
ABSTRACT
Endocrine disrupting chemicals (EDCs) pose a public health risk through disruption of normal biological processes. Identifying toxicoepigenetic mechanisms of developmental exposure-induced effects for EDCs, such as phthalates or bisphenol A (BPA), is essential. Here, we investigate whether maternal exposure to EDCs is predictive of infant DNA methylation at candidate gene regions. In the Michigan Mother-Infant Pairs (MMIP) cohort, DNA was extracted from cord blood leukocytes for methylation analysis by pyrosequencing (n = 116) and methylation changes related to first trimester levels of 9 phthalate metabolites and BPA. Growth and metabolism-related genes selected for methylation analysis included imprinted (IGF2, H19) and non-imprinted (PPARA, ESR1) genes along with LINE-1 repetitive elements. Findings revealed decreases in methylation of LINE-1, IGF2, and PPARA with increasing phthalate concentrations. For example, a log unit increase in ΣDEHP corresponded to a 1.03 [95% confidence interval (CI): −1.83, −0.22] percentage point decrease in PPARA methylation. Changes in DNA methylation were also inversely correlated with PPARA gene expression determined by RT-qPCR (r = −0.34, P = 0.02), thereby providing evidence in support of functional relevance. A sex-stratified analysis of EDCs and DNA methylation showed that some relationships were female-specific. For example, urinary BPA exposure was associated with a 1.35 (95%CI: −2.69, −0.01) percentage point decrease in IGF2 methylation and a 1.22 (95%CI: −2.27, −0.16) percentage point decrease in PPARA methylation in females only. These findings add to a body of evidence suggesting epigenetically labile regions may provide a conduit linking early exposures with disease risk later in life and that toxicoepigenetic susceptibility may be sex specific.
Introduction
Endocrine disrupting chemicals (EDCs) are ubiquitous in modern society due to their widespread use in consumer products (e.g., food and beverage containers), personal care products (e.g., lotion), and medical supplies (e.g., plastic tubing). Phthalates and bisphenol A (BPA) are two of the most commonly studied EDCs, and both human and animal studies suggest these chemicals can disrupt normal biological processes [Citation1]. BPA can impact endocrine function by acting as an estrogenic [Citation2] and anti-androgenic compound [Citation3]. In addition, BPA and some phthalates can alter the thyroid hormone balance [Citation4], which can contribute to metabolic dysfunction in adults [Citation5]. While exposure at any life stage may be impactful, the developing human fetus is particularly vulnerable to EDC exposure as this is a critical period of rapid growth, and perturbations during this time can have lasting effects on disease susceptibility [Citation6]. To date, the exposure-induced phenotypes reported in the literature, with regard to BPA and phthalates, have been inconsistent with the direction of association differing by population, age, and sex [Citation7]. Nevertheless, multiple cohort studies have found that prenatal exposures to these toxicants are associated with growth-related outcomes [Citation8–Citation11]. A growing body of animal data bolsters these epidemiological findings. Specifically, studies in rodents have demonstrated that perinatal exposure to BPA is associated with persistent changes in body weight, food intake or preference, and hormone levels [Citation12–Citation16]. Therefore, with a global obesity epidemic as a backdrop, it is imperative to identify aberrant exposure-induced programing events that have the potential to shape metabolic outcomes later in life.
Epigenetic modifications such as DNA methylation are mitotically heritable alterations capable of modulating gene expression without changing the underlying DNA sequence [Citation17]. As such, DNA methylation is at the interface of genetics and the fetal environment with the ability to act as an adaptive layer of regulatory control for gene expression and retrotransposon repression [Citation18,Citation19]. Further, evidence suggests that toxicoepigenetic changes can occur in response to BPA and phthalates [Citation20]. Using the viable yellow agouti mouse model, we have demonstrated that perinatal BPA exposure alters coat color distribution with associated shifts in DNA methylation profiles in the offspring [Citation21,Citation22]. Observations from human cohort studies suggest BPA can influence epigenetic programming of fetal liver enzymes [Citation23] and imprinted genes [Citation24]. Interestingly, perinatal BPA-induced epigenetic effects are commonly sex specific [Citation25] and linked to DNA methylation changes at a number of loci including genes involved in liver beta oxidation [Citation26], energy homeostasis [Citation27], and growth and metabolism [Citation24,Citation28]. Maternal exposure to phthalates averaged across pregnancy has been found to be inversely associated with methylation profiles of retrotransposons Alu and LINE-1 in a population of Mexican-American children [Citation29]. Studies with the ELEMENT Mexico City birth cohort also found maternal phthalate exposure in the third trimester is associated with altered methylation of H19 [Citation24], which is involved in body composition and growth [Citation30]. Given the growing evidence demonstrating the toxicoepigenetic potential of EDCs along with the potential developmental contribution to metabolic-related disease risk, there exists a clear need to identify molecular biomarkers of exposure. Identification of EDC sensitive epigenetic biomarkers will aid in elucidating the developmental origins of metabolic diseases. Therefore, using a biologically accessible source of surrogate DNA and RNA, the goal of this study was to determine whether first trimester maternal exposures to EDCs are predictive of newborn DNA methylation and whether DNA methylation is associated with gene expression levels at growth and metabolism-related candidate regions.
Methods
Study population
Women were recruited between 2010 and 2015 during their first trimester of pregnancy as part of the Michigan Mother Infant-Pairs (MMIP) project, an ongoing birth cohort. Prospective participants were informed of the study during their first prenatal visit at a University of Michigan clinic, and were eligible if they were 18 years of age or older, conceived naturally, and had a singleton pregnancy. Women provided spot urine and venous blood samples during their first trimester prenatal visit (8–14 weeks). Infant cord blood was collected at time of birth. The University of Michigan Medical School Institutional Review Board approved this study, and all women provided written informed consent prior to participation.
Phthalate metabolite and BPA measurement
Spot urine samples were collected into polypropylene urine collection containers, aliquoted into glass vials, and frozen at −80°C until analysis. Urinary BPA (56 out of the cohort of 116) and nine phthalate metabolites (109 out of the cohort of 116), comprising monoethyl phthalate (MEP), mono-n-butyl phthalate (MnBP), monoisobutyl phthalate (MiBP), monobenzyl phthalate (MBzP), mono-3-carboxypropyl phthalate (MCPP), mono-2-ethylhexyl phthalate (MEHP), mono-2-ethyl- 5-hydroxyhexyl phthalate (MEHHP), mono-2-ethyl-5-oxohexyl phthalate (MEOHP), and mono-2-ethyl-5-carboxypentyl phthalate (MECPP), were measured at NSF International in two batches (Ann Arbor, MI) using isotope dilution–liquid chromatography–tandem mass spectrometry (ID–LC–MS/MS) as previously described [Citation31]. Summary measures for parent compounds di-(2-ethylhexyl) phthalate (ΣDEHP) and dibutyl phthalate (ΣDBP) for each sample were calculated by dividing their respective individual metabolite concentrations by their molar mass and summing them. The ΣDEHP measure comprised MEHP, MEHHP, MEOHP, and MECPP, while the ΣDBP measure comprised MnBP and MiBP. Specific gravity (SG) was measured using a handheld digital refractometer (Atago Co., Ltd., Tokyo, Japan) at the time of sample analysis. Values below the limit of detection (LOD) were replaced with LOD/√2. While the maternal participants recruited in the second half of the MMIP study had total urinary BPA measured at NSF International, the participants in the first half of the study had unconjugated plasma BPA (n = 60 out of the cohort of 116) measured in plasma at the Wadsworth Center (Albany, NY) as previously described [Citation32]. Plasma BPA processing was performed following collection and analysis methods developed and validated by four independent laboratories [Citation33]. The geometric mean and standard deviation for BPA concentrations measured in the two matrices were similar (); however, because no subject had both matrices analyzed, we have no way of doing a head-to-head comparison. Therefore, plasma BPA and urinary BPA were considered distinct exposure measurements and not one combined exposure measurement.
Table 1. Population statistics.
Data collection for covariates and potentially confounding variables
The infant's sex, gestational age, and birth weight as well as maternal pre-pregnancy weight, height, and age were taken from the medical records. Maternal pre-pregnancy weight and height were used to calculate maternal body mass index (BMI). Self-reported ethnicity, maternal smoking status, as well as household income were collected via survey.
DNA isolation and methylation analysis
Infant cord blood was collected into Paxgene Blood DNA and RNA tubes (PreAnalytix) at the time of birth and stored at −80°C until processing. Total genomic DNA was isolated using the Paxgene Blood DNA kit. Genomic DNA was bisulfite converted using the EZ-96 DNA Methylation Kit (Zymo). Briefly, sodium bisulfite was added to approximately 500 ng of genomic DNA, converting unmethylated cytosines to uracil, which are replaced with thymine during PCR; methylated cytosines remain unchanged [Citation34]. For this study, we selected imprinted (IGF2, H19) and non-imprinted (PPARA, ESR1) genes along with LINE-1 repetitive elements as potential candidate regions of epigenetic lability. Given its prevalence across the genome, LINE-1 can be used as a surrogate for global DNA methylation levels [Citation35]. Apart from LINE-1, all of the other interrogated genes play a role in metabolism, growth, or development. Insulin-like growth factor II (IGF2) and H19 are well-characterized imprinted genes, in which parent-of-origin monoallelic expression is involved in the regulation of body composition and growth [Citation30,Citation36–Citation38]. PPARA is a non-imprinted gene that encodes the peroxisome proliferator-activated receptor alpha (PPAR-α) protein, a nuclear receptor that regulates fatty acid metabolism [Citation39–Citation41]. ESR1 is a non-imprinted gene that encodes estrogen receptor alpha (ER-α), a transcription factor involved in regulation of energy homeostasis [Citation42]. The gene-specific PCR and pyrosequencing primers and conditions for the target regions are listed in Supplemental Table 1. PCR amplification was performed after bisulfite conversion using HotStarTaq master mix (Qiagen), forward primer (50 pmol), and reverse biotinylated primer (50 pmol) in a 30 μl reaction. PCR fragments were analyzed by gel electrophoresis or automated capillary electrophoresis using the Qiaxcel Advanced System (Qiagen). DNA methylation quantification of CpG sites was performed using pyrosequencing on a PyroMark ID instrument (Qiagen). To determine percent methylation, PyroMark software calculated the fraction of methylated cytosines (%mC) among the total sum of methylated and unmethylated cytosines. For quality assurance, all pyrosequencing plates included 0, 50, and 100% methylated bisulfite converted human control DNA (Qiagen), as well as at least one no DNA template control. A subset of the samples from every 96-well plate was run in technical duplicate to calculate a coefficient of variation (CV) and if the average %CV was greater than 5% the plate was repeated.
Real-time quantitative PCR (RT-qPCR)
RNA was extracted from whole cord blood samples using the Paxgene Blood RNA kit (PreAnalytix). RNA concentration and purity were checked using a NanoDrop 2000 spectrophotometer (Thermo Scientific, Wilmington, DE). Complementary DNA (cDNA) synthesis was performed on 1 µg RNA template for each sample with the Bio Rad iScript cDNA Synthesis kit (Hercules, CA) according to manufacturer's instructions. Each cDNA sample was diluted 1:2 and added to a mixture of gene-specific forward and reverse primer, nuclease-free water, and iQ SYBR Green Supermix followed by 2-step PCR + melt detection on a Bio-Rad CFx96 system (Hercules, CA) using the following parameters: one cycle of 95°C for 3 minutes, followed by 45 cycles of [95°C for 10 seconds, 55°C for 30 seconds, plate read], and finally one cycle of 95°C for 10 seconds. The melt curve for each plate was 65°C - 95°C; 5°C increment for 5 seconds, with plate read at each temperature. For RT-qPCR analysis, individual samples were run in triplicate for PPARA and three housekeeping genes [Beta-actin (B-actin), Glyceraldehyde 3-phosphate dehydrogenase (GAPDH), and Tyrosine 3-Monooxygenase/Tryptophan 5-Monooxygenase Activation Protein Zata (YWHAZ)]. In addition to housekeeping genes, an inter-plate control and genomic DNA control were included to improve quality assurance and facilitate the calculation of relative expression using the 2−ΔΔ Ct method [Citation43]. The gene-specific primer sets used and their respective literature sources are listed in Supplementary Table 2. Primer pair specificity for all designed primers was checked using the NCBI Primer-BLAST online tool (https://www.ncbi.nlm.nih.gov/tools/primer-blast/).
Statistical methods
DNA methylation analysis was conducted using the average methylation value for all measured CpGs for each candidate gene. To adjust for heterogeneity in variance, BPA and phthalate were ln-transformed prior to regression analysis. The distribution of ln-transformed EDCs was examined across categories of sociodemographic and perinatal characteristics using simple linear regression for continuous variables and ANOVA with post-hoc Tukey HSD for categorical variables to identify potential confounders. To adjust for urine dilution in linear regression analyses, urinary SG was included as a covariate in models using continuous urinary EDC variables, as we have done previously [Citation31]. In fully adjusted models, the child‘s sex, maternal pre-pregnancy body mass index (BMI) and maternal age were included as potential confounders based on a priori expectations informed by previous studies and current literature. This method of analysis was used to evaluate the relationship between EDC exposure and DNA methylation. Other covariates that were considered included household income and smoking status. When income was included in the model, the effect sizes did not change appreciably (>10%) and therefore was not included in the final model. Similarly, smoking status was not included because the majority of women were non-smokers. The results of the multivariable linear regression model for DNA methylation are presented as an absolute percentage point change in DNA methylation outcome [95% confidence interval (CI)] per log unit increase in continuous EDC value. PPARA relative expression values were log-normally distributed and ln-transformed. Spearman's rank correlation test was used to evaluate the relationship between PPARA methylation and PPARA gene expression. Given that the literature suggests EDC-induced effects may be sexually dimorphic, sex-stratified associations between exposure and methylation outcomes were also investigated.
Results
Demographics, exposure distribution, and DNA methylation
summarizes maternal and infant population characteristics, maternal first trimester EDC levels, and infant cord blood DNA methylation. Women in the present analysis represent 116 participants in the MMIP cohort study. This population of women was 31 years of age on average, mostly white, slightly overweight, and non-smoking. The average infant birth weight and gestational age was 3510 grams and 39.7 weeks, respectively. Overall, EDC measurements varied widely across this population. Plasma BPA had a wider range (0.14 to 96.43 ng/ml) relative to urinary BPA (0.04 to 4.76 ng/ml), while the geometric means were comparable [0.57 ng/ml (SD 4.72) and 0.78 ng/ml (5.47), respectively]. Measured levels of MBzP, MCPP, and MEP ranged from 0.25 to 78.8 ng/ml, 0.35 to 351.85 ng/ml, and 1.28 to 1,598.00 ng/ml, respectively. After conversion to the sum of their phthalate metabolites, the geometric mean of ΣDEHP and ΣDBP were similar [0.09 nMol (SD 0.11) and 0.08 nMol (0.09), respectively]. The inter-individual variation across CpG loci for each of the candidate genes [ESR1 (4 sites), PPARA (2 sites), H19 (4 sites), IGF2 (3 sites), and LINE-1 (4 sites)] was consistent; therefore, average methylation level across CpGs for each gene was used in all analysis. The mean DNA methylation of LINE-1, ESR1, IGF2 PPARA, and H19 was 79.97 (SD 3.22), 4.31 (0.96), 47.85 (3.68), 17.29 (2.66), and 55.60 (4.94), respectively. The mean DNA methylation of LINE-1, ESR1, IGF2 or PPARA did not significantly vary by infant sex (Δ relative to males, P value for t test; Δ = −0.07, P = 0.91; Δ = −0.13, P = 0.52; Δ = 1.32, P = 0.08; Δ = 0.22, P = 0.68, respectively). However, the mean DNA methylation of H19 was 2.53 percentage points higher (P = 0.01) in females relative to males.
Maternal EDC levels are related to cohort-wide infant DNA methylation
Several significant relationships were observed between maternal first trimester EDC levels and infant DNA methylation profiles, adjusting for child's sex, maternal pre-pregnancy BMI, maternal age, and urinary SG (). Interestingly, all of the significant associations were observed for phthalate metabolites, and these associations were all negative. For instance, a log unit increase in MCPP was associated with a 0.60 (95%CI: −1.14, −0.06) percentage point decrease in LINE-1 methylation and a 0.83 (95%CI: −1.52, −0.15) percentage point decrease in IGF2 methylation. Similarly, MBzP, MCPP and ΣDEHP were each negatively associated with PPARA methylation. For example, a log unit increase in ΣDEHP corresponded to 1.03 (95%CI: −1.83, −0.22) percentage point decrease in PPARA methylation with similar associations observed for MBzP and MCPP (). Similar to the direction of effect for the urinary phthalate metabolites, there was a borderline significant inverse relationship between urinary BPA and ESR1 methylation [−0.38 (95%CI: −0.78, 0.01)]. However, BPA measured in either urine or plasma was largely not associated (P > 0.05) with all other candidate gene nor LINE-1 methylation.
Table 2. Absolute change in infant DNA methylation per log unit increase in maternal EDC exposure.
Infant methylation patterns are correlated with candidate gene expression
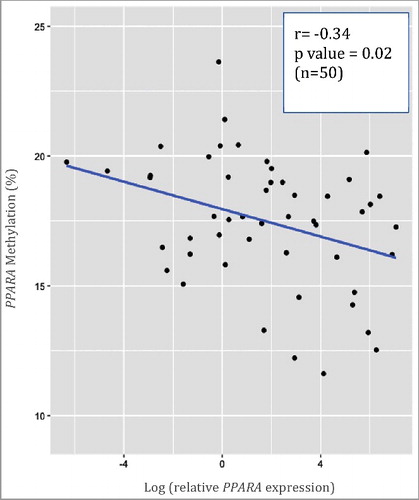
Due to the observed hypomethylation in infant PPARA methylation with increasing concentrations of several maternal phthalate metabolites, RT-qPCR was performed to determine if PPARA methylation corresponded with altered levels of gene expression. A subset of MMIP cohort RNA samples (n = 50) was available for expression analysis and also had a DNA methylation value that met stringent quality assurance standards. depicts a plot of the unadjusted DNA methylation and gene expression data along with the associated r and P values for the Spearman Rank Correlation Test. DNA methylation levels at PPARA inversely correlate with PPARA gene expression (r = −0.34, P = 0.02).
Figure 1. Correlation between PPARA gene expression and PPARA DNA methylation in infant cord blood. The unadjusted data are plotted, and the associated r and p values for the Spearman Rank Correlation Test are in upper right. Here, we show that PPARA expression is inversely correlated with PPARA methylation.
Sex-stratified analysis reveals sex-specific EDC effects
In sex-stratified analyses sex-specific BPA-related effects were observed for multiple gene regions. Urinary BPA was associated with a 1.35 (95%CI: −2.69, −0.01) percentage point decrease in IGF2 methylation and a 1.22 (95%CI: −2.27, −0.16) percentage point decrease in PPARA methylation in females () but not males (). Female-specific inverse associations were also observed between MEP and IGF2 as well as MCPP and ESR1 (). In contrast to the female-specific inverse associations, plasma BPA and urinary BPA in males tended to be positively related to LINE-1 and ESR1 (plasma BPA only), but these relationships were only borderline significant ().
Table 3a. Absolute change in infant DNA methylation per log unit increase in maternal EDC exposure (females).
Table 3b. Absolute change in infant DNA methylation per log unit increase in maternal EDC exposure (males).
Discussion
Recently, considerable emphasis has been placed on determining the contribution of early life exposures to the rising tide of obesity [Citation44]. However, there have been few epidemiological studies focused on assessing the epigenetic impact of exposures to EDCs in early pregnancy within the context of growth and metabolic programming. This may in part be due to the difficulty of recruiting women in the first trimester of pregnancy and highlights a unique feature of the MMIP cohort. Since EDCs can profoundly impact developmental pathways during critical windows of development and potentially lead to adult disease, identifying differential epigenetic profiles that act not only as biomarkers of exposure but also as potential markers of disease susceptibility is of great importance.
Using a hypothesis driven candidate gene approach focused on genes related to growth and development as well as the repetitive element, LINE-1, we observed hypomethylation of LINE-1, IGF2, and PPARA with increasing phthalate concentrations. Two individual phthalate metabolites (MBzP and MCPP) as well as the sum of DEHP metabolites were inversely associated with PPARA methylation, and DNA methylation was inversely correlated with PPARA gene expression, providing evidence of functional relevance. Additionally, a sex-stratified analysis of EDCs and DNA methylation showed that some relationships, including the association of urinary or plasma BPA with methylation, were female-specific.
The negative relationship observed between PPARA methylation and ΣDEHP is particularly notable. In addition to being one of the most commonly identified EDCs in the US population [Citation45], observed changes in DNA methylation and expression in adult rats resulting from in utero ΣDEHP exposure have raised speculation that the PPAR pathway, and specifically PPARA, may participate in epigenetic disruption [Citation46]. The findings from the present study support this hypothesis, as human PPARA promoter was inversely associated with ΣDEHP levels. Moreover, epigenetic modification in this region was also correlated with PPARA expression in infant cord blood DNA. Given that PPARA is a master regulator of lipid metabolism in the liver and predominant PPAR subtype in this organ [Citation47], the identification of a functionally relevant region measured in a surrogate easily obtainable matrix would be extremely valuable as a biomarker, provided it reflects the profile of the liver. Other studies have found that rats perinatally exposed to BPA displayed increased PAPRA activation in the liver [Citation48], but this group did not evaluate PPARA expression simultaneously in blood. Moving forward, studies investigating PPARA expression should consider investigating matched tissue specimens (e.g., liver and a bioavailable tissue) as well as address the issue of cellular heterogeneity, which we did not account for here. Together, these future studies will aid in determining the biological relevance of our findings.
Our findings in a human birth cohort characterized for EDC levels in maternal first trimester urine and blood adds to a relatively sparse, but growing, body of data on prenatal phthalate-induced epigenetic effects [Citation20]. The finding that MCPP had a consistent negative impact on 3 of the 5 candidate loci evaluated, including methylation patterns of the imprinted gene IGF2 is consistent with the findings of impact from phthalate exposure. A study with 196 women from the Boston, MA area found first trimester MCPP had an impact on epigenetic patterns of imprinted genes IGF2 and H19 in the placenta [Citation49]. When the sum of all phthalates or the sum of low molecular weight phthalates were considered, both were found to be inversely associated with IGF2 DMR methylation [Citation49]. Findings from the present study also revealed an inverse relationship between MCPP and methylation of LINE-1 repetitive elements. Aberrant repetitive element methylation patterns can impact genome stability and gene expression [Citation50], and previous reports have suggested these repetitive loci are targets of developmental phthalate exposure. For instance, increasing concentrations of MEP in early and late pregnancy in the CHAMACOS cohort were found to be inversely associated with Alu methylation in infant cord blood [Citation29]. Zhao et al. found that maternal third trimester levels of ΣDEHP were inversely associated with LINE-1 measured in DNA collected from pooled samples from 4 within individual placental biopsies [Citation51]. Solomon et al. were recently the first to assess epigenome-wide effects in humans from prenatal phthalate exposure. Using the Infinium 450K BeadChip they identified 27 differentially methylated regions (DMRs) in infant cord blood of which more than half of the DMRs were associated with ΣDEHP and some DMRs were found in genes involved in endocrine function [Citation52]. Taken together, the prenatal epigenomic landscape, from gene to genome-wide, appears to be sensitive to several different phthalates or phthalate mixtures.
Because it is well known that EDCs can have sexually dimorphic phenotypic effects, we also performed a sex-stratified analysis for DNA methylation. Interestingly, we found female-specific EDC-induced effects, which were consistently negative. Sex-specific BPA-induced epigenetic effects have been observed previously in human cohorts [Citation24] and rodent models [Citation26–Citation28]. In some instances the sexually dimorphic BPA-induced epigenetic changes have been linked to metabolic phenotypes. For example, Anderson et al. showed that BPA-induced epigenetic effects in female mice mediated metabolic outcomes including body weight and body fat [Citation53]. Although the sex-specific findings from the present study should be considered preliminary in view of the small sample size, they highlight the need to include both sexes in animal and human studies when evaluating EDC-related effects.
A logistical challenge for epidemiological studies is precisely capturing and quantifying environmental exposures. Relating infant cord blood DNA methylation changes to maternal EDC levels in spot urine sample is one limitation of our study. However, while urinary EDC concentrations may vary throughout pregnancy [Citation54,Citation55], it has been suggested that a single urinary measurement may reasonably represent several months of maternal exposure and thus potential fetal exposure [Citation56]. By contrast, a major strength of our study is the exposure assessment of EDCs in the first trimester of pregnancy as this is a critical window of sexual differentiation and a period when perturbation of normal and necessary epigenetic patterns is particularly impactful. Due to the number of comparisons made, we note that the possibility of chance findings in our assessment of the effect of early EDC exposures on candidate gene DNA methylation cannot be ruled out. Finally, the potential biological relevance of such small changes in DNA methylation to offspring health outcome remains to be elucidated; a recent review of literature suggests that such small epigenetic changes during critical windows of differentiation have the potential to induce short and long term gene expression changes [Citation57].
Conclusion
Results from this study further contribute to a growing body of evidence suggesting that in utero exposure to EDCs can perturb normal biological functions via the epigenome. The epigenetically labile regions described here, along with others yet to be identified, may act as biomarkers of disease susceptibility if they are linked to childhood or adult phenotypes in subsequent studies. There is a great need to discover such biomarkers and exploit them in an effort to break the cycle of intergenerational obesity and metabolic disorders.
Montrose__MMIP_Table_S2_qPCR_primers_final.docx
Download MS Word (55.8 KB)Montrose_MMIP_Table_S1_primers_resubmit.docx
Download MS Word (19.3 KB)Additional information
Funding
Related Research Data
References
- Wong KH, Durrani TS. Exposures to endocrine disrupting chemicals in consumer products-a guide for pediatricians. Curr Probl Pediatr Adolesc Health Care. 2017;47(5):107–118. Epub 2017/05/21. doi:10.1016/j.cppeds.2017.04.002. PubMed PMID: 28526231
- Gould JC, Leonard LS, Maness SC, et al. Bisphenol A interacts with the estrogen receptor alpha in a distinct manner from estradiol. Mol Cell Endocrinol. 1998;142(1–2):203–214. Epub 1998/10/23. doi:10.1016/S0303-7207(98)00084-7. PubMed PMID: 9783916
- Kruger T, Long M, Bonefeld-Jorgensen EC. Plastic components affect the activation of the aryl hydrocarbon and the androgen receptor. Toxicology. 2008;246(2-3):112–123. Epub 2008/02/26. doi:10.1016/j.tox.2007.12.028. PubMed PMID: 18294747
- Kim S, Kim S, Won S, et al. Considering common sources of exposure in association studies - Urinary benzophenone-3 and DEHP metabolites are associated with altered thyroid hormone balance in the NHANES 2007–2008. Environ Int. 2017;107:25–32. Epub 2017/06/27. doi:10.1016/j.envint.2017.06.013. PubMed PMID: 28651165
- Gore AC, Chappell VA, Fenton SE, et al. Executive Summary to EDC-2: the endocrine society's second scientific statement on ?endocrine-disrupting chemicals. Endocr Rev. 2015;36(6):593–602. Epub 2015/09/29. doi:10.1210/er.2015-1093. PubMed PMID: 26414233; PubMed Central PMCID: PMC4702495
- Wadhwa PD, Buss C, Entringer S, et al. Developmental origins of health and disease: brief history of the approach and current focus on epigenetic mechanisms. Semin Reprod Med. 2009;27(5):358–368. Epub 2009/08/28. doi:10.1055/s-0029-1237424. PubMed PMID: 19711246; PubMed Central PMCID: PMC2862635
- Yang TC, Peterson KE, Meeker JD, et al. Bisphenol A and phthalates in utero and in childhood: association with child BMI z-score and adiposity. Environ Res. 2017;156:326–333. Epub 2017/04/09. doi:10.1016/j.envres.2017.03.038. PubMed PMID: 28390300; PubMed Central PMCID: PMC5482500
- Buckley JP, Engel SM, Braun JM, et al. Prenatal phthalate exposures and body mass index among 4- to 7-year-old children: a pooled analysis. Epidemiology. 2016;27(3):449–458. Epub 2016/01/09. doi:10.1097/EDE.0000000000000436. PubMed PMID: 26745610; PubMed Central PMCID: PMC4821741
- Buckley JP, Engel SM, Mendez MA, et al. Prenatal phthalate exposures and childhood fat mass in a New York city cohort. Environ Health Perspect. 2016;124(4):507–513. Epub 2015/08/27. doi:10.1289/ehp.1509788. PubMed PMID: 26308089; PubMed Central PMCID: PMC4829985
- Vafeiadi M, Roumeliotaki T, Myridakis A, et al. Association of early life exposure to bisphenol A with obesity and cardiometabolic traits in childhood. Environ Res. 2016;146:379–387. Epub 2016/01/29. doi:10.1016/j.envres.2016.01.017. PubMed PMID: 26821262
- Valvi D, Casas M, Romaguera D, et al. Prenatal phthalate exposure and childhood growth and blood pressure: evidence from the Spanish INMA-Sabadell Birth Cohort Study. Environ Health Perspect. 2015;123(10):1022–1029. Epub 2015/04/08. doi:10.1289/ehp.1408887. PubMed PMID: 25850106; PubMed Central PMCID: PMC4590754
- Angle BM, Do RP, Ponzi D, et al. Metabolic disruption in male mice due to fetal exposure to low but not high doses of bisphenol A (BPA): evidence for effects on body weight, food intake, adipocytes, leptin, adiponectin, insulin and glucose regulation. Reprod Toxicol. 2013;42:256–268. Epub 2013/07/31. doi:10.1016/j.reprotox.2013.07.017. PubMed PMID: 23892310; PubMed Central PMCID: PMC3886819
- Xu X, Tan L, Himi T, et al. Changed preference for sweet taste in adulthood induced by perinatal exposure to bisphenol A-A probable link to overweight and obesity. Neurotoxicol Teratol. 2011;33(4):458–463. Epub 2011/06/28. doi:10.1016/j.ntt.2011.06.002. PubMed PMID: 21704699
- Wei J, Lin Y, Li Y, et al. Perinatal exposure to bisphenol A at reference dose predisposes offspring to metabolic syndrome in adult rats on a high-fat diet. Endocrinology. 2011;152(8):3049–3061. Epub 2011/05/19. doi:10.1210/en.2011-0045. PubMed PMID: 21586551
- van Esterik JC, Dolle ME, Lamoree MH, et al. Programming of metabolic effects in C57BL/6JxFVB mice by exposure to bisphenol A during gestation and lactation. Toxicology. 2014;321:40–52. Epub 2014/04/15. doi:10.1016/j.tox.2014.04.001. PubMed PMID: 24726836
- Alonso-Magdalena P, Vieira E, Soriano S, et al. Bisphenol A exposure during pregnancy disrupts glucose homeostasis in mothers and adult male offspring. Environ Health Perspect. 2010;118(9):1243–1250. Epub 2010/05/22. doi:10.1289/ehp.1001993. PubMed PMID: 20488778; PubMed Central PMCID: PMC2944084
- Wolffe AP, Matzke MA. Epigenetics: regulation through repression. Science. 1999;286(5439):481–486. Epub 1999/10/16. doi:10.1126/science.286.5439.481. PubMed PMID: 10521337
- Nilsson E, Ling C. DNA methylation links genetics, fetal environment, and an unhealthy lifestyle to the development of type 2 diabetes. Clin Epigenetics. 2017;9:105. Epub 2017/10/14. doi:10.1186/s13148-017-0399-2. PubMed PMID: 29026446; PubMed Central PMCID: PMC5627472
- Cheng Y, Xie N, Jin P, et al. DNA methylation and hydroxymethylation in stem cells. Cell Biochem Funct. 2015;33(4):161–173. Epub 2015/03/18. doi:10.1002/cbf.3101. PubMed PMID: 25776144; PubMed Central PMCID: PMC4687961
- Singh S, Li SS. Epigenetic effects of environmental chemicals bisphenol A and phthalates. Int J Mol Sci. 2012;13(8):10143–10153. Epub 2012/09/06. doi:10.3390/ijms130810143. PubMed PMID: 22949852; PubMed Central PMCID: PMC3431850
- Dolinoy DC, Huang D, Jirtle RL. Maternal nutrient supplementation counteracts bisphenol A-induced DNA hypomethylation in early development. Proc Natl Acad Sci U S A. 2007;104(32):13056–13061. Epub 2007/08/03. doi:10.1073/pnas.0703739104. PubMed PMID: 17670942; PubMed Central PMCID: PMC1941790
- Anderson OS, Nahar MS, Faulk C, et al. Epigenetic responses following maternal dietary exposure to physiologically relevant levels of bisphenol A. Environ Mol Mutagen. 2012;53(5):334–342. Epub 2012/04/03. doi:10.1002/em.21692. PubMed PMID: 22467340; PubMed Central PMCID: PMC3570056
- Nahar MS, Kim JH, Sartor MA, et al. Bisphenol A-associated alterations in the expression and epigenetic regulation of genes encoding xenobiotic metabolizing enzymes in human fetal liver. Environ Mol Mutagen. 2014;55(3):184–195. Epub 2013/11/12. doi:10.1002/em.21823. PubMed PMID: 24214726; PubMed Central PMCID: PMC3999958
- Goodrich JM, Dolinoy DC, Sánchez BN, et al. Adolescent epigenetic profiles and environmental exposures from early life through peri-adolescence. Environ Epigenetics Environ Epigenetics. 2016;2(3): dvw018.
- McCabe C, Anderson OS, Montrose L, et al. Sexually dimorphic effects of early-life exposures to endocrine disruptors: sex-specific epigenetic reprogramming as a potential mechanism. Curr Environ Health Reports. 2017;4(4):426–438. Epub 2017/10/06. doi:10.1007/s40572-017-0170-z. PubMed PMID: 28980159
- Strakovsky RS, Wang H, Engeseth NJ, et al. Developmental bisphenol A (BPA) exposure leads to sex-specific modification of hepatic gene expression and epigenome at birth that may exacerbate high-fat diet-induced hepatic steatosis. Toxicol Appl Pharmacol. 2015;284(2):101–112. Epub 2015/03/10. doi:10.1016/j.taap.2015.02.021. PubMed PMID: 25748669; PubMed Central PMCID: PMC4520316
- Kundakovic M, Gudsnuk K, Franks B, et al. Sex-specific epigenetic disruption and behavioral changes following low-dose in utero bisphenol A exposure. Proc Natl Acad Sci USA. 2013;110(24):9956–9961. Epub 2013/05/30. doi:10.1073/pnas.1214056110. PubMed PMID: 23716699; PubMed Central PMCID: PMC3683772
- Mao Z, Xia W, Huo W, et al. Pancreatic impairment and Igf2 hypermethylation induced by developmental exposure to bisphenol A can be counteracted by maternal folate supplementation. J Appl Toxicol. 2017;37(7):825–835. Epub 2017/02/07. doi:10.1002/jat.3430. PubMed PMID: 28165156
- Huen K, Calafat AM, Bradman A, et al. Maternal phthalate exposure during pregnancy is associated with DNA methylation of LINE-1 and Alu repetitive elements in Mexican-American children. Environ Res. 2016;148:55–62. Epub 2016/03/29. doi:10.1016/j.envres.2016.03.025. PubMed PMID: 27019040; PubMed Central PMCID: PMC4874877
- Huang RC, Galati JC, Burrows S, et al. DNA methylation of the IGF2/H19 imprinting control region and adiposity distribution in young adults. Clin Epigenetics. 2012;4(1):21. Epub 2012/11/15. doi:10.1186/1868-7083-4-21. PubMed PMID: 23148549; PubMed Central PMCID: PMC3507742
- Watkins DJ, Milewski S, Domino SE, et al. Maternal phthalate exposure during early pregnancy and at delivery in relation to gestational age and size at birth: a preliminary analysis. Reprod Toxicol. 2016;65:59–66. Epub 2016/06/30. doi:10.1016/j.reprotox.2016.06.021. PubMed PMID: 27352641; PubMed Central PMCID: PMC5067196
- Veiga-Lopez A, Kannan K, Liao C, et al. Gender-specific effects on gestational length and birth weight by early pregnancy BPA exposure. J Clin Endocrinol Metab. 2015;100(11):E1394–E1403. Epub 2015/09/26. doi:10.1210/jc.2015-1724. PubMed PMID: 26406292; PubMed Central PMCID: PMC4702459
- Vandenberg LN, Gerona RR, Kannan K, et al. A round robin approach to the analysis of bisphenol A (BPA) in human blood samples. Environ Health. 2014;13(1):25. Epub 2014/04/03. doi:10.1186/1476-069X-13-25. PubMed PMID: 24690217; PubMed Central PMCID: PMC4066311
- Grunau C, Clark SJ, Rosenthal A. Bisulfite genomic sequencing: systematic investigation of critical experimental parameters. Nucleic Acids Res. 2001;29(13):E65. Epub 2001/07/04. PubMed PMID: 11433041; PubMed Central PMCID: PMC55789.
- Yang AS, Estecio MR, Doshi K, et al. A simple method for estimating global DNA methylation using bisulfite PCR of repetitive DNA elements. Nucleic Acids Res. 2004;32(3):e38. Epub 2004/02/20. doi:10.1093/nar/gnh032. PubMed PMID: 14973332; PubMed Central PMCID: PMC373427
- St-Pierre J, Hivert MF, Perron P, et al. IGF2 DNA methylation is a modulator of newborn's fetal growth and development. Epigenetics. 2012;7(10):1125–1132. Epub 2012/08/22. doi:10.4161/epi.21855. PubMed PMID: 22907587; PubMed Central PMCID: PMC3469454
- Soubry A, Schildkraut JM, Murtha A, et al. Paternal obesity is associated with IGF2 hypomethylation in newborns: results from a Newborn Epigenetics Study (NEST) cohort. BMC medicine. 2013;11:29. Epub 2013/02/08. doi:10.1186/1741-7015-11-29. PubMed PMID: 23388414; PubMed Central PMCID: PMC3584733
- Lui JC, Finkielstain GP, Barnes KM, et al. An imprinted gene network that controls mammalian somatic growth is down-regulated during postnatal growth deceleration in multiple organs. Am J Physiol Regul Integr Comp Physiol. 2008;295(1):R189–R196. Epub 2008/05/02. doi:10.1152/ajpregu.00182.2008. PubMed PMID: 18448610; PubMed Central PMCID: PMC2494817
- Yoon M. The role of PPARalpha in lipid metabolism and obesity: focusing on the effects of estrogen on PPARalpha actions. Pharmacol Res. 2009;60(3):151–159. Epub 2009/08/04. doi:10.1016/j.phrs.2009.02.004. PubMed PMID: 19646654
- Rees WD, McNeil CJ, Maloney CA. The roles of PPARs in the fetal origins of metabolic health and disease. PPAR Res. 2008;2008:459030. Epub 2008/02/22. doi:10.1155/2008/459030. PubMed PMID: 18288289; PubMed Central PMCID: PMC2234254
- Lillycrop KA, Phillips ES, Torrens C, et al. Feeding pregnant rats a protein-restricted diet persistently alters the methylation of specific cytosines in the hepatic PPAR alpha promoter of the offspring. Br J Nutr. 2008;100(2):278–282. Epub 2008/01/12. doi:10.1017/S0007114507894438. PubMed PMID: 18186951; PubMed Central PMCID: PMC2564112
- Mauvais-Jarvis F. Estrogen and androgen receptors: regulators of fuel homeostasis and emerging targets for diabetes and obesity. Trends Endocrinol Metab: TEM. 2011;22(1):24–33. Epub 2010/11/27. doi:10.1016/j.tem.2010.10.002. PubMed PMID: 21109497; PubMed Central PMCID: PMC3011051
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25(4):402–408. Epub 2002/02/16. doi:10.1006/meth.2001.1262. PubMed PMID: 11846609
- Bianco-Miotto T, Craig JM, Gasser YP, et al. Epigenetics and DOHaD: from basics to birth and beyond. J Dev Orig Health Dis. 2017;8(5):513–519. Epub 2017/09/12. doi:10.1017/S2040174417000733. PubMed PMID: 28889823
- Nadal A, Quesada I, Tuduri E, et al. Endocrine-disrupting chemicals and the regulation of energy balance. Nat Rev Endocrinol. 2017;13(9):536–546. Epub 2017/05/20. doi:10.1038/nrendo.2017.51. PubMed PMID: 28524168
- Martinez-Arguelles DB, Papadopoulos V. Prenatal phthalate exposure: epigenetic changes leading to lifelong impact on steroid formation. Andrology. 2016;4(4):573–584. Epub 2016/04/05. doi:10.1111/andr.12175. PubMed PMID: 27044004
- Reddy JK. Nonalcoholic steatosis and steatohepatitis. III. Peroxisomal beta-oxidation, PPAR alpha, and steatohepatitis. Am J Physiol Gastrointest Liver Physiol. 2001;281(6):G1333–G1339. Epub 2001/11/14. PubMed PMID: 11705737
- Wei J, Sun X, Chen Y, et al. Perinatal exposure to bisphenol A exacerbates nonalcoholic steatohepatitis-like phenotype in male rat offspring fed on a high-fat diet. J Endocrinol. 2014;222(3):313–325. Epub 2014/08/13. doi:10.1530/JOE-14-0356. PubMed PMID: 25112833
- LaRocca J, Binder AM, McElrath TF, et al. The impact of first trimester phthalate and phenol exposure on IGF2/H19 genomic imprinting and birth outcomes. Environ Res. 2014;133:396–406. Epub 2014/06/29. doi:10.1016/j.envres.2014.04.032. PubMed PMID: 24972507; PubMed Central PMCID: PMC4155603
- Kitkumthorn N, Mutirangura A. Long interspersed nuclear element-1 hypomethylation in cancer: biology and clinical applications. Clin Epigenetics. 2011;2(2):315–330. Epub 2012/06/19. doi:10.1007/s13148-011-0032-8. PubMed PMID: 22704344; PubMed Central PMCID: PMC3365388
- Zhao Y, Shi HJ, Xie CM, et al. Prenatal phthalate exposure, infant growth, and global DNA methylation of human placenta. Environ Mol Mutagen. 2015;56(3):286–292. Epub 2014/10/21. doi:10.1002/em.21916. PubMed PMID: 25327576
- Solomon O, Yousefi P, Huen K, et al. Prenatal phthalate exposure and altered patterns of DNA methylation in cord blood. Environ Mol Mutagen. 2017;58(6):398–410. Epub 2017/05/31. doi:10.1002/em.22095. PubMed PMID: 28556291
- Anderson OS, Kim JH, Peterson KE, et al. Novel epigenetic biomarkers mediating bisphenol A exposure and metabolic phenotypes in female mice. Endocrinology. 2017;158(1):31–40. Epub 2016/11/09. doi:10.1210/en.2016-1441. PubMed PMID: 27824486; PubMed Central PMCID: PMC5412976
- Meeker JD, Cantonwine DE, Rivera-Gonzalez LO, et al. Distribution, variability, and predictors of urinary concentrations of phenols and parabens among pregnant women in Puerto Rico. Environ Sci Technol. 2013;47(7):3439–3447. Epub 2013/03/09. doi:10.1021/es400510g. PubMed PMID: 23469879; PubMed Central PMCID: PMC3638245
- Philippat C, Wolff MS, Calafat AM, et al. Prenatal exposure to environmental phenols: concentrations in amniotic fluid and variability in urinary concentrations during pregnancy. Environ Health Perspect. 2013;121(10):1225–1231. Epub 2013/08/15. doi:10.1289/ehp.1206335. PubMed PMID: 23942273; PubMed Central PMCID: PMC3801458
- Smith KW, Braun JM, Williams PL, et al. Predictors and variability of urinary paraben concentrations in men and women, including before and during pregnancy. Environ Health Perspect. 2012;120(11):1538–1543. Epub 2012/06/23. doi:10.1289/ehp.1104614. PubMed PMID: 22721761; PubMed Central PMCID: PMC3556607
- Breton CV, Marsit CJ, Faustman E, et al. Small-magnitude effect sizes in epigenetic end points are important in children's environmental health studies: the children's environmental health and disease prevention research center's epigenetics working group. Environ Health Perspect. 2017;125(4):511–526. Epub 2017/04/01. doi:10.1289/EHP595. PubMed PMID: 28362264; PubMed Central PMCID: PMC5382002