
ABSTRACT
Augmented maternal care during the first postnatal week promotes life-long stress resilience and improved memory compared with the outcome of routine rearing conditions. Recent evidence suggests that this programming commences with altered synaptic connectivity of stress sensitive hypothalamic neurons. However, the epigenomic basis of the long-lived consequences is not well understood. Here, we employed whole-genome bisulfite sequencing (WGBS), RNA-sequencing (RNA-seq), and a multiplex microRNA (miRNA) assay to examine the effects of augmented maternal care on DNA cytosine methylation, gene expression, and miRNA expression. A total of 9,439 differentially methylated regions (DMRs) associated with augmented maternal care were identified in male offspring hypothalamus, as well as a modest but significant decrease in global DNA methylation. Differentially methylated and expressed genes were enriched for functions in neurotransmission, neurodevelopment, protein synthesis, and oxidative phosphorylation, as well as known stress response genes. Twenty prioritized genes were identified as highly relevant to the stress resiliency phenotype. This combined unbiased approach enabled the discovery of novel genes and gene pathways that advance our understanding of the epigenomic mechanisms underlying the effects of maternal care on the developing brain.
Introduction
Early postnatal care in mammals is critical for survival as well as neurodevelopment. Specifically, early maternal care can epigenetically program the behavior of offspring for their entire lifetime. Rodent models that manipulate maternal care and maternal stress have been used to examine the molecular and cellular mechanisms underlying the long-term behavioral changes observed in the offspring [Citation1–Citation3]. In the augmented maternal care (AMC) paradigm, daily separation of rat dams from their pups for 15 minutes from postnatal day 2 (PND2) to postnatal day 8 (PND8) triggers the increased maternal behaviors of arched-back nursing, as well as elevated licking and grooming of the pups by the dam [Citation3,Citation4]. Subsequently, it was found that multiple, continuous days of pup handling within the first postnatal week of life were required for the AMC response, suggesting that AMC initiates changes during this critical period of neurodevelopment in the offspring [Citation5,Citation6]. The repeated, daily sessions of enhanced maternal care triggered by the AMC paradigm result in offspring that show reduced plasma corticosterone levels in response to an acute restraint stress [Citation7–Citation11], reduced anxiety related behaviors [Citation11,Citation12], resilience to depressive-like behaviors [Citation11], and enhanced performance on several cognitive tasks as adults [Citation12,Citation13].
AMC appears to alter brain function for the lifetime of the animal [Citation14]. Adult males experiencing early-life AMC are stress resilient, which is manifested as reduced corticosterone response to stress [Citation6,Citation13] and improved cognitive function [Citation6]. The current study focused on epigenetic processes that underlie a well-established phenotypic outcome of AMC. To date, our group has established the phenotype in males only [Citation6,Citation13]. Therefore, we focused our investigations to male hypothalamus to understand the molecular and epigenetic mechanisms that drive the cognitive and emotional phenotype observed in males. Previous work has demonstrated that this mechanism depends upon the temporally coordinated regulation of both corticotrophin releasing hormone (CRH) in the paraventricular nucleus (PVN) of the hypothalamus [Citation5,Citation13,Citation16] and glucocorticoid receptor (GR, encoded by Nr3c1) [Citation6] in the hippocampus [Citation13,Citation15]. Specifically, Crh transcripts in the hypothalamus are significantly reduced immediately following AMC at PND9 [Citation5,Citation6] and remain repressed throughout adulthood [Citation9]. However, increased Nr3c1 expression in the hippocampus was delayed until PND23 [Citation9], suggesting that early gene expression changes in the PVN of the hypothalamus drive the initial molecular mechanisms underlying AMC. Together, the elevated hippocampal Nr3c1 expression and reduced hypothalamic Crh expression [Citation2,Citation5,Citation11,Citation16] in AMC adult offspring suggest that increased negative feedback signaling may underlie the observed hypo-responsive HPA axis. These studies have raised an important question about how hypothalamic reprogramming is initiated and maintained by AMC induced early-life gene expression changes. AMC initiates transient structural changes in CRH-expressing cells in the hypothalamus of pups [Citation12,Citation16]. By PND9 there is decreased excitatory synaptic drive onto hypothalamic Crh expressing neurons in AMC offspring, that returns to baseline by adulthood (PND30-45) [Citation16], suggesting that the AMC induced reduction in Crh expression is maintained long-term by additional mechanisms [Citation14]. In vitro studies have suggested that the synaptic changes initiate transcriptional regulation in these CRH positive cells [Citation12]. Specifically, recent work has implicated the neuron restrictive silencing factor (NRSF) in establishing long-term epigenetic gene expression patterns resulting from AMC [Citation14,Citation17]. NRSF expression and binding to the Crh gene is increased in hypothalamus of AMC offspring at PND9 but is no longer present in adulthood [Citation12,Citation16]. Thus, there is little information about the comprehensive epigenomic processes by which augmented sensory signals from the mother result in enduring, large-scale reprogramming of gene expression in the hypothalamus. In hippocampus, prior studies have described epigenetic DNA methylation alterations and H3K9 acetylation in response to differences in maternal care at Nr3c1 [Citation18]. The search for additional genes underlying AMC effects on adult hippocampus revealed alterations to gene expression of more than 900 genes related to cellular metabolism and energy production, signal transduction, protein synthesis (mainly ribosomal genes), and neurodevelopment [Citation19]. Targeted analysis of a seven megabase (Mb) region of chromosome 18 flanking Nr3c1 revealed that the clustered protocadherins (Pcdh) showed the highest differential response in DNA methylation [Citation20].
Together, these combined results indicate that NRSF binding in PND9 hypothalamus [Citation12,Citation16] as well as differential gene expression and DNA methylation in adult hippocampus are involved in regulating long-term behavioral changes induced by AMC. Whereas NRSF ChIP-seq has identified gene sets contributing to AMC-induced cellular changes [Citation12], a multi-methodology genome-wide approach has not been undertaken in the hypothalamus [Citation21]. To address this gap in knowledge we performed an integrated, genome-wide characterization of epigenetic and gene expression differences in hypothalamus from PND9 pups in response to AMC. These experiments identified that the response to AMC occurs not only in the genes previously identified, such as Crh, but also in thousands of genes throughout the genome. In addition to a modest but significant global reduction in hypothalamic DNA methylation levels, AMC induces differential methylation and expression in mRNA transcripts related to neurodevelopment, synaptic signaling, ribosome function, and cellular stress. The results provide strong evidence for the role of epigenomic modifications in regulating gene expression profiles related to resilience and offer a suite of prioritized genes and gene pathways for the development of diagnostic biomarkers and therapies for neuropsychiatric disorders in humans.
Methods
Detailed methods are provided in the Supplemental Information section. All animal studies were performed in accordance with UC Davis IACUC protocol #18745. Augmented Maternal Care (AMC) was performed as described previously [Citation13,Citation16]. Briefly, pups were gathered at birth and five male and five female pups were randomly assigned to dams. Pups were removed for 15 min at 9 AM (PST) each day from PND2-PND9 to a separate cage in a different room equipped with a heating pad. Levels of maternal care varied between dams with three dams averaging 1069 ± 129 (SEM) minutes licking and grooming their pups in AMC litters compared with three control dams that averaged 482.66 ± 35 minutes licking and grooming their pups. Molecular analyses were performed on samples from one AMC and one control litter. Since the stress-resilient phenotype has been robustly established for males only in the literature [Citation6,Citation12,Citation13,Citation17], only male hypothalami were evaluated for genome-wide scans. Further details can be found in the Supplemental Information (SI) Materials and Methods. For WGBS DNA methylation analysis, genomic DNA was converted by bisulfite treatment using a Zymo kit followed by Illumina sequencing library preparation. WGBS libraries were sequenced on an Illumina Hi-Seq 2500. DMR identification and PMD analysis was performed as detailed in Schroeder [Citation22] and Dunaway [Citation23]. The SI Materials and Methods section contain an in depth description of WGBS methods. Bisulfite converted genomic DNA was amplified using custom, gene specific primers; amplicons were analyzed by pyrosequencing on a Biotage PSQ-96MA Pyrosequencer. A detailed description of pyrosequencing methods is contained in the SI Materials and Methods. For RNA-seq total RNA was prepared from tissue using a Qiagen RNeasy Kit (Qiagen). Total RNA was depleted of ribosomal RNA for creation of stranded RNA-seq libraries (See SI Materials and Methods). Bar coded RNA-seq libraries were sequenced on an Illumina Hi-Seq 2500 platform. Analysis of RNA-seq reads is described in SI Materials and Methods. Validation for differential RNA-seq identified genes was performed on samples from the same cohort of PND9 male and female hypothalami (SI Materials and Methods). For miRNA transcript analysis a multiplex miRNA assay of miRNA transcripts was performed using an nCounter Rat miRNA expression assay (NanoString). A detailed description of miRNA analysis is available in SI Materials and Methods. All sequencing data will be available through the NIH GEO database accession number GSE99282.
Results
AMC induces large-scale hypomethylation in the PND9 hypothalamus
To investigate genome-wide DNA methylation changes associated with AMC, hypothalami from AMC and control male rat pups were chosen for whole-genome bisulfite sequencing (WGBS) based on significant (P = 0.0042) differential AMC (total licking and grooming) at postnatal day 9 (Supplemental Figure 1). Principal component analysis (PCA) was performed on 20,000 base (20-Kb) windows of percent DNA methylation with CpG islands masked ( A) to explore genome-wide patterns of methylation between treatment conditions in the individual rats. Methylation levels in individual PND9 hypothalami were separated by AMC condition for principal component 1 (30.2% of variance explained), indicating differences in methylation levels between conditions. Unexpectedly, hypothalami from AMC pups displayed significant global hypomethylation (P = 0.015) at PND 9 ( B). Specifically, global CpG methylation levels for augmented hypothalami averaged 76.08% while controls averaged 79.20%, yielding to a 3.12% reduction (Supplemental Table 1). Non-CpG methylation levels in augmented and control hypothalami ranged from 0.86 to 1.46% of total cytosines (Supplemental Table 1) consistent with previous findings of normal neurodevelopment [Citation24]. CpG hypomethylation was significant across all chromosomes with the exception of chromosome 13 and X, indicating a genome-wide impact of AMC on CpG methylation levels ( C). Interestingly, windowing over different genomic features revealed hypomethylation specifically in the region 5 kb upstream of the transcriptional start site (TSS) of CpG island promoters (P = 0.0057) in AMC hypothalamus (Supplemental Figure 2). To identify potential hypothalamic genes and pathways contributing to AMC, large-scale differential DNA methylation patterns were identified from the WGBS data sets. Partially methylated domains (PMDs) were mapped from AMC and control WGBS data sets using a hidden Markov model (HMM) previously described for detecting partially methylated domains in neuronal cell lines [Citation22]. AMC PMDs averaged 7.3% of the genome and control PMDs averaged 6.7% of the genome. Although consistent with genome-wide hypomethylation the increased PMD coverage with AMC was not significant (P = 0.158) (Supplemental Table 2). While PMDs were found throughout the genomes of both groups, only five PMDs were differential by treatment group (). Consistent with genome-wide hypomethylation shown in B, these five PMDs were found in AMC but not control hypothalami. AMC PMDs covered 674,924 bases and overlapped with seven genes. These PMD genes included Dbn1, Prr7, Foxo3, and Cnksr2, which function in neuronal development, and Pak6, which functions in the MAP kinase pathway to regulate cytoskeletal dynamics underlying multiple brain processes [Citation25–Citation29] (). AMC PMDs also contained AY383691 and Ankrd63, which are gene products without well-established functions.
Figure 1. Reduced DNA methylation in hypothalamus is associated with AMC. (A) Principal components 1 and 2 of the average DNA methylation in 20 kb windows with masking of CpG Islands separately groups AMC samples (red outline, n = 3) from control samples (blue outline, n = 3). Outlines represent 95% confidence intervals. (B) Average genome wide CpG methylation is significantly reduced (P = 0.015) by 3.12% in AMC samples (n = 3) compared with control samples (n = 3). (C) DNA hypomethylation in the AMC hypothalami at PND9 was significant (*P < 0.05) on every chromosome except chromosome 13 and X (P < 0.52 and 0.054, respectively). RM ANOVA AMC group X-chromosome interaction F (20.80) = 21.89, P < 0.0001; * Benjamini-Hochberg corrected t-tests for each chromosome all P < 0.05.
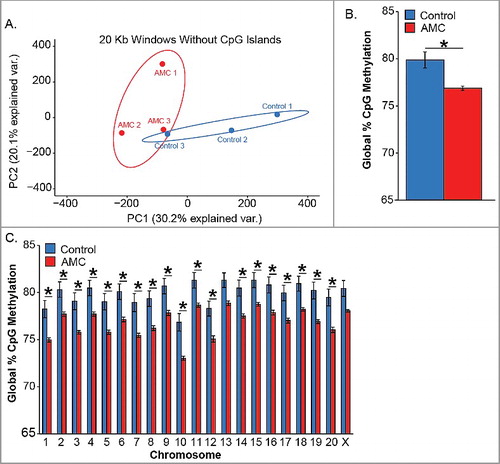
Table 1. Gene transcripts located within AMC PMDs.
AMC DMRs are linked to neurodevelopmental signaling and neurotransmission
While PMDs highlight broad regions of differential methylation associated with AMC, few genes are located within these loci. Therefore, to identify small scale (<3 kb) methylation changes, differentially methylated regions (DMRs) were identified from WGBS data [Citation23,Citation30]. With this bioinformatics approach, a total of 9,439 DMRs between AMC and Control PND9 hypothalami were identified (Supplemental Table 3). Of these 9,439 AMC DMRs, 6,228 had reduced methylation levels in the AMC samples, which is consistent with the genome-wide hypomethylation shown in . To investigate the relationship between large-scale PMDs and small-scale DMRs, the two genomic features were compared. Surprisingly, of the 9,439 DMRs identified, only one in Pak6 was found within the five AMC PMDs. Visualization of the DMRs using hierarchical clustering analysis (HCA) of significant (P < 0.05) DMRs from hypothalami clustered each rat by treatment ( A; Supplemental Table 3). Both PCA of 20 kb windows ( A) and HCA ( A) independently partitioned the AMC and control hypothalami into separate groups, indicating that distinct methylation patterns distinguish the AMC from control hypothalamic samples at two different resolutions. Motif enrichment analysis of the DMRs revealed the top significant (q < 0.0001) enrichment was for CTCF binding sites, which were present in 4% of DMRs ( B; Supplemental Table 4). CTCF binds these sites in a methylation sensitive manner and functions as a genomic insulator critical to chromatin architecture [Citation31]. Of the 9,439 total DMRs identified, 5,284 were between 5 kb upstream and 1 kb to downstream of 4,023 coding and noncoding RefSeq genes in the rat genome (Rn6). Forty-four percent of the DMRs were intergenic and 35.3% were localized to introns. Fourteen percent of DMRs were immediately upstream of genes and only 0.4% and 1.1% were located in the 5’ and 3’ prime untranslated regions (UTRs) respectively (Supplemental Figure 3A). For the DMRs annotated to within genes, the majority (93.6%) were localized to coding genes and 4.1% were located in long noncoding RNAs (lncRNAs) (Supplemental Figure 3B).
Figure 2. Differences in DNA methylation between AMC (n = 3) and control (n = 3) P9 hypothalami. (A) Hierarchical clustering of significant (P < 0.05) DMRs. Individual percent methylation values are normalized to the mean of each DMR. (B) CTCF motif binding motif was significantly (q < 0.0001) enriched in DMRs with 4.24% of DMRs containing the binding motif compared to 3.15% of background sequences with the motif (q < 0.00001). (C) Significant (q < 0.05) enriched gene ontologies and pathways of genes mapped to significant (P < 0.05) DMRs. Overlap corresponds to genes observed compared with the total genes in the pathway.
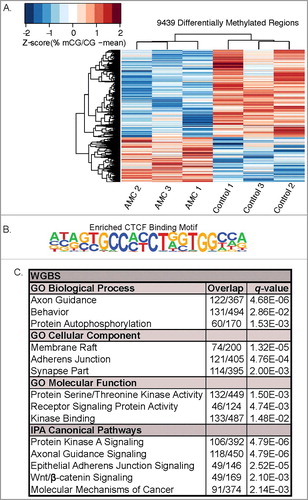
To identify the functions associated with the 4,023 genes mapped to AMC DMRs, gene ontology (GO) and pathways analyses were performed ( C). Gene ontology analysis revealed a significant (q < 0.05) enrichment for the biological processes of axon guidance, behavior, and protein auto-phosphorylation. Membrane raft, adherens junction, and synapse were the significantly (q < 0.05) enriched cellular components. Protein serine/threonine kinase activity, receptor signaling protein activity, and kinase binding were the most unique significant (q < 0.05) molecular functions. Pathway analyses revealed a significant (q < 0.05) enrichment for protein kinase A signaling, axonal guidance signaling, epithelial adherens junction signaling, Wnt/β-catenin signaling, and molecular mechanisms of cancer. Notably, the molecular mechanisms of cancer pathway represent a collection of developmental signaling pathways. Overall, all of the above GO terms and pathways are related to neurodevelopment as well as synaptic and cellular signaling ( C).
To further examine the DMRs for relevance to AMC, the 9,439 DMRs were subject to permutation testing, which yielded seven high confidence or “gold” DMRs (family-wise error rate (FWER) <0.05) (Supplemental Table 5). These gold DMRs were annotated to LOC00911370, Tuba3b, Glb1l, Papolb, Grb10, Tenm3, and Zfp332a (Supplemental Table 5). Zfp322a and Tenm3 both exhibited hypomethylation and increased gene expression in AMC pups. Interestingly, Tenm3 plays a critical role in axon guidance and Zfp322a is a transcription factor that inhibits cellular differentiation [Citation32,Citation33]. The hypomethylation of the Tenm3 associated DMR was validated (P = 0.01) by an independent pyrosequencing assay in a validation cohort (Supplemental Figure 4).
Differential AMC gene expression influences translation and oxidative phosphorylation
To investigate the regulatory relationship between DMRs and gene expression, RNA-seq analysis was performed on postnatal day 9 (PND9) male hypothalami ( A and B & Supplemental Table 6). Examination of the top 500 most variably expressed genes revealed clustering according to AMC condition ( A). For differential analysis, a total of 2,464 significant (q < 0.05) differentially expressed (DE) transcripts were identified between AMC and control hypothalami ( B, C & Supplemental Table 7A & 7B). There were 1,411 genes with reduced expression and 1,053 with increased expression following AMC. Log2 fold changes in gene transcripts ranged from 2.67 to -2.08 (Supplemental Table 7A & 7B) and the top 20 significant (q < 0.05) genes ranked by fold change are shown in . Consistent with previous studies that have established a key role for stress-induced neuropeptide Crh [Citation5,Citation11], we also found that Gal, Oxt, and Ucn3, transcripts encoding galanin, oxytocin, and the Crh homologue urocortin, respectively, were significantly reduced in handled pup hypothalamus as were transcripts for the Crh1 receptor (P < 0.05)( & B).
Figure 3. Differences in gene expression between AMC (n = 3) and control (n = 3) PND9 hypothalami. (A) Principal components analysis (PCA) of the top 500 most variably expressed genes. Outlines represent 95% confidence intervals. (B) Scatter plot DE transcripts with the top DE transcripts labeled. Grey indicates no significant difference in expression, while red indicates a significant (q < 0.05) difference. (C) Hierarchal clustering analysis (HCA) of significant (q < 0.05) DE transcripts. (D) Significant (q < 0.05) enriched gene ontologies and pathways from significantly (q < 0.05) DE transcripts. (E) Most significant (q < 0.0001) transcription factor binding site identified by motif enrichment analysis.
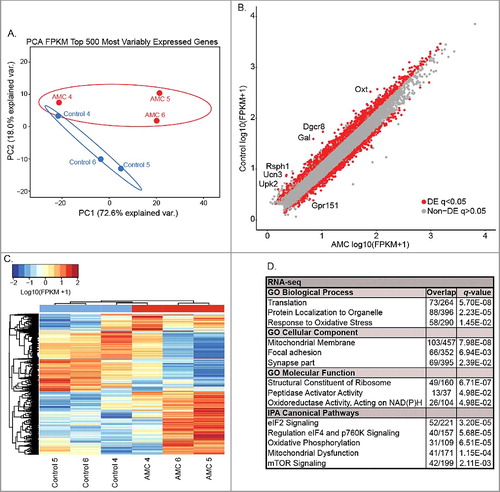
Table 2. The top 20 elevated and repressed AMC RNA transcripts.
To identify the functions associated with the 2,464 transcripts, gene ontology (GO) and pathways analyses were performed ( D). Gene ontology analysis revealed a significant (q < 0.05) enrichment for the biological processes of translation, protein localization to organelle, and response to oxidative stress. Mitochondrial membrane, focal adhesion, and synapse were the significantly (q < 0.05) enriched cellular components. Structural constituent of ribosome, peptidase activator activity, and oxidoreductase activity acting on NAD(P)H were the most unique significant (q < 0.05) molecular functions. Pathway analyses revealed a significant (q < 0.05) enrichment for eIF2 signaling, regulation of eIF4 and p760K signaling, oxidative phosphorylation, mitochondrial dysfunction, and mTOR signaling ( D). Notably, most of the ontologies and pathways are related to the output of PI3K/AKT/mTOR/PTEN signaling [Citation34], particularly translation by the ribosome (eIF2 and eIF4 signaling) and oxidative phosphorylation in the mitochondria. Motif enrichment analysis identified several transcription factor binding sites, with the top significant (q < 0.0001) enrichment representing binding sites for the transcription factor Elk1 that were present in 31% of transcripts (Supplemental Table 7). Elk1 is part of an insulin signaling network that cross-talks with PI3K/AKT/mTOR/PTEN signaling [Citation34] and is also involved in synaptic plasticity, learning, and neurodegenerative disease [Citation35]. Together, these results reveal differential regulation of a variety of cellular processes, and signaling pathways not previously associated with AMC, which are consistent with neurodevelopmental events occurring in PND9 hypothalamus during AMC [Citation34].
To validate and expand on DE genes identified by genome-wide RNA-seq analysis, reverse transcriptase quantitative PCR (RT-qPCR) analysis was performed on select genes of interest in both males and females as well as a second, independent cohort of AMC offspring. RT-qPCR analysis confirmed that Crh (Supplemental Figure 5A) was significantly reduced in AMC hypothalamus (P = 0.0006). There was a trend (P = 0.07) for an increased expression of Ube3a ligase domain containing isoforms (Supplemental Figure 5B), which were discovered using RNA-seq. There was also a significant (P < 0.0001) reduction in transcripts containing the alternative 3’UTR of Ube3a1 (Supplemental Figure 5C), which has the noncoding function of a competing endogenous RNA (ceRNA) or “miRNA sponge” that is critical to neurodevelopment [Citation36].
Reciprocal differences in miRNA and target gene expression
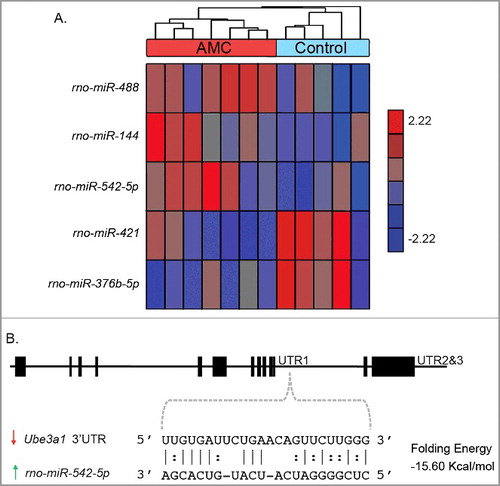
One of the top gene expression changes was decreased expression of the Dgcr8 microprocessor complex subunit gene (). Dgcr8, in combination with Drosha, is involved in the processing of primary miRNA (pri-miRNA) transcripts into precursor miRNAs (pre-miRNA) [Citation37]. The decreased expression of Dgcr8 suggests alterations to miRNA processing. Also suggestive of alterations to miRNA regulation was the decrease in the Ube3a1 ceRNA. Since the RNA-seq data from PND9 hypothalamus did not accurately profile miRNA transcripts, we next evaluated the impact of AMC on miRNA expression using a multiplex miRNA expression assay on PND9 hypothalamus from handled and control pups. A total of 156 miRNAs were expressed at levels above background, with five showing significant (P < 0.05) differential expression between AMC and control samples. Of these five, transcript levels for rno-miR-488, rno-miR-144, and rno-miR-542-5p were increased while transcript levels for rno-miR-421 and rno-miR-376b-5p were reduced ( A). The five significant (P < 0.05) DE miRNAs were then analyzed alongside the significant (q < 0.05) DE mRNAs for both a bioinformatically predicted match and reciprocal DE relationship in order to produce a list of 127 putative target genes (Supplemental Table 9). Among the 127 putative mRNA targets of DE miRNAs are the stress response genes Gal (rno-miR-144), Pomc (rno-miR-488), and Pvalb (rno-miR-488) [Citation38–Citation40]. Other miRNA targets include the sialidase Neu1 (rno-miR-542-5p) and the Amyloid beta precursor protein (APP) protease Adam10 (rno-miR-421), and transcripts for the neuronal regulators Pcdh9 (rno-miR-376b-5p) and Avpr1a (rno-miR-488) (Supplemental Table 9) [Citation41–Citation43]. Finally, rno-mir-542-5p was predicted to target the Ube3a1 ceRNA and the two showed a reciprocal relationship in expression ( B).
Figure 4. Differences in miRNA expression between AMC (n = 7) and control (n = 5) PND9 hypothalami. (A) Hierarchal clustering analysis of significant (P < 0.05) differential miRNA expression between. The expression of each gene was normalized to a mean of 0 and a standard deviation of 1. The values range from -2.22 to 2.22. Downregulated genes with AMC have negative values (blue), genes with no change have a value of zero (grey), and upregulated genes with AMC have a positive value (red). (B) Base-pairing of the predicted relationship between rno-mir-542-5p and the Ube3a1 3’UTR.
Integrative epigenomic prioritization of differentially regulated genes
To identify a convergent set of genes regulated by AMC in hypothalamus we integrated genes identified by the DNA methylation, RNA-seq, and multiplex miRNA assay analyses. For this combined analysis the 127 unique genes potentially regulated by the five differential miRNAs from Supplemental Table 9 were compared with the 4,023 unique genes in proximity to DMRs shown in . The overlap between these three gene lists resulted in 20 unique genes ( A). The presence of factors from diverse processes suggests that many pathways contribute to the AMC response in hypothalamus (). For example, Avrp1a functions in the stress pathway and regulates social behaviors, Kcnh5 is an ion channel that regulates hormone and neurotransmitter release, and Ring1 is representative of several factors on this list that repress gene expression levels () [Citation44–Citation46]. Ring1, a component of the Polycomb Repressive Complex 1 (PRC1) complex, together with Ube3a, another ubiquitin E3 ligase, has been recently shown to regulate genes associated with autism [Citation23]. The integration of genome-wide RNA-seq, DMRs, and miRNA target data sets produces a refined set of gene transcripts ( A), while also demonstrating the genome-wide nature of the differential expression and epigenetic modification that follows AMC ( B).
Figure 5. AMC is associated with genome-wide differences in DNA methylation and gene expression. (A) Integration of DMR, RNA-seq, and miRNA analyses yields 20 prioritized genes (inset). (B) Circos plot of the different data sets. The outer heatmap represents the percent differences in methylation of DMRs, the inner heatmap represents the log2 fold change of DE transcripts, and the links represent miRNAs and their predicted target genes.
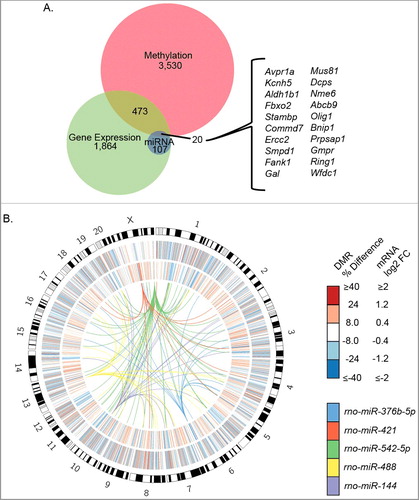
Table 3. Integrated DMR and miRNA analysis.
Discussion
Early life experience has tremendous power to shape neurodevelopment with long-term consequences [Citation1–Citation3]. In humans, disjointed or reduced maternal care that occurs in cases of poverty, maternal depression, or addiction is associated with increased risk for developing cognitive and emotional deficits, neuropsychiatric disorders, and cognitive decline later in life [Citation47,Citation48]. On the opposite end of the spectrum, studies in humans suggest that a positive early-life environment can produce resilience to affective disorders [Citation49–Citation51]. In animal models, enhancements in maternal care can positively impact the developing brain leading to reduced anxiety, resilience to depression-like behaviors, and enhanced learning and memory [Citation10,Citation11,Citation15]. In the AMC rat paradigm, while the brief removal of the pups is a stressful event for both the dam and pups, it triggers increased maternal licking and grooming as well as nursing of the pups. Together, these actions have been shown to re-program neonatal HPA circuits (14). The programming of the stress response in offspring receiving augmented maternal care can last a lifetime [Citation52] and help offset age-related cognitive deficits [Citation14]. Much of the previous work on AMC has focused on regulation of stress response genes such as Crh and the Nr3c1. While critical for the AMC phenotype [Citation11] these two components of the stress system are likely not the only important factors for such large-scale and diverse reprogramming of the developing brain.
Together, our results provide the first integrative, genome-wide overview of the genes and pathways that contribute to the “reprogrammed” cognitive and emotional phenotype resulting from AMC. The results offer five novel observations. First, we identified a significant (P = 0.015) brain region specific, reduction of global DNA methylation in the hypothalamus, as well as five large-scale PMDs. Second, we identified 9,439 significant (P < 0.05) small-scale DMRs associated with 4,023 unique genes, which function in neurodevelopment and neurotransmission and may contribute to the establishment and maintenance of the behavioral phenotype. Third, we identified 2,464 significant (q < 0.05) DE transcripts by RNA-seq analysis that extends our understanding of the gene expression differences to neurotransmission and other crucial neurologic functions by highlighting a role for translation and oxidative phosphorylation. Fourth, we identified the predicted targets of 5 significantly (P < 0.05) altered miRNAs and also a putative interaction between mir-542-5p and the Ube3a1 3’UTR. Finally, we integrated these diverse data sets and identified a list of 20 genes with three lines of evidence (methylation, expression, and altered miRNA target) that are highly relevant to the stress resiliency phenotype resulting from AMC.
Consistent with prior descriptions for reduced methylation together with reduced CRH expression in the hypothalamus of AMC rats [Citation21], the connection between genome-wide DNA methylation and gene expression was complex, and more apparent at the level of pathways than at individual genes. At the level of gene ontology, the cellular component term “synapse part” is significantly (q < 0.05) enriched in both datasets. Reactive oxygen species (ROS) generated by oxidative phosphorylation in the mitochondria are not only a key signaling molecule behind synaptic plasticity and memory formation [Citation53], but are also critical to neurodevelopment since they regulate PI3K/AKT/mTOR/PTEN signaling [Citation54]. Furthermore, the hypomethylated Grb10 gold DMR (FWER <0.05) belongs to a genomically-imprinted gene encoding for a receptor-binding protein that interacts with insulin and insulin-like growth factor receptors of the PI3K/AKT/mTOR/PTEN signaling pathway [Citation55]. Although a CpG island in the promoter of Grb10 is hypomethylated in AMC, RNA-seq analysis does not show altered transcript abundance. This disconnect could reflect an ongoing regulatory process where transcriptional changes are not yet or no longer apparent or may be a limitation of our analysis. Translational control is achieved via eukaryotic initiation factors (eIFs) that interact with the ribosome to drive the protein synthesis needed for synaptic plasticity [Citation56]. Additionally, PI3K/AKT/mTOR/PTEN signaling, Wnt/β-catenin signaling, and protein kinase A (PKA) signaling converge to shape neuroplasticity, neurogenesis, cell survival, and resilience [Citation57]. Finally, protein kinase A signaling is activated by G-protein-coupled receptors (GPCRs) that have neurotransmitter and hormone ligands, which include CRH and URN3 [Citation58]. The GPCRs of PKA signaling utilize cyclic adenosine monophosphate (cAMP) as a secondary messenger to activate PKA, which is also affected by handling to alter relevant gene expression profiles, potentially including the glucocorticoid receptor gene Nr3c1 [Citation52]. Ultimately, the overlapping pathways between the methylation and gene expression suggest a broad and potentially long-term impact on numerous cellular and molecular functions important for hypothalamic function.
The genes and pathways identified from our unbiased analysis of AMC-induced hypothalamic changes may be used towards the development of biomarkers and therapeutics. Suderman et al. have shown that epigenetic programming of the HPA axis by maternal care is conserved in humans at a select locus [Citation59]. Given the potential conservation, the suite of new candidate loci identified may be examined in humans towards the development of novel epigenetic biomarkers of resiliency [Citation60]. Such a diagnostic may then be used to identify individuals that would benefit from corrective measures, which include environmental enrichment [Citation61] and pharmacologic agents. Other studies have shown that the transcriptomic profile and epigenetic modifications of altered maternal care can be reprogrammed at adulthood by the global epigenetic modifiers, specifically the methyl donor L-methionine or the histone deacetylase inhibitor trichostatin A (TSA) [Citation18,Citation19,Citation62,Citation63]. More recently, it has been shown that pharmacologic agents can mimic some of the effects of AMC in vitro [Citation12]. Therefore, some of the epigenomic profile established by maternal care is plastic and can be manipulated later in life. However, these previously examined modifiers result in attenuations that are global in nature and may have off-target effects. Our results could be used to overcome this limitation since the suite of therapeutic candidates identified can be utilized as prioritized targets for somatic epigenetic editing studies [Citation64–Citation67]. Ultimately, our results could form the molecular foundation for developing effective interventions for deficient early maternal care in humans or be developed as new treatments for psychiatric disorders [Citation67].
Author contributions
A.V.C., B.I.L., D.S.S., T.Z.B., J.M.L., and D.H.Y. designed research; A.V.C., B.I.L., T.S.T., J.F., A.S-T, and D.H.Y. performed research; K.W.D. and C.E.M. contributed new reagents/analytic tools; A.V.C., B.I.L., K.W.D., R.L.C., and D.H.Y. analyzed data; and A.V.C., B.I.L., and D.H.Y wrote the paper.
Disclosure of Potential Conflicts of Interest
The authors declare that no financial or scientific conflicts of interest exist.
AMC_SupplementalTables_Epigenetics.xlsx
Download MS Excel (3.9 MB)Vogel_Ciernia_et_al_Supplemental_Information_Epigenetics.docx
Download MS Word (235 KB)Acknowledgments
This work used the Vincent J. Coates Genomics Sequencing Laboratory at UC Berkeley, supported by NIH [S10 OD018174] Instrumentation Grant.
Additional information
Funding
Related Research Data
References
- Levine S. Infantile experience and resistance to physiological stress. Science. 1957;126:405. doi:10.1126/science.126.3270.405. PMID:13467220
- Francis D, Diorio J, Liu D, et al. Nongenomic transmission across generations of maternal behavior and stress responses in the rat. Science. 1999;286:1155–8. doi:10.1126/science.286.5442.1155. PMID:10550053
- Barnett SA, Burn J. Early stimulation and maternal behaviour. Nature. 1967;213:150–2. doi:10.1038/213150a0. PMID:6030570
- Meaney M, Aitken D, van Berkel C, et al. Effect of neonatal handling on age-related impairments associated with the hippocampus. Science. 1988;239:766–8. doi:10.1126/science.3340858. PMID:3340858
- Avishai-Eliner S, Eghbal-Ahmadi M, Tabachnik E, et al. Down-regulation of hypothalamic corticotropin-releasing hormone messenger ribonucleic acid (mRNA) precedes early-life experience-induced changes in hippocampal glucocorticoid receptor mRNA. Endocrinology. 2001;142:89–97. doi:10.1210/endo.142.1.7917. PMID:11145570
- Fenoglio KA, Chen Y, Baram TZ. Neuroplasticity of the hypothalamic-pituitary-adrenal axis early in life requires recurrent recruitment of stress-regulating brain regions. J Neurosci. 2006;26:2434–42. doi:10.1523/JNEUROSCI.4080-05.2006. PMID:16510721
- Meaney MJ, Viau V, Bhatnagar S, et al. Cellular mechanisms underlying the development and expression of individual differences in the hypothalamic-pituitary-adrenal stress response. J Steroid Biochem Mol Biol. 1991;39:265–74. doi:10.1016/0960-0760(91)90072-D. PMID:1888687
- Caldji C, Tannenbaum B, Sharma S, et al. Maternal care during infancy regulates the development of neural systems mediating the expression of fearfulness in the rat. Proc Natl Acad Sci U S A. 1998;95:5335–40. doi:10.1073/pnas.95.9.5335. PMID:9560276
- Liu D, Diorio J, Tannenbaum B, et al. Maternal care, hippocampal glucocorticoid receptors, and hypothalamic-pituitary-adrenal responses to stress. Science. 1997;277:1659–62. doi:10.1126/science.277.5332.1659. PMID:9287218
- Viau V, Sharma S, Plotsky P, et al. Increased plasma ACTH responses to stress in nonhandled compared with handled rats require basal levels of corticosterone and are associated with increased levels of ACTH secretagogues in the median eminence. J Neurosci. 1993;13:1097–105. doi:10.1523/JNEUROSCI.13-03-01097.1993. PMID:8382733
- Plotsky PM, Meaney MJ. Early, postnatal experience alters hypothalamic corticotropin-releasing factor (CRF) mRNA, median eminence CRF content and stress-induced release in adult rats. Mol Brain Res. 1993;18:195–200. doi:10.1016/0169-328X(93)90189-V. PMID:8497182
- Singh-Taylor A, Molet J, Jiang S, et al. NRSF-dependent epigenetic mechanisms contribute to programming of stress-sensitive neurons by neonatal experience, promoting resilience. Mol Psychiatry. 2018;23:648–57.
- Fenoglio KA, Brunson KL, Avishai-Eliner S, et al. Enduring, handling-evoked enhancement of hippocampal memory function and glucocorticoid receptor expression involves activation of the corticotropin-releasing factor type 1 receptor. Endocrinology. 2005;146:4090–6. doi:10.1210/en.2004-1285. PMID:15932935
- Korosi A, Baram TZ. The pathways from mother's love to baby's future. Front Behav Neurosci. 2009;3:27. doi:10.3389/neuro.08.027.2009. PMID:19826614
- Meaney MJ, Aitken DH. The effects of early postnatal handling on hippocampal glucocorticoid receptor concentrations: Temporal parameters. Dev Brain Res. 1985;22:301–4. doi:10.1016/0165-3806(85)90183-X.
- Korosi A, Shanabrough M, McClelland S, et al. Early-life experience reduces excitation to stress-responsive hypothalamic neurons and reprograms the expression of corticotropin-releasing hormone. J Neurosci. 2010;30:703–13. doi:10.1523/JNEUROSCI.4214-09.2010. PMID:20071535
- Singh-Taylor A, Korosi A, Molet J, et al. Synaptic rewiring of stress-sensitive neurons by early-life experience: A mechanism for resilience? Neurobiol Stress. 2015;1:109–15. doi:10.1016/j.ynstr.2014.10.007. PMID:25530985
- Weaver ICG, Cervoni N, Champagne FA, et al. Epigenetic programming by maternal behavior. Nat Neurosci. 2004;7:847–54. doi:10.1038/nn1276. PMID:15220929
- Weaver ICG, Meaney MJ, Szyf M. Maternal care effects on the hippocampal transcriptome and anxiety-mediated behaviors in the offspring that are reversible in adulthood. Proc Natl Acad Sci U S A. 2006;103:3480–5. doi:10.1073/pnas.0507526103. PMID:16484373
- McGowan PO, Suderman M, Sasaki A, et al. Broad epigenetic signature of maternal care in the brain of adult rats. PLoS One. 2011;6:e14739. doi:10.1371/journal.pone.0014739. PMID:21386994
- McClelland S, Korosi A, Cope J, et al. Emerging roles of epigenetic mechanisms in the enduring effects of early-life stress and experience on learning and memory. Neurobiol Learn Mem. 2011;96:79–88. doi:10.1016/j.nlm.2011.02.008. PMID:21338703
- Schroeder DI, Lott P, Korf I, et al. Large-scale methylation domains mark a functional subset of neuronally expressed genes. Genome Res. 2011;21:1583–91. doi:10.1101/gr.119131.110. PMID:21784875
- Dunaway KW, Islam MS, Coulson RL, et al. Cumulative impact of polychlorinated biphenyl and large chromosomal duplications on DNA methylation, chromatin, and expression of autism candidate genes. Cell Rep. 2016;17:3035–48. doi:10.1016/j.celrep.2016.11.058. PMID:27974215
- Lister R, Mukamel EA, Nery JR, et al. Global epigenomic reconfiguration during mammalian brain development. Science. 2013;341:1237905. doi:10.1126/science.1237905. PMID:23828890
- Geraldo S, Khanzada UK, Parsons M, et al. Targeting of the F-actin-binding protein drebrin by the microtubule plus-tip protein EB3 is required for neuritogenesis. Nat Cell Biol. 2008;10:1181–9. doi:10.1038/ncb1778. PMID:18806788
- Murata Y, Doi T, Taniguchi H, et al. Proteomic analysis revealed a novel synaptic proline-rich membrane protein (PRR7) associated with PSD-95 and NMDA receptor. Biochem Biophys Res Commun. 2005;327:183–91. doi:10.1016/j.bbrc.2004.11.154. PMID:15629447
- Brunet A, Bonni A, Zigmond MJ, et al. Akt promotes cell survival by phosphorylating and inhibiting a forkhead transcription factor. Cell. 1999;96:857–68. doi:10.1016/S0092-8674(00)80595-4. PMID:10102273
- Houge G, Rasmussen IH, Hovland R. Loss-of-function CNKSR2 mutation is a likely cause of non-syndromic X-linked intellectual disability. Mol Syndromol. 2012;2:60–3. doi:10.1159/000335159. PMID:22511892
- Lee SR, Ramos SM, Ko A, et al. AR and ER interaction with a p21-activated kinase (PAK6). Mol Endocrinol. 2002;16:85–99. doi:10.1210/mend.16.1.0753. PMID:11773441
- Hansen KD, Langmead B, Irizarry RA. BSmooth: From whole genome bisulfite sequencing reads to differentially methylated regions. Genome Biol. 2012;13:R83. doi:10.1186/gb-2012-13-10-r83. PMID:23034175
- Ong C-T, Corces VG. CTCF: An architectural protein bridging genome topology and function. Nat Publ Gr. 2014;15:234–46.
- Antinucci P, Nikolaou N, Meyer MP, et al. Teneurin-3 specifies morphological and functional connectivity of retinal ganglion cells in the vertebrate visual system. Cell Rep. 2013;5:582–92. doi:10.1016/j.celrep.2013.09.045. PMID:24183672
- Ma H, Ng HM, Teh X, et al. Zfp322a regulates mouse ES cell pluripotency and enhances reprogramming efficiency. PLoS Genet. 2014;10:e1004038. doi:10.1371/journal.pgen.1004038. PMID:24550733
- Nyman E, Rajan MR, Fagerholm S, Brännmark C, Cedersund G, Strålfors P. A single mechanism can explain network-wide insulin resistance in adipocytes from obese patients with type 2 diabetes. J Biol Chem. 2014;289:33215–33230.
- Besnard A, Galan-Rodriguez B, Vanhoutte P, et al. Elk-1 a transcription factor with multiple facets in the brain. Front Neurosci. 2011;5:35. doi:10.3389/fnins.2011.00035. PMID:21441990
- Valluy J, Bicker S, Aksoy-Aksel A, et al. A coding-independent function of an alternative Ube3a transcript during neuronal development. Nat Neurosci. 2015;18:666–73. doi:10.1038/nn.3996. PMID:25867122
- Han J, Lee Y, Yeom K-H, et al. Molecular basis for the recognition of primary microRNAs by the drosha-DGCR8 complex. Cell. 2006;125:887–901. doi:10.1016/j.cell.2006.03.043. PMID:16751099
- Pritchard LE, Turnbull AV, White A. REVIEW Pro-opiomelanocortin processing in the hypothalamus: Impact on melanocortin signalling and obesity. J Endocrinol. 2002;172:411–21. doi:10.1677/joe.0.1720411. PMID:11874690
- Khoshbouei H, Cecchi M, Dove S, et al. Behavioral reactivity to stress: Amplification of stress-induced noradrenergic activation elicits a galanin-mediated anxiolytic effect in central amygdala. Pharmacol Biochem Behav. 2002;71:407–17. doi:10.1016/S0091-3057(01)00683-9. PMID:11830175
- Hu W, Zhang M, Czéh B, et al. Stress impairs GABAergic network function in the hippocampus by activating nongenomic glucocorticoid receptors and affecting the integrity of the parvalbumin-expressing neuronal network. Neuropsychopharmacology. 2010;35:1693–707. doi:10.1038/npp.2010.31. PMID:20357756
- Lammich S, Kojro E, Postina R, et al. Constitutive and regulated alpha-secretase cleavage of Alzheimer's amyloid precursor protein by a disintegrin metalloprotease. Proc Natl Acad Sci U S A. 1999;96:3922–7. doi:10.1073/pnas.96.7.3922. PMID:10097139
- Marshall CR, Noor A, Vincent JB, et al. Structural variation of chromosomes in autism spectrum disorder. Am J Hum Genet. 2008;82:477–88. doi:10.1016/j.ajhg.2007.12.009. PMID:18252227
- Murgatroyd C, Spengler D. Epigenetic programming of the HPA axis: Early life decides. Stress. 2011;14:581–9. doi:10.3109/10253890.2011.602146. PMID:21854166
- Pappa I, St Pourcain B, Benke K, et al. A genome-wide approach to children's aggressive behavior: The EAGLE consortium. Am J Med Genet Part B Neuropsychiatr Genet. 2016;171:562–72. doi:10.1002/ajmg.b.32333.
- Ju M, Wray D. Molecular identification and characterisation of the human eag2 potassium channel. FEBS Lett. 2002;524:204–10. doi:10.1016/S0014-5793(02)03055-7. PMID:12135768
- Satijn DP, Gunster MJ, van der Vlag J, et al. RING1 is associated with the polycomb group protein complex and acts as a transcriptional repressor. Mol Cell Biol. 1997;17:4105–13. doi:10.1128/MCB.17.7.4105. PMID:9199346
- Lupien SJ, King S, Meaney MJ, et al. Child's stress hormone levels correlate with mother's socioeconomic status and depressive state. Biol Psychiatry. 2000;48:976–80. doi:10.1016/S0006-3223(00)00965-3. PMID:11082471
- Baram TZ, Davis EP, Obenaus A, et al. Fragmentation and unpredictability of early-life experience in mental disorders. Am J Psychiatry. 2012;169:907–15. doi:10.1176/appi.ajp.2012.11091347. PMID:22885631
- Gunnar MR, Frenn K, Wewerka SS. Van Ryzin MJ. Moderate versus severe early life stress: Associations with stress reactivity and regulation in 10-12-year-old children. Psychoneuroendocrinology. 2009;34:62–75. doi:10.1016/j.psyneuen.2008.08.013. PMID:18835102
- Halligan SL, Herbert J, Goodyer I, et al. Disturbances in morning cortisol secretion in association with maternal postnatal depression predict subsequent depressive symptomatology in adolescents. Biol Psychiatry. 2007;62:40–6. doi:10.1016/j.biopsych.2006.09.011. PMID:17188253
- Wilson RS, Schneider JA, Boyle PA, et al. Chronic distress and incidence of mild cognitive impairment. Neurology. 2007;68:2085–92. doi:10.1212/01.wnl.0000264930.97061.82. PMID:17562829
- Gourion D, Arseneault L, Vitaro F, et al. Early environment and major depression in young adults: A longitudinal study. Psychiatry Res. 2008;161:170–6. doi:10.1016/j.psychres.2007.07.026. PMID:18849082
- Kishida KT, Klann E. Sources and targets of reactive oxygen species in synaptic plasticity and memory. Antioxid Redox Signal. 2006;0:61121054212009. doi:10.1089/ars.2007.9.ft-8.
- Ostrakhovitch EA, Semenikhin OA. The role of redox environment in neurogenic development. Arch Biochem Biophys. 2013;534:44–54. doi:10.1016/j.abb.2012.08.002. PMID:22910298
- Yu Y, Yoon S-O, Poulogiannis G, et al. Phosphoproteomic Analysis Identifies Grb10 as an mTORC1 Substrate That Negatively Regulates Insulin Signaling. Science. 2011;332:1322–6. doi:10.1126/science.1199484. PMID:21659605
- Santini E, Huynh TN, Klann E. Mechanisms of translation control underlying long-lasting synaptic plasticity and the consolidation of long-term memory. Progr Mol Biol Transl Sci. 2014;122:131–67.
- Charney DS, Manji HK. Life stress, genes, and depression: Multiple pathways lead to increased risk and new opportunities for intervention. Sci Signal. 2004;2004:re5. doi:10.1126/stke.2252004re5.
- Henckens MJ, Deussing JM, Chen A. Region-specific roles of the corticotropin-releasing factor–urocortin system in stress. Nat Rev Neurosci. 2016;17:636–51.
- Suderman M, McGowan PO, Sasaki A, et al. Conserved epigenetic sensitivity to early life experience in the rat and human hippocampus. Proc Natl Acad Sci U S A. 2012;109:17266–72. doi:10.1073/pnas.1121260109. PMID:23045659
- consortium TB. Quantitative comparison of DNA methylation assays for biomarker development and clinical applications. Nat Biotech. 2016;34:726–37. doi:10.1038/nbt.3605.
- Francis DD, Diorio J, Plotsky PM, et al. Environmental enrichment reverses the effects of maternal separation on stress reactivity. J Neurosci. 2002;22:7840–3. PMID:12223535
- Champagne FA, Francis DD, Mar A, et al. Variations in maternal care in the rat as a mediating influence for the effects of environment on development. Physiol Behav. 2003;79:359–71. doi:10.1016/S0031-9384(03)00149-5. PMID:12954431
- Weaver ICG, Champagne FA, Brown SE, et al. Reversal of maternal programming of stress responses in adult offspring through methyl supplementation: Altering epigenetic marking later in life. J Neurosci. 2005;25:11045–54. doi:10.1523/JNEUROSCI.3652-05.2005. PMID:16306417
- Laufer BI, Singh SM. Strategies for precision modulation of gene expression by epigenome editing: An overview. Epigenet Chromatin. 2015;8:34. doi:10.1186/s13072-015-0023-7. PMID:26388942
- Stelzer Y, Wu H, Song Y, et al. Parent-of-origin DNA methylation dynamics during mouse development. Cell Rep. 2016;16:3167–80. doi:10.1016/j.celrep.2016.08.066. PMID:27653683
- Morita S, Noguchi H, Horii T, et al. Targeted DNA demethylation in vivo using dCas9–peptide repeat and scFv–TET1 catalytic domain fusions. Nat Biotechnol. 2016;34:1060–5. doi:10.1038/nbt.3658. PMID:27571369
- Stepper P, Kungulovski G, Jurkowska RZ, et al. Efficient targeted DNA methylation with chimeric dCas9–Dnmt3a–Dnmt3L methyltransferase. Nucleic Acids Res. 2017;45:1703–13. doi:10.1093/nar/gkw1112. PMID:27899645