicroRNA-29 family reverts aberrant methylation in lung cancer by targeting DNA methyltransferases 3A and 3B. Proc Natl Acad Sci U S A 2007; 104(40):15805-10; PMID:17890317; https://doi.org/10.1073/pnas.0707628104
- Shortt J, Ott CJ, Johnstone RW, Bradner JE. A chemical probe toolbox for dissecting the cancer epigenome. Nat Rev Cancer 2017; 17(3):160-183; PMID:28228643; https://doi.org/10.1038/nrc.2016.148
- Raynal NJ-M, Issa JP. DNA methyltransferase inhibitors. In: Egger G, Arimondo P, editors. Drug Discovery in Cancer Epigenetics. Waltham, MA: Academic Press; 2016. p 169-190
- Jones PA, Taylor SM. Cellular differentiation, cytidine analogs and DNA methylation. Cell 1980; 20(1):85-93; PMID:6156004; https://doi.org/10.1016/0092-8674(80)90237-8
- Chen JC, Goldhamer DJ. Transcriptional mechanisms regulating MyoD expression in the mouse. Cell Tissue Res 1999; 296(1):213-9; PMID:10199981; https://doi.org/10.1007/s004410051282
- Ghoshal K, Datta J, Majumder S, Bai S, Kutay H, Motiwala T, Jacob ST. 5-Aza-deoxycytidine induces selective degradation of DNA methyltransferase 1 by a proteasomal pathway that requires the KEN box, bromo-adjacent homology domain, and nuclear localization signal. Mol Cell Biol 2005; 25(11):4727-41; PMID:15899874; https://doi.org/10.1128/MCB.25.11.4727-4741.2005
- Christman JK. 5-Azacytidine and 5-aza-2′-deoxycytidine as inhibitors of DNA methylation: mechanistic studies and their implications for cancer therapy. Oncogene 2002; 21(35):5483-95; PMID:12154409; https://doi.org/10.1038/sj.onc.1205699
- Zhou L, Cheng X, Connolly BA, Dickman MJ, Hurd PJ, Hornby DP. Zebularine: a novel DNA methylation inhibitor that forms a covalent complex with DNA methyltransferases. J Mol Biol 2002; 321(4):591-9; PMID:12206775; https://doi.org/10.1016/S0022-2836(02)00676-9
- Cheng JC, Yoo CB, Weisenberger DJ, Chuang J, Wozniak C, Liang G, Marquez VE, Greer S, Orntoft TF, Thykjaer T, et al. Preferential response of cancer cells to zebularine. Cancer Cell 2004; 6(2):151-8; PMID:15324698; https://doi.org/10.1016/j.ccr.2004.06.023
- Cheng JC, Matsen CB, Gonzales FA, Ye W, Greer S, Marquez VE, Jones PA, Selker EU. Inhibition of DNA methylation and reactivation of silenced genes by zebularine. J Natl Cancer Inst 2003; 95(5):399-409; PMID:12618505; https://doi.org/10.1093/jnci/95.5.399
- Cheng JC, Weisenberger DJ, Gonzales FA, Liang G, Xu GL, Hu YG, Marquez VE, Jones PA. Continuous zebularine treatment effectively sustains demethylation in human bladder cancer cells. Mol Cell Biol 2004; 24(3):1270-8; PMID:14729971; https://doi.org/10.1128/MCB.24.3.1270-1278.2004
- Dote H, Cerna D, Burgan WE, Carter DJ, Cerra MA, Hollingshead MG, Camphausen K, Tofilon PJ. Enhancement of in vitro and in vivo tumor cell radiosensitivity by the DNA methylation inhibitor zebularine. Clin Cancer Res 2005; 11(12):4571-9; PMID:15958643; https://doi.org/10.1158/1078-0432.CCR-05-0050
- Holleran JL, Parise RA, Joseph E, Eiseman JL, Covey JM, Glaze ER, Lyubimov AV, Chen YF, D'Argenio DZ, Egorin MJ. Plasma pharmacokinetics, oral bioavailability, and interspecies scaling of the DNA methyltransferase inhibitor, zebularine. Clin Cancer Res 2005; 11(10):3862-8; PMID:15897587; https://doi.org/10.1158/1078-0432.CCR-04-2406
- Chuang JC, Warner SL, Vollmer D, Vankayalapati H, Redkar S, Bearss DJ, Qiu X, Yoo CB, Jones PA. S110, a 5-Aza-2′-deoxycytidine-containing dinucleotide, is an effective DNA methylation inhibitor in vivo and can reduce tumor growth. Mol Cancer Ther 2010; 9(5):1443-50; PMID:20442312; https://doi.org/10.1158/1535-7163.MCT-09-1048
- Yoo CB, Jeong S, Egger G, Liang G, Phiasivongsa P, Tang C, Redkar S, Jones PA. Delivery of 5-aza-2′-deoxycytidine to cells using oligodeoxynucleotides. Cancer Res 2007; 67(13):6400-8; PMID:17616700; https://doi.org/10.1158/0008-5472.CAN-07-0251
- Kuang Y, El-Khoueiry A, Taverna P, Ljungman M, Neamati N. Guadecitabine (SGI-110) priming sensitizes hepatocellular carcinoma cells to oxaliplatin. Mol Oncol 2015; 9(9):1799-814; PMID:26160429; https://doi.org/10.1016/j.molonc.2015.06.002
- Issa JP, Roboz G, Rizzieri D, Jabbour E, Stock W, O'Connell C, Yee K, Tibes R, Griffiths EA, Walsh K, et al. Safety and tolerability of guadecitabine (SGI-110) in patients with myelodysplastic syndrome and acute myeloid leukaemia: a multicentre, randomised, dose-escalation phase 1 study. Lancet Oncol 2015; 16(9):1099-110; PMID:26296954; https://doi.org/10.1016/S1470-2045(15)00038-8
- Chuang JC, Yoo CB, Kwan JM, Li TW, Liang G, Yang AS, Jones PA. Comparison of biological effects of non-nucleoside DNA methylation inhibitors versus 5-aza-2′-deoxycytidine. Mol Cancer Ther 2005; 4(10):1515-20; PMID:16227400; https://doi.org/10.1158/1535-7163.MCT-05-0172
- Velicescu M, Weisenberger DJ, Gonzales FA, Tsai YC, Nguyen CT, Jones PA. Cell division is required for de novo methylation of CpG islands in bladder cancer cells. Cancer Res 2002; 62(8):2378-84; PMID:11956100
- Baylin SB. DNA methylation and gene silencing in cancer. Nat Clin Pract Oncol 2005; 2 Suppl 1:S4-11; PMID:16341240; https://doi.org/10.1038/ncponc0354
- Hegi ME, Diserens AC, Gorlia T, Hamou MF, de Tribolet N, Weller M, Kros JM, Hainfellner JA, Mason W, Mariani L, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med 2005; 352(10):997-1003; PMID:15758010; https://doi.org/10.1056/NEJMoa043331
- Tsai HC, Li H, Van Neste L, Cai Y, Robert C, Rassool FV, Shin JJ, Harbom KM, Beaty R, Pappou E, et al. Transient low doses of DNA-demethylating agents exert durable antitumor effects on hematological and epithelial tumor cells. Cancer Cell 2012; 21(3):430-46; PMID:22439938; https://doi.org/10.1016/j.ccr.2011.12.029
- Kasinathan S, Henikoff S. 5-Aza-CdR delivers a gene body blow. Cancer Cell 2014; 26(4):449-51; PMID:25314073; https://doi.org/10.1016/j.ccell.2014.09.004
- Chiappinelli KB, Strissel PL, Desrichard A, Li H, Henke C, Akman B, Hein A, Rote NS, Cope LM, Snyder A, et al. Inhibiting DNA methylation causes an interferon response in cancer via dsRNA including endogenous retroviruses. Cell 2016; 164(5):1073; PMID:27064190; https://doi.org/10.1016/j.cell.2015.10.020
- Roulois D, Loo Yau H, Singhania R, Wang Y, Danesh A, Shen SY, Han H, Liang G, Jones PA, Pugh TJ, et al. DNA-Demethylating agents target colorectal cancer cells by inducing viral mimicry by endogenous transcripts. Cell 2015; 162(5):961-73; PMID:26317465; https://doi.org/10.1016/j.cell.2015.07.056
- Liu M, Ohtani H, Zhou W, Orskov AD, Charlet J, Zhang YW, Shen H, Baylin SB, Liang G, Gronbaek K, et al. Vitamin C increases viral mimicry induced by 5-aza-2′-deoxycytidine. Proc Natl Acad Sci U S A 2016; 113(37):10238-44; PMID:27573823; https://doi.org/10.1073/pnas.1612262113
- Ikehata M, Ogawa M, Yamada Y, Tanaka S, Ueda K, Iwakawa S. Different effects of epigenetic modifiers on the cytotoxicity induced by 5-fluorouracil, irinotecan or oxaliplatin in colon cancer cells. Biol Pharm Bull 2014; 37(1):67-73; PMID:24172061; https://doi.org/10.1248/bpb.b13-00574
- Wrangle J, Wang W, Koch A, Easwaran H, Mohammad HP, Vendetti F, Vancriekinge W, Demeyer T, Du Z, Parsana P, et al. Alterations of immune response of non-small cell lung cancer with azacytidine. Oncotarget 2013; 4(11):2067-79; PMID:24162015; https://doi.org/10.18632/oncotarget.1542
- Muvarak NE, Chowdhury K, Xia L, Robert C, Choi EY, Cai Y, Bellani M, Zou Y, Singh ZN, Duong VH, et al. Enhancing the cytotoxic effects of PARP Inhibitors with DNA demethylating agents - a potential therapy for cancer. Cancer Cell 2016; 30(4):637-650; PMID:27728808; https://doi.org/10.1016/j.ccell.2016.09.002
- Ahuja N, Sharma AR, Baylin SB. Epigenetic therapeutics: a new weapon in the war against cancer. Annu Rev Med 2016; 67:73-89; PMID:26768237; https://doi.org/10.1146/annurev-med-111314-035900
- Diesch J, Zwick A, Garz AK, Palau A, Buschbeck M, Gotze KS. A clinical-molecular update on azanucleoside-based therapy for the treatment of hematologic cancers. Clin Epigenetics 2016; 8:71; PMID:27330573; https://doi.org/10.1186/s13148-016-0237-y
- Fenaux P, Mufti GJ, Hellstrom-Lindberg E, Santini V, Gattermann N, Germing U, Sanz G, List AF, Gore S, Seymour JF, et al. Azacitidine prolongs overall survival compared with conventional care regimens in elderly patients with low bone marrow blast count acute myeloid leukemia. J Clin Oncol 2010; 28(4):562-9; PMID:20026804; https://doi.org/10.1200/JCO.2009.23.8329
- Pleyer L, Burgstaller S, Girschikofsky M, Linkesch W, Stauder R, Pfeilstocker M, Schreder M, Tinchon C, Sliwa T, Lang A, et al. Azacitidine in 302 patients with WHO-defined acute myeloid leukemia: results from the Austrian Azacitidine Registry of the AGMT-study group. Ann Hematol 2014; 93(11):1825-38; PMID:24951123; https://doi.org/10.1007/s00277-014-2126-9
- Thepot S, Itzykson R, Seegers V, Recher C, Raffoux E, Quesnel B, Delaunay J, Cluzeau T, Marfaing Koka A, Stamatoullas A, et al. Azacitidine in untreated acute myeloid leukemia: a report on 149 patients. Am J Hematol 2014; 89(4):410-6; PMID:24375487; https://doi.org/10.1002/ajh.23654
- Dombret H, Seymour JF, Butrym A, Wierzbowska A, Selleslag D, Jang JH, Kumar R, Cavenagh J, Schuh AC, Candoni A, et al. International phase 3 study of azacitidine vs conventional care regimens in older patients with newly diagnosed AML with >30% blasts. Blood 2015; 126(3):291-9; PMID:25987659; https://doi.org/10.1182/blood-2015-01-621664
- Juergens RA, Wrangle J, Vendetti FP, Murphy SC, Zhao M, Coleman B, Sebree R, Rodgers K, Hooker CM, Franco N, et al. Combination epigenetic therapy has efficacy in patients with refractory advanced non-small cell lung cancer. Cancer Discov 2011; 1(7):598-607; PMID:22586682; https://doi.org/10.1158/2159-8290.CD-11-0214
- Connolly R, Li H, Jankowitz RC, Zhang Z, Rudek MA, Jeter SC, Slater S, Powers P, Wolff AC, Fetting JH, et al. Combination epigenetic therapy in advanced breast cancer with 5-azacitidine and entinostat: a phase ii national cancer institute/stand up to cancer study. Clin Cancer Res 2016; PMID:27979916; https://doi.org/10.1158/1078-0432.CCR-16-1729
- Fu S, Hu W, Iyer R, Kavanagh JJ, Coleman RL, Levenback CF, Sood AK, Wolf JK, Gershenson DM, Markman M, et al. Phase 1b-2a study to reverse platinum resistance through use of a hypomethylating agent, azacitidine, in patients with platinum-resistant or platinum-refractory epithelial ovarian cancer. Cancer 2011; 117(8):1661-9; PMID:21472713; https://doi.org/10.1002/cncr.25701
- Scartozzi M, Bearzi I, Mandolesi A, Giampieri R, Faloppi L, Galizia E, Loupakis F, Zaniboni A, Zorzi F, Biscotti T, et al. Epidermal growth factor receptor (EGFR) gene promoter methylation and cetuximab treatment in colorectal cancer patients. Br J Cancer 2011; 104(11):1786-90; PMID:21559018; https://doi.org/10.1038/bjc.2011.161
- Garrido-Laguna I, McGregor KA, Wade M, Weis J, Gilcrease W, Burr L, Soldi R, Jakubowski L, Davidson C, Morrell G, et al. A phase I/II study of decitabine in combination with panitumumab in patients with wild-type (wt) KRAS metastatic colorectal cancer. Invest New Drugs 2013; 31(5):1257-64; PMID:23504398; https://doi.org/10.1007/s10637-013-9947-6
- Azad NS, El-Khoueiry A, Yin J, Oberg AL, Flynn P, Adkins D, Sharma A, Weisenberger DJ, Brown T, Medvari P, et al. Combination epigenetic therapy in metastatic colorectal cancer (mCRC) with subcutaneous 5-azacitidine and entinostat; a phase 2 consortium/stand Up 2 cancer study. Oncotarget 2017; PMID:28186961; https://doi.org/10.18632/oncotarget.15108