
ABSTRACT
Periodontal disease (PD) afflicts 46% of Americans with no effective adjunctive therapies available. While most pharmacotherapy for PD targets bacteria, the host immune response is responsible for driving tissue damage and bone loss in severe disease. Herein, we establish that the histone demethylase KDM4B is a potential drug target for the treatment of PD. Immunohistochemical staining of diseased periodontal epithelium revealed an increased abundance of KDM4B that correlates with inflammation. In murine calvarial sections exposed to Aggregatibacter actinomycetemcomitans lipopolysaccharide (Aa-LPS), immunohistochemical staining revealed a significant increase in KDM4B protein expression. The 8-hydroxyquinoline ML324 is known to inhibit the related demethylase KDM4E in vitro, but has not been evaluated against any other targets. Our studies indicate that ML324 also inhibits KDM4B (IC50: 4.9 μM), and decreases the pro-inflammatory cytokine response to an Aa-LPS challenge in vitro. Our results suggest that KDM4B inhibition-induced immunosuppression works indirectly, requiring new protein synthesis. In addition, fluorescence-stained macrophages exhibited a significant decrease in global monomethyl histone 3 lysine 4 (H3K4me) levels following an Aa-LPS challenge that was prevented by KDM4B inhibition, suggesting this effect is produced through KDM1A-mediated demethylation of H3K4. Finally, ML324 inhibition of KDM4B in osteoclast progenitors produced a significant reduction in Aa-LPS-induced osteoclastogenesis. These data link histone methylation with host immune response to bacterial pathogens in PD, and suggest a previously unreported, alternative mechanism for epigenetic control of the host inflammatory environment. As such, KDM4B represents a new therapeutic target for treating hyper-inflammatory diseases that result in bone destruction.
Introduction
Periodontal disease (PD) causes irreversible tissue damage and bone loss, and affects 46% of adult Americans. PD is a common chronic inflammatory disease characterized by destruction of the supporting structures of the teeth [Citation1,Citation2]. Current traditional therapies focus on mechanical debridement through scaling and root planing but are not always effective [Citation3,Citation4]. This procedure disrupts plaque biofilms, thus enabling the host immune system to abate, and consequently reduces collateral tissue damage due to the exaggerated host immune response. Unfortunately, complete removal of subgingival plaque and calculus in PD patients, even by highly skilled clinicians, is impossible, and patients who present with PD are more likely to suffer from progressive disease (3). Localized disease sites are commonly non-responsive to this treatment alone, highlighting the need for development of effective adjunctive therapeutics to treat PD. Traditional and even emerging treatments seldom target the host immune response, which is the true source of tissue damage in PD (3). Pharmacology research in the field of periodontics is relatively stagnant and the current focus is aimed towards antimicrobial drugs as an adjunct treatment to scaling and root planing (3). This manuscript describes a novel approach that targets epigenetic control of gene expression in the host to resolve the pro-inflammatory immune response driving PD.
The first histone lysine demethylase (KDM) enzyme, KDM1A (also known as lysine-specific demethylase 1, LSD1), was discovered in 2004 [Citation5]. Since that time, 16 histone demethylases (2 FAD+ dependent and 14 JmjC-domain-containing) have been characterized [Citation6,Citation7]. To date, most efforts to target KDMs have been related to cancer drug discovery; however, dysregulated epigenetic modulation has more recently been identified as a factor in diseases other than cancer, such as neurological disease [Citation8,Citation9], blood disorders [Citation10,Citation11], viral infection [Citation12], diabetes[Citation13,Citation14], and fibrosis [Citation15]. More recently, the KDM4 family of epigenetic modifying enzymes have been linked to positive regulation of many immunological processes, and therefore serve as an interesting target for development of therapeutics to treat hyperinflammatory or autoimmune disorders.
The KDM4 family of epigenetic modifiers catalyze the demethylation of di- and tri-methyl histone 3 lysine 9 (H3K9me2 and H3K9me3), di- and tri-methyl histone 3 lysine 36 (H3K36me2 and H3K36me3), di- and tri-methyl histone1.4 lysine 26 (H1.4K26me2 and H1.4K26me3), and trimethyl histone 3 lysine 56 (H3K56me3). Each member of the KDM4 family contains a Jumonji domain, and requires Fe2+, 2-oxoglutarate, and O2 as cofactors for demethylase activity [Citation16]. Only KDM4 family members A-C contain double PHD and Tudor domains, and these differences are thought to attribute to the variable specificity between KDM4A-C vs. KDM4D-F. KDM4A-C have a 5-fold specificity for H3K9 over that of H3K36 and H1.4K26 [Citation17]. KDM4D-F are half the size of other KDM4 family members and are unable to demethylate H3K36 [Citation18].
The pro-inflammatory cytokines TNF-α and IL-6 are classically upregulated in gingival connective tissues of PD patients [Citation19], and these cytokines are secreted from periopathogen-activated macrophages through toll like receptor (TLR) signaling [Citation20]. TLR4 binding by periopathogenic LPS activates a signaling cascade that drives both cytokine and chemokine production. These signals in turn result in production of matrix metalloproteinases (MMPs) that break down tissue and osteoclast activation, which breaks down bone, the major hallmarks of PD [Citation21]. KDM4B and its major substrate, H3K9, have been linked to this process by several research groups. For example, TNF-α treatment induces leukocyte attachment and migration through induction of KDM4B and a subsequent decrease in H3K9 methylation [Citation22]. Conversely, TNF-α transcription is repressed following H3K9 methylation during the process of endotoxin tolerance [Citation23]. Trimethylation at H3K9 contributes to the repression of TLR4 expression [Citation24] and decreased levels of this mark are associated with increased TLR4-mediated expression of pro-inflammatory cytokines such as IL-6. This occurs through recruitment of NF-KB p65 to the proximal promoter of these genes [Citation25]. Also, H3K9me3 levels are decreased in macrophages following exposure to high glucose, and this occurs with a simultaneous increase in inflammatory cytokine production [Citation26]. H3K9me3 levels have also been found to increase in response to hypoxia, which downregulates mRNA expression for chemokine Ccl2 and the chemokine receptors Ccr1 and Ccr5 [Citation27]. A minor substrate of KDM4B, H3K36, has also been associated with macrophage polarization and increased methylation at this mark results in suppressed production of IL-6 and TNF-α by macrophages [Citation28].These data implicate KDM4B as a mediator of PD progression, and demonstrate that its demethylation activity is a signature of several pro-inflammatory processes.
It is well known that histone demethylase enzymes are conserved throughout species, and that these enzymes are commonly redundant, share substrate specificity among different classes and families, and are coordinated in their activity. Multiple studies support the idea that H3K4 and H3K9 methylation are mutually exclusive states [Citation29,Citation30]. For example, cross talk between KDM4B and KDM1A enzymes leads to a balanced system wherein lysine 9 methylation serves as a prerequisite to lysine 4 demethylation by KDM1A [Citation30]. Because it is known that the demethylation activity of KDM1A on Histone 3 lysine 4 leads to repression of pro-inflammatory cytokine gene transcription [Citation31], we postulate that KDM4B is a positive regulator of the pro-inflammatory cytokine response through an indirect mechanism by inhibiting KDM1A.
The current study aims to interrogate the activity of KDM4B as it relates to the immune response in periodontal disease through the use of the JMJD2 demethylase inhibitor ML324 [Citation32]. We hypothesize that KDM4B inhibition using this inhibitor will result in a reduced immune response to bacterial LPS, and that ML324 could prove useful as a chemical tool and lead compound for future studies on PD or other hyper-inflammatory or autoimmune diseases.
Results
KDM4B and KDM4E protein abundance is increased in areas of periodontal inflammatory infiltrate
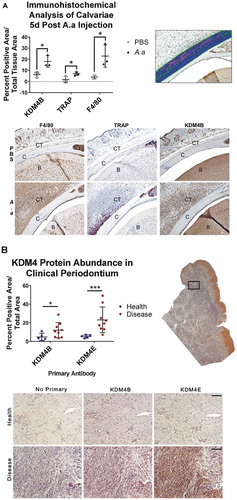
To test the hypothesis that KDM4B is overexpressed upon LPS stimulation, histological sections from the calvariae of mice that had been injected daily for 5 days with fixed Aggregatibacter actinomycetemcomitans (Aa) or PBS were stained for KDM4B protein. An increase in resorption pits due to osteoclast activity was observed in calvariae treated with A.a, confirming this as a viable model for periodontal disease [Citation33]. Staining for tartrate resistant acid phosphatase (TRAP) and F4/80 marked the area in the calvarial sections most active with inflammatory infiltration. This region of interest showed a significantly higher concentration of KDM4B protein ()), indicating that KDM4B protein levels correlate with immune activation in periodontal disease. The experimental inhibitor ML324 has also been shown to inhibit the related demethylase KDM4E [Citation32], but this protein is not found in mice. Therefore, tissue sections from periodontally diseased human patients and healthy controls were stained for both KDM4E and KDM4B using immunohistochemistry. The connective tissue underlying the oral epithelium was chosen as the region of interest for analysis. A statistically significant increase in both KDM4B and KDM4E abundance was observed in diseased vs. healthy tissues ()), demonstrating that KDM4 enzymes are implicated in periodontal disease status.
Figure 1. KDM4B abundance is significantly increased in periodontal diseased versus healthy tissues. Live Aa was injected subcutaneously into 12-week old C57BL/6 mice at the mid-sagittal region of the calvarium every day for 5d. Paraffin embedded sections were stained for F4/80, TRAP and KDM4B using immunohistochemistry, all of which were significantly upregulated in diseased versus healthy calvariae. 10x Images presented are representative of the data set. (a) In clinical periodontal specimens, the region of interest was defined as the connective tissue underlying the oral epithelium. Paraffin embedded sections were stained for KDM4E and KDM4B using immunohistochemistry, both of which were upregulated in diseased versus healthy patient tissues. 20x images are representative of the data set. (b) positive pixels quantified using color thresholding in imagej. Data are presented as mean ± SD. Significance was determined using a one-tailed Wilcoxon ranked sum test. Epithelium (E) Calvarial Bone (C) Brain (B) *P < 0.05, ***P < 0.001. Scale bars, 100 μm.
ML324, previously defined as a KDM4E inhibitor, shows inhibitory activity against KDM4B
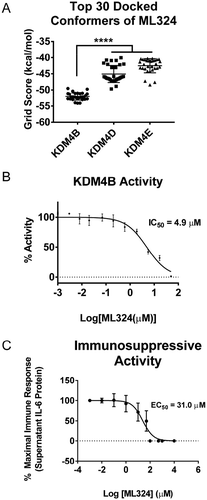
ML324 was docked into the active site of the KDM4A-E family in flexible mode, and was found to have high computational selectivity for the KDM4B active site ()). ML324 exhibited a lower average binding energy to the KDM4B active site (−53.04 kcal/mol) for the top 30 conformers vs. KDM4E (−42.52 kcal/mol). Additionally, we used an Alphascreen® assay to determine the effect of ML324 on the demethylation activity of KDM4B. Here we show that ML324 has inhibitory activity towards KDM4B, with an IC50 value of 4.9 μM ()). Additionally, we have defined the EC50 of ML324 for translational immunosuppression in primary macrophages to be 31 μM ()). IL-6 production drives periodontal disease pathogenesis; therefore, the ability of ML324 to effectively reduce the production of this cytokine emphasizes the potential of KDM4B inhibitors as therapeutics for PD treatment.
Figure 2. ML324 demonstrates inhibitory activity towards KDM4B and causes dose dependent immunosuppression. The KDM4A-E protein crystal structures were subjected to unbiased docking of ML324 where the top 30 conformers in the active site were used for analysis (a). KDM4A and C had no poses of ML324 dock into the active site of these enzymes, therefore this data is not displayed. Inhibition of KDM4B was assessed using an 11-point IC50 determination using the histone demethylase AlphaScreen (PerkinElmer) assay in triplicate resulting in an IC50 of 4.9 μM (b). The EC50 for immunosuppression using ML324 was determined to be 31 μM by measuring supernatant IL-6 protein following a 24h Aa-LPS stimulation with variable concentrations of ML324 in primary BMDM cells (c). Cells were treated for 1h with each indicated concentration of ML324, followed by Aa-LPS challenge for 24h. The data were normalized as a percentage of the maximal IL-6 response in response to LPS. Data for all panels are represented as mean ± SD. n = 4.
KDM4B inhibition using ML324 results in a significantly reduced cytokine immune response to Aa-LPS in macrophages
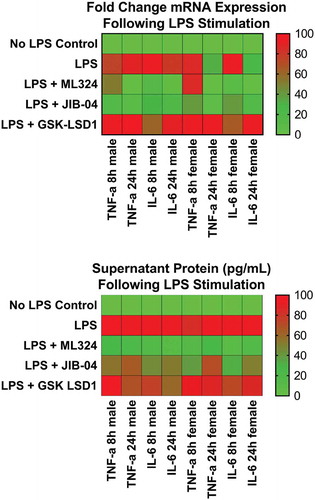
After a 1-hour pre-treatment with ML324 (50 μM) followed by an inflammatory Aa-LPS challenge (100 ng/mL), ELISA and PCR analysis revealed that the KDM4B inhibitor ML324 significantly reduced the levels of inflammatory cytokines in primary murine macrophages (). At 8- and 24-hour time points and in both male and female cells, ML324 was able to significantly reduce IL-6 and TNF-α transcription and translation compared to LPS treatment with vehicle control (DMSO). A pan-selective KDM4 family inhibitor, JIB-04, was also able to produce this effect in most groups, but with a more variable response. As expected, GSK-LSD1, a KDM1A inhibitor, produced either no change or a significant increase compared to vehicle controls in the majority of groups (Figure S1). These data demonstrate not only that KDM4B inhibition reduces inflammatory cytokine production but also that this effect is specific to KDM4B over the KDM4 family as a whole. It is important to note, however, that kinetics and mode of binding of ML324 to KDM4B have not been described, although the in vivo pharmacokinetics and ADME properties of this compound are extremely favorable [Citation32]. Additionally, this data shows that, as hypothesized, the activity of KDM4B and KDM1A are negatively correlated, and that inhibiting KDM1A via GSK-LSD1 gives an opposing effect on inflammatory cytokine production compared to KDM4B inhibition using ML324.
Figure 3. KDM4B inhibition significantly reduces the Aa-LPS-induced immune response in primary macrophages. Male and female murine bone marrow derived macrophages were pre-treated for 1h with the selective KDM4B inhibitor, ML324, a family-wide KDM4 inhibitor, JIB-04, and a KDM1A inhibitor, GSK-LSD1 or DMSO vehicle. Following drug treatment, cells were challenged with Aa-LPS for 8 and 24h, where gene expression and supernatant protein concentration were measured via rt-qPCR relative to GAPDH and ELISA relative to a standard curve. Data was normalized as a percentage of the maximal response (red) in each group for display. Refer to figure S1 for raw data and statistical significance. n = 4 per experiment, data is representative of 3 experiments.
KDM4B inhibition using ML324 results in a significant reduction in osteoclastogenesis
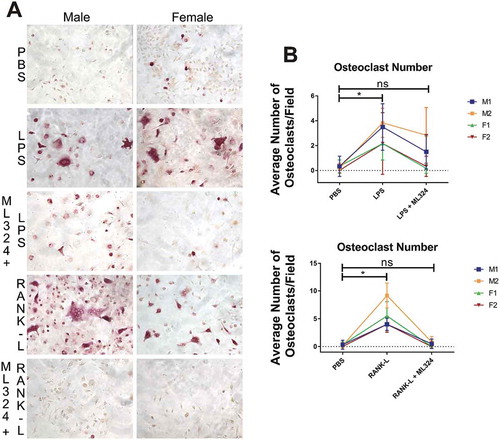
After 5 days of priming bone marrow-derived hematopoietic stem cells into pre-osteoclasts using macrophage colony stimulating factor (M-CSF) and receptor activator of nuclear factor kappa β ligand (RANK-L), osteoclastogenesis was significantly increased in cells treated with RANK-L or Aa-LPS for 3 days compared to PBS treated cells, and this effect was lost in cells pre-treated with ML324 (10 μM). TRAP+, multinucleated cells were significantly increased in both cells treated with Aa-LPS as well as RANK-L compared to PBS treated control cells. By contrast, in cells that were pre-treated with ML324, there was no significant difference in osteoclast formation compared to control groups, regardless of whether cells were stimulated with Aa-LPS or RANK-L. Additionally, formed osteoclasts appeared smaller, and had less intense TRAP staining ().
Figure 4. KDM4B inhibition via ML324 prevents osteoclast formation induced by either Aa-LPS or RANK-L. Murine bone marrow was differentiated into osteoclasts by supplementation of the hematopoietic compartment with M-CSF for 3 days and RANK-L + M-CSF for 2 days, where cells were rinsed and pre-treated for 1h with M-CSF and ML324 or DMSO vehicle followed by supplementation with RANK-L or Aa-LPS. After 72 hours, cells were fixed and stained for tartrate resistant acid phosphatase. 3 representative images were taken of each well, and TRAP+, multinucleated cells were counted in each field. 10x representative images of M1 and F2 are displayed (a). Each mouse (M1, M2 = male; F1, F2 = female) independently showed a significant increase in osteoclast formation in response to LPS or RANK-L alone compared to PBS controls, but no significant difference in osteoclast formation was observed between PBS and ML324 + LPS or ML324 + RANK-L treated cells (b). Data is presented as the mean number of osteoclasts in each field ± SD. Statistical significance was determined using a paired Friedman test with multiple comparisons at an α = 0.05. *P < 0.03 n = 3 fields per well, 2 wells per group, per mouse.
Immunosuppressive effects of KDM4B inhibition act indirectly, through demethylation of H3K4 by KDM1A
To understand the mechanism for KDM4B inhibition induced immunosuppression, we used the RAW264.7 macrophage cell line, which is the best representation of primary bone marrow derived macrophages [Citation34]. Interestingly, when cycloheximide (5 μg/mL) was co-administered with ML324 (50 μM) prior to Aa-LPS challenge in these cells, the ability of ML324 to reduce LPS-induced cytokine production is lost ()). Because cycloheximide inhibits eukaryotic translation, this data indicates that ML324-induced immunosuppression relies on new protein synthesis, and that the reduced cytokine response to LPS is not a direct effect of KDM4B inhibition, but rather requires intracellular signaling.
Figure 5. KDM4B inhibition mediated immunosuppressive effect is indirect, requiring new protein synthesis and increased KDM1A activity. New protein synthesis is required for the immunosuppressive effects of ML324 on the LPS-induced immune response (a). Cells were pre-treated with cycloheximide (5ug/mL) for 4 h with or without ML324 (50 μM) followed by Aa-LPS challenge (100 ng/mL) for 16h where RNA was collected and analyzed using qRT-PCR to measure IL-6 expression compared to GAPDH as an endogenous control. Statistical significance was determined using a repeated measures ANOVA with multiple comparisons. Demethylation at H3K4 is significantly decreased in cells following LPS treatment, but the effect is reversed upon addition of ML324. (b) RAW264.7 cells were pre-treated for 1h with ML324 followed by Aa-LPS challenge for 24h where H3K4 mono-methylation was measured in fixed cells using immunofluorescent antibody to H3K4me at 10x. KDM1A activity is significantly decreased following Aa-LPS treatment alone, but in combination with ML324 pretreatment, KDM1A activity is increased. n = 3 wells, 48 fields/well, **P < 0.01. Data are represented as mean ± SD. Statistical significance was determined using one-way ANOVA with multiple comparisons where the outcome was log transformed.
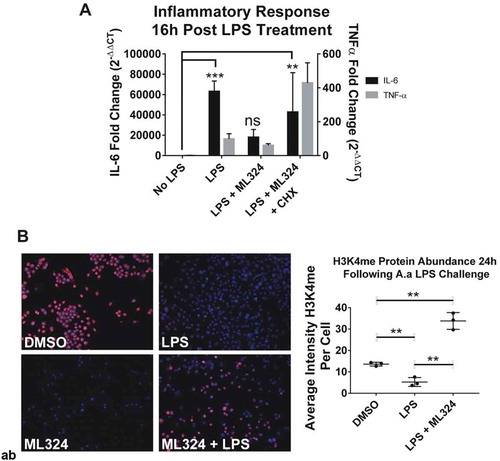
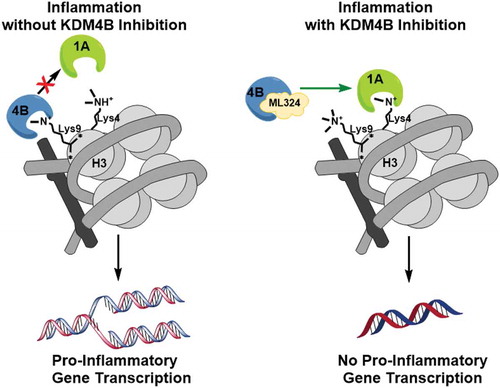
Figure 6. A schematic illustration of the mechanism of KDM4B inhibition-mediated immunosuppression. KDM4B activity inactivates KDM1A and allows for active transcription of pro-inflammatory cytokines while KDM4B inhibition allows for active demethylation by KDM1A, reducing pro-inflammatory cytokine production.
To determine if the cellular response to LPS altered KDM4B expression levels, we used a bioinformatics database [NCBI Gene Expression Omnibus (GEO) profiles]. There was no change in mRNA expression of KDM4B in response to LPS, suggesting that the activity of KDM4B as opposed to its expression level drives the immune response to LPS. Further, KDM4A, KDM4C, and KDM4D also show no significant differences in expression in response to LPS. To confirm that the activity of KDM4B was altered in response to LPS, the histone methylation marks H3K4me and H3K9me3 were monitored by immunofluorescence following ML324 pre-treatment (50 μM) and Aa-LPS challenge. Concurrent with the absence of change in mRNA levels of KDM4B, there was no significant difference in KDM4B protein levels between groups (Figure S2A). In contrast, the activity of KDM4B was decreased following ML324 pre-treatment as evidenced by a significant increase in H3K9me3. Interestingly, there was not a significant difference between control and LPS treated cells (Figure S2B). Conversely, H3K4me levels significantly decreased following Aa-LPS challenge, but ML324 pre-treatment not only reversed this effect, but also caused a significant increase in H3K4me levels compared to PBS controls (). These data together suggest that H3K4 methylation is differentially regulated by inflammatory stimuli in macrophages, and this activity can be modulated indirectly through pharmacological inhibition of KDM4B.
Discussion
To date, there is limited mechanistic data concerning the epigenetic modulation of periodontal inflammation. A 2014 study by Meng et al. used a novel BET bromodomain inhibitor, JQ1, in an experimental periodontal disease model and found that a decrease in BRD4 recruitment led to a reduction in periodontal inflammation and subsequent bone loss [Citation35]. Importantly, there have been no studies that suggest that any of the histone demethylases play a direct role in the progression or persistence of periodontal disease, despite the fact that numerous links between the KDM4 family of epigenetic modifiers and inflammation have been published. The study described herein demonstrates that inhibition of KDM4B reduces the pro-inflammatory cytokine immune response to bacterial lipopolysaccharide in macrophages. This effect occurs through new protein synthesis and a subsequent overactivation of KDM1A. In the absence of KDM4B, macrophages do not produce these inflammatory signals, and thus modulation of KDM4B activity could be utilized to locally suppress the immune response to plaque and microbial biofilms in PD patients for the resolution of inflammatory disease states.
To determine whether KDM4B inhibition could be used as a therapeutic strategy to manage periodontal disease, we first demonstrated that the abundance of KDM4B protein is significantly increased in areas of inflammatory infiltrate marked by increased F4/80+ macrophage cells and increased tartrate resistant acid phosphatase (TRAP)+ osteoclastic cells following live A.a. subcutaneous injection into murine calvariae ()) [Citation36]. The murine genome does not express KDM4E [Citation37], leading us to believe that the ML324-induced immunomodulatory effect is entirely dependent on KDM4B. However, it is likely that ML324 would also interact with KDM4E when administered in humans. Thus, we sought to determine whether one or both of these enzymes were overproduced in periopathogen-activated immune cells using human clinical PD tissues. Our results demonstrate that the abundance of both KDM4B and KDM4E protein is significantly increased in the oral epithelium of patients with periodontal disease, compared to healthy controls. ()).
Based on computational studies, ML324 demonstrated promising selectivity for KDM4B ()). Within the first 500 least energy docked poses, ML324 did not enter the active site of KDM4A or KDM4C. ML324 is a methyl derivative of the 8-hydroxyquinoline compounds developed as selective KDM4B inhibitors; yet, thus far, the drug has only been published as an inhibitor of KDM4E with an IC50 of 920 nM. The synthetic route used to produce JIB-04 is depicted in
We thus reasoned that the periodontal immune response might be attenuated in the absence of KDM4B activity. Macrophages have been established as the primary mediator of the acute PD inflammatory response [Citation39–Citation41]. For this reason, murine bone marrow derived macrophages (BMDMs) have been utilized extensively to model oral inflammatory responses. The pro-inflammatory cytokines TNF-α and IL-6 are classically up regulated in gingival connective tissues of PD patients [Citation19], and these cytokines are secreted from periopathogen-activated macrophages through toll like receptor (TLR) signaling [Citation20]. Lipopolysaccharide (LPS) is a predominate surface antigen that activates this pathway [Citation42,Citation43]; therefore, Aa-LPS, a well characterized periopathogen [Citation42,Citation43] was used to simulate the immune challenge present in PD. We anticipate that KDM4B inhibition will be useful clinically following standard of care treatment [scaling and root planing (SRP)], where cells are primed for responding to inflammatory stimuli but are temporarily halted due to the elimination of the plaque biofilm. Introduction of an adjuvant therapy directly after SRP allows for modulation of the host immune response that can prevent future episodes of hyper-inflammation that drives tissue damage and bone loss. Since most patients resume building biofilms back almost immediately after clinical intervention, we felt that a 1 hour pre-treatment would recapitulate the clinical setting most likely encountered by PD patients. Our results show that ML324-induced inhibition of KDM4B significantly suppresses the inflammatory response to bacterial LPS (). TNF-α mRNA at 8 hours after Aa-LPS challenge was the only time point tested that did not produce a statistically significant effect, and this was consistent between both male and female cells (Figure S1). This suggests that TNF-α and IL-6 are epigenetically regulated differently due to the differences seen between groups in these cytokines.
An additional component of the pathogenesis of periodontal disease is the imbalance in osteoimmunological mediators, resulting in a net loss of alveolar bone. KDM4B was recently shown to drive mesenchymal stem cells towards an osteogenic lineage preferentially over adipogenesis [Citation44], but it is unknown how KDM4B regulates osteoclastic cell types. Because we have seen a decrease in inflammatory mediators required for endogenous osteoclastogenesis, we hypothesized that inhibition of KDM4B would also reduce osteoclastogenesis. Our data demonstrates that osteoclastogenesis proceeds normally with supplementation of either Aa-LPS or RANK-L but, when KDM4B is inhibited in pre-osteoclasts using ML324, neither of these additives induce significant osteoclast formation compared to vehicle control (). This effect is seen in cells from both sexes, although there is a significantly higher number of osteoclasts formed in male cells compared to female cells, consistent with previous literature[Citation45] (Figure S3).
The mechanism by which KDM4B inhibition promotes immunosuppression is unknown. A study by Whetstine et al. demonstrated that KDM4B is structurally distinct from its other family members, and has the lowest demethylase activity of the KDM4 family, for reasons that are not clear [Citation46]. KDM4B has also been recognized for its ability to demethylate non-histone proteins, many of which are transcriptional repressors [Citation47]. We used the NCBI Gene Expression Omnibus databank to determine the differences in expression of the KDM4 family of enzymes following lipopolysaccharide treatment in primary BMDMs. We found that the expression level of these enzymes doesn’t reflect their activity, as there are no significant differences in the expression of any KDM4 family gene between LPS treated cells and vehicle treated cells (Figure S4). Therefore, we probed methylation marks rather than expression of KDM4B itself to determine the mechanism of action of ML324. The RAW264.7 macrophage cell line is the best representation of primary bone marrow derived macrophages [Citation34], but exhibit increased resiliency for use with glass plates for fluorescent imaging and highly cytotoxic chemicals such as cycloheximide. Herein we used immunofluorescent staining for methylation marks as well as cycloheximide treatment to show that KDM4B-induced immunosuppression acts via an indirect mechanism [Citation48]. After ML324 pre-treatment and Aa-LPS challenge in the presence of cycloheximide, the immunosuppressive effects of ML324 are completely abolished ()). Additionally, it appears that when ML324 is added, even more IL-6 and TNF-α are being transcribed in response to Aa-LPS ()). We postulate that because the cells are not able to effectively propagate intra- and inter-cellular signaling events, the transcripts are aberrantly abundant. Although the immunosuppressive activity of KDM4B inhibition requires new protein synthesis, an epigenetic mechanism is still at play, as evidenced by the increase in H3K9me3 and H3K4me levels that are reversed when KDM4B is inhibited ()).
These data together demonstrate that newly synthesized protein signals KDM1A following KDM4B inhibition, resulting in reduced transcriptional processing of pro-inflammatory cytokines as well as reduced osteoclast formation ().
Conclusion
ML324 is an effective inhibitor of KDM4B, which acts through binding to KDM4B and initiating de novo protein synthesis, subsequent KDM1A activation followed by H3K4 demethylation. This results in a reduced cytokine inflammatory response and decreased osteoclastogenesis in primary bone marrow cells. These data together provide a novel mechanism of immunomodulation through epigenetic modification, which can be used for further development of therapeutics for treatment of hyper-inflammatory disorders such as periodontal disease.
Methods
Human periodontal tissue procurement
This study was approved by the Institutional Review Board for the Health Sciences at the Medical University of South Carolina, USA. Samples used for the present study represent a subset of samples used for a larger study. Informed consent was obtained with all patients prior to initiating study. Prior to surgery, clinical parameters were measured at the same sites where tissues were harvested including: plaque Index (PI) on a scale of 0–3 (0-no plaque, 1-w/probe, 2-visible, 3-abundant) [Citation49], gingival Index (GI) on a scale of 0–3 (0-no inflammation, 1-mild, 2-moderate with spontaneous bleeding on proving (BOP), 3-severe with BOP, pocket depth (PD), BOP, gingival recession (REC), and clinical attachment level (CAL). Based on these parameters the inclusion criteria for the diseased group consisted of at least 1 site with PD >4 mm, GI 1–3, and PI 1–3. For the healthy controls acceptable parameters were: PD ≤4 mm, GI ≤1, and PI ≤2. The exclusion criteria for both groups included: smokers, unstable systemic diseases or chronic disorders (diabetes, rheumatoid arthritis), patients using steroids, antibiotics, NSAIDS and/or other host modulators. The procured samples were from tissues that would have been otherwise discarded after periodontal surgery and or extraction sites. When clinically indicated, procured tissues included connective tissue near the sulcular epithelium.
Immunohistochemistry
Calvariae from mice with periodontal disease, collected as previously described [Citation33] were formalin fixed, decalcified using 0.5 M EDTA, pH 8.0 for 2 weeks, paraffin embedded and cut into 7 μm sections following standard protocols. Human periodontal tissues were formalin fixed, paraffin embedded and cut into 7 μm sections following standard protocols. Sections were permeabilized using 0.2 M boric acid (Sigma, Cat# B6768). Tartrate resistant acid phosphatase (TRAP) was measured as described in BD Bioscience Technical Bulletin No. 445 following a 30-minute incubation with TRAP buffer. KDM4B was visualized following blocking with 3% normal goat serum (SeraCare, Cat# 5560–0007) using rabbit anti-KDM4B (AbCam, Cat# ab191434, 1:125) or rabbit anti-KDM4E (Novus, Cat# NBP2-49,124) overnight at 4°C. The sample was then incubated for 1 hour with biotinylated goat anti-rabbit (Vectorlabs, Cat# BA-1000, 1:500). VECTASTAIN Elite ABC HRP Kit (Vectorlabs, 1:500, Cat# PK-6100), and a DAB Peroxidase (HRP) Substrate Kit (Vectorlabs, Cat# SK-4100) was then used for development. Fifteen percent Hematoxylin (Sigma, Cat# H3136) was employed as a counterstain. Images were captured using a Nikon 80i Eclipse microscope equipped with a DS-Fi1 camera. Region of interest selection and subsequent quantification was performed using visiopharm software (n = 3) for calvarial tissues. Human periodontal tissues were analyzed using imagej (n = 5–9).
Computational chemistry
KDM4 family crystal structures (KDM4A: PDB 5F3C; KDM4B: PDB 4LXL; KDM4C: PDB 5KR7; KDM4D: PDB 5FP7; KDM4E: PDB 2W2I) were prepared using UCSF Chimera following DOCK6.5 protocols. Ligands were removed and ML324 was docked in an unbiased manner by using the entire enzyme as the active site for docking. Five hundred conformers of ML324 were written using DOCK6.5 in flexible mode with 1000 maximum orientations per computation. The top 30 conformers that docked in the active site were used for analysis.
JMJD2B enzyme assay
Inhibition of JMJD2B was assayed by BPS Biosciences using an 11-point IC50 determination using the histone demethylase AlphaScreen (PerkinElmer) assay (BPS Bioscience, Cat# 50,414). Briefly, enzymatic reactions were conducted in triplicate at room temperature for 60 minutes in a 10 µl mixture containing assay buffer (BPS Bioscience, Cat# 52,407), histone H3 peptide substrate, demethylase enzyme (BPS Bioscience, Cat# 50,111), and ML324 or 2,4-pyridine dicarboxylic acid as a reference inhibitor. All wells had a final DMSO concentration of 1%. After the enzymatic reactions were complete, anti-Mouse Acceptor beads (PerkinElmer, Cat# AL105C, 1:500) and Primary H3K9me3 antibody (BPS Biosciences, Cat# 52140E, 1:200) were added and samples were mixed. The reactions were incubated for an additional 30 minutes followed by addition of AlphaScreen Streptavidin-conjugated donor beads (PerkinElmer,Cat# 6760002S, 1:125). Thirty minutes later the samples were measured using an AlphaScreen microplate reader (EnSpire Alpha 2390 Multilabel Reader, PerkinElmer). In the absence of the compound, the intensity (Ce) in each data set was defined as 100% activity. In the absence of enzyme, the intensity (C0) in each data set was defined as 0% activity. The percent activity in the presence of each compound was calculated according to the following equation: %activity = (C-C0)/(Ce-C0), where C is the A-screen intensity in the presence of the compound. A plot of % activity vs. concentration was then constructed using non-linear regression analysis of the Sigmoidal dose-response curve generated with the equation Y = B+(T-B)/1 + 10((LogEC50-X)×Hill Slope), where Y is percent activity, B is the minimum percent activity, T is the maximum percent activity, X is the logarithm of compound concentration and Hill Slope is the slope factor/Hill coefficient. The IC50 value was determined as the concentration causing half-maximal percent activity.
Primary bone marrow culture
Primary bone marrow was cultured into macrophages as described previously [Citation50]. Briefly, bone marrow from left and right femurs and tibiae of 12–14-week-old wild type C57BL/6 mice was flushed into α-MEM (Corning, Cat# 10–022-CV) supplemented with 10% fetal bovine serum (Hyclone, Cat# SH30071.03HI) and 1% penicillin streptomycin (Sigma, Cat# P4333) and plated overnight at 37°C in 5% CO2. Cells remaining in suspension were differentiated into experimental wells at a concentration of 2E6 cells/mL for 7 days or until 80% confluency and homogeneity was achieved using macrophage colony stimulating factor (R&D Systems, Cat# 416-ML-500), reconstituted in PBS + 1% BSA (Sigma, Cat# A8806) supplementation (10 ng/mL/48 h). Cells at this point are referred to as bone marrow derived macrophages (BMDMs).
Synthetic chemistry
5-Chloro-2-[(E)-2-[phenyl(pyridin-2-yl)methylidene]hydrazin-1-yl]pyridine (JIB-04). Hydrazine hydrate (5.48 g, 171 mmol, 5.34 mL) was added to a solution of 2,5-dichloropyridine 1 (0.21 g, 1.42 mmol) in pyridine (10 mL) and the reaction mixture was refluxed for 6 h. The resulting suspension was dried in vacuo (rotary evaporator), dissolved in dichloromethane and washed with a 50 mL portion of 1.0 N NaOH and three 50 mL portions of water. The organic layer was dried over anhydrous magnesium sulfate, filtered, and the solvent was removed in vacuo to yield 5-chloro-2-hydrazinylpyridine 2 as a white crystalline solid (0.097 g, 47%) 1H NMR (CDCl3) δ: 3.795 (s, 2H), 5.797 (s, 1H), 6.697 (d, 1H, J = 8.8 Hz), 7.422 – 7.450 (dd, 1H, J = 2.5, 8.8 Hz), 8.058 (d, 1H, J = 2.3 Hz). MS calcd for C5H6ClN3 144.03 [M + H+], found 144.02 [M + H+].
A 20 mg portion of 2 (20 mg, 1.4 mmol) and benzoyl pyridine 3 (25.5 mg, 1.4 mmol) were refluxed overnight in methanol (10 mL) with a traces of acetic acid. The resulting solution was dried via rotary evaporator and crystallized from ethyl acetate to yield 5-Chloro-2-[(E)-2-[phenyl(pyridin-2-yl)methylidene]hydrazin-1-yl]pyridine (JIB-04) 4 as fine yellow needles (0.099 g, 23%). 1H NMR (CDCl3) δ: 7.260–7.356 (m, 2H), 7.397–7.466 (m, 3H), 7.536–7.587 (m, 4H), 7.506 (td, 1H, J = 1.6, 7.8 Hz), 8.136 (d, 1H, J = 2.2 Hz), 8.823 (dd, 1H, J = 4.9, 0.7 Hz), 13.301 (s, 1H). MS calcd for C17H13ClN4 309.09 [M + H+], found 309.41 [M + H+]
In vitro periodontal disease inflammation model
BMDMs were pretreated with 50 μM ML324 (SelleckChem, Cat# S7296) JIB-04 (See Synthetic Chemistry methods, 25 μM), GSK-LSD1 (SelleckChem, Cat# S7574, 1 μM), or DMSO vehicle control for 1 hour. Pre-treated macrophages were challenged with Aa LPS for 8 and 24 hours where supernatant protein was analyzed using DuoSet TNF-α and IL-6 ELISA kits (R&D Systems, Cat# DY406, DY410) and cells were collected for qRT-PCR analysis.
qRT-PCR
Media was rinsed with PBS and cells were lysed using TriZol Reagent (Invitrogen, Cat# 15,596,026). mRNA was isolated according to manufacturer’s protocols and purity confirmed using a Nanodrop-1000 spectrophotometer (Thermo Fisher). Quantitative reverse-transcription real time polymerase chain (qRT-PCR) reaction was run using a High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Cat# 4,368,814) followed by TaqMan® Fast Advanced Master Mix (Applied Biosystems, Cat# 4,444,557) with TaqMan® Gene Expression Assay Primers (Applied biosystems, listed below) using a StepOne Plus instrument (Thermo Fisher). TNF-α, IL-6 and the internal control GAPDH were then quantitated for each sample in triplicate. Results are reported as fold change (2−^^CT).Primers: TaqMan® Gene Expression Assay Primers:GAPDH:Mm99999915_g1IL-6:Mm00446190_m1TNF-α: Mm00443258_m1
In vitro osteoclastogenesis model
Bone marrow stem cells were isolated from C57BL/6 mice, and differentiated into osteoclasts as previously described [Citation51,Citation52]. In brief, bone marrow isolated from femurs and tibiae of two male and two female 12-week-old wild type C57BL/6 mice was plated overnight into phenol red free α-MEM (Gibco, Cat# 41,061–029). Adherent cells were discarded and cells that remained in suspension were plated into the wells of a 96-well culture plate (Corning, Cat#3598) at a density of 15,000 cells/well. Cells were supplemented with macrophage colony stimulating factor (M-CSF, R&D Systems, Cat# 416-ML-500) (15ng/mL/48h) for 3 days followed by supplementing with M-CSF (15 ng/mL/48 h) and receptor activator of nuclear factor kappa b ligand (RANK-L) (R&D Systems, Cat# 462-TEC-010) (50ng/mL/48h) for 2 days. On day 5, wells were rinsed to remove RANK-L and replacement media was supplemented with M-CSF (15 ng/mL) with or without Aa-LPS (100 ng/mL) or RANK-L (R&D Systems, Cat# 462-TEC-010) (50 ng/mL) and ML324 (SelleckChem, Cat# S7296, 10μM) or DMSO vehicle control for 72 hours. Cells were rinsed twice, fixed with 10% glutaraldehyde (Fisher, Cat# O2957-1) and stained for tartrate resistant acid phosphatase (TRAP) as described in BD Bioscience Technical Bulletin No. 445 using a 10-minute incubation with TRAP buffer. Subsequently, 3 representative images per well were captured using an Eclipse TS100 microscope (Nikon) equipped with an Evolution MP camera (Media Cybernetics). Osteoclasts were quantified as multinucleated, TRAP+ cells and statistically significant differences were computed using a paired one-way ANOVA with multiple comparisons with an alpha of 0.05.
Cycloheximide treatment
RAW264.7 macrophages were grown to 80% confluence in α-MEM supplemented with 10% FBS (Hyclone, Cat# SH30071.03HI) and 1% penicillin/streptomycin (Sigma, Cat# P4333). Cells were then pre-treated with ML324 (SelleckChem, Cat# S7296, 50μM) or DMSO vehicle control with or without cycloheximide (Sigma, Cat# C7698, 10 ug/mL) for 4 h. DMSO remained constant in each group at a level of 0.1% to eliminate interference with the assay. Cells were challenged with Aa-LPS (100 ng/mL) or PBS vehicle control for 24 hours and collected for qRT-PCR analysis.
Immunofluorescence
RAW264.7 macrophages were grown to 80% confluence in phenol red free α-MEM (Gibco, Cat# 41,061–029) supplemented with 10% FBS (Hyclone, Cat# SH30071.03HI) and 1% penicillin/streptomycin (Sigma, Cat# P4333) on Sensoplate Plus glass bottom plates (Grenier Bio-One, Cat# 655,892). Cells were pre-treated for 1 hour with ML324 (SelleckChem, Cat# S7296, 50 μM) or DMSO vehicle control followed by Aa-LPS (100 ng/mL) for 24 hours. Cells were then rinsed with PBS and fixed using 4% paraformaldehyde (Sigma) at 37°C for 10 minutes. Cells were permeablized using 0.1% Triton X-100 (Amresco, Cat# 0694) for 10 minutes and blocked using 3% Bovine Serum Albumin (Sigma, Cat# A8806) in PBS for 30 minutes. Cells were then incubated for 1 hour with rabbit anti-H3K4me (Active Motif, Cat# 39,297, 1:750) in 3% BSA. Cells were rinsed and incubated with fluorescent goat anti-rabbit antibody (AbCam, Cat# ab150078, 1:500) in 3% serum for 1 hour. Cells were counterstained with DAPI (VWR, Cat# 95,059–474, 30uM) and AlexaFluor Phalloidin 488 (Invitrogen, A12379). Images were captured using a Wiscan imaging system (Hermes). 35 Images per well were used for analysis (n = 3 wells/group).
Supplemental Material
Download Zip (1.2 MB)Acknowledgments
Special thanks to Bethany A. Harbin, of the MUSC Department of Oral Health Sciences for providing calvarial tissue slides, and to Abby Lauer, M.S. for statistical consultation.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplemental data
Supplemental data for this article can be accessed here.
Additional information
Funding
Related Research Data
References
- Silva N, Abusleme L, Bravo D, et al. Host response mechanisms in periodontal diseases. J Appl Oral Sci. 2015 May-Jun;23(3):329–355. PubMed PMID: 26221929; PubMed Central PMCID: PMCPMC4510669. eng.
- Eke PI, Dye BA, Wei L, et al. Update on prevalence of periodontitis in adults in the United States: NHANES 2009 to 2012. J Periodontol. 2015 May;86(5):611–622. PubMed PMID: 25688694; PubMed Central PMCID: PMCPMC4460825. Eng.
- Eke PI, Dye BA, Wei L, et al. Prevalence of periodontitis in adults in the United States: 2009 and 2010. J Dent Res. 2012 Oct 1 2012;91(10):914–920.
- Krayer JW, Leite RS, Kirkwood KL. Non-surgical chemotherapeutic treatment strategies for the management of periodontal diseases. Dent Clin North Am. 2010 Jan;54(1):13–33. PubMed PMID: 20103470; PubMed Central PMCID: PMCPMC3086469. eng. .
- Shi Y, Lan F, Matson C, et al. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell. 2004 Dec 29;119(7):941–953. PubMed PMID: 15620353; eng.
- Pachaiyappan B, Woster PM. Design of small molecule epigenetic modulators. Bioorg Med Chem Lett. 2014 Jan 1;24(1):21–32. PubMed PMID: 24300735; PubMed Central PMCID: PMC3879946.
- Woster PM. Novel therapeutics targeting epigenetics. In: Chacklamannil S, Rotella DP, Ward S, editors. Comprehensive medicinal chemistry III. Vol. 2. 3rd ed. London: Elsevier, Ltd.; 2017. p. 297–328.
- Gottesfeld JM, Pandolfo M. Development of histone deacetylase inhibitors as therapeutics for neurological disease. Future Neurol. 2009 Nov 1;4(6):775–784. PubMed PMID: 20177429; PubMed Central PMCID: PMC2824892. Eng.
- Lakowski B, Roelens I, Jacob S. CoREST-like complexes regulate chromatin modification and neuronal gene expression. J Mol Neurosci. 2006;29(3):227–239. PubMed PMID: 17085781; eng.
- Ginder GD. Epigenetic regulation of fetal globin gene expression in adult erythroid cells. Transl Res. 2015 May 11;165:115–125.
- Shi L, Cui S, Engel JD, et al. Lysine-specific demethylase 1 is a therapeutic target for fetal hemoglobin induction. Nat Med. 2013 Mar;19(3):291–294. PubMed PMID: 23416702.
- Liang Y, Quenelle D, Vogel JL, et al. A novel selective LSD1/KDM1A inhibitor epigenetically blocks herpes simplex virus lytic replication and reactivation from latency. MBio. 2013;4(1):e00558–00512. PubMed PMID: 23386436; PubMed Central PMCID: PMC3565832.
- Mishra M, Zhong Q, Kowluru RA. Epigenetic modifications of Nrf2-mediated glutamate-cysteine ligase: implications for the development of diabetic retinopathy and the metabolic memory phenomenon associated with its continued progression. Free Radic Biol Med. 2014 Jul 9;75:129–139.
- Pan D, Mao C, Wang YX Suppression of gluconeogenic gene expression by LSD1-mediated histone demethylation. PLoS ONE. 2013;8(6):e66294. PubMed PMID: 23755305; PubMed Central PMCID: PMC3673910.
- Yang IV, Schwartz DA. Epigenetics of idiopathic pulmonary fibrosis. Transl Res. 2014 Mar 31;165:48–60.
- Labbé RM, Holowatyj A, Yang ZQ. Histone lysine demethylase (KDM) subfamily 4: structures, functions and therapeutic potential. Am J Transl Res. 2014;6(1):1–15. PubMed PMID: 24349617; PubMed Central PMCID: PMCPmc3853420. eng.
- Berry WL, Janknecht R. KDM4/JMJD2 histone demethylases: epigenetic regulators in cancer cells. Cancer Res. 2013 May 15;73(10):2936–2942. PubMed PMID: 23644528; PubMed Central PMCID: PMCPMC3655154. eng.
- Su Z, Wang F, Lee JH, et al. Reader domain specificity and lysine demethylase-4 family function. Nat Commun. 2016 Nov 14;7:13387. PubMed PMID: 27841353; PubMed Central PMCID: PMCPMC5114558 declare no competing financial interests. eng.
- Noh MK, Jung M, Kim SH, et al. Assessment of IL-6, IL-8 and TNF-α levels in the gingival tissue of patients with periodontitis. Exp Ther Med. 2013 Sep;6(3):847–851. PubMed PMID: 24137277; PubMed Central PMCID: PMCPMC3786859. eng.
- Park SR, Kim DJ, Han SH, et al. Diverse toll-like receptors mediate cytokine production by fusobacterium nucleatum and aggregatibacter actinomycetemcomitans in macrophages. Infect Immun. 2014 May;82(5):1914–1920. PubMed PMID: 24566622; PubMed Central PMCID: PMCPMC3993440. eng.
- Hienz SA, Paliwal S, Ivanovski S Mechanisms of bone resorption in periodontitis. J Immunol Res. 2015;2015. PubMed PMID: 26065002; PubMed Central PMCID: PMCPMC4433701. eng. doi: 10.1155/2015/615486
- Choi JY, Yoon SS, Kim SE, et al. KDM4B histone demethylase and G9a regulate expression of vascular adhesion proteins in cerebral microvessels. Sci Rep. 2017;7. PubMed PMID: 28327608; PubMed Central PMCID: PMCPMC5361151. eng. DOI:10.1038/srep45005
- Gazzar ME, Yoza BK, Chen X, et al. G9a and HP1 couple histone and DNA methylation to TNFα transcription silencing during endotoxin tolerance. J Biol Chem. 2008 Nov 21 2008;283(47):32198–32208.
- Kim TW, Lee SJ, Oh BM, et al. Epigenetic modification of TLR4 promotes activation of NF-κB by regulating methyl-CpG-binding domain protein 2 and Sp1 in gastric cancer. Oncotarget. 2016 Jan 26;7(4):4195–4209. PubMed PMID: 26675260; PubMed Central PMCID: PMCPMC4826199. eng.
- Hachiya R, Shiihashi T, Shirakawa I, et al. The H3K9 methyltransferase Setdb1 regulates TLR4-mediated inflammatory responses in macrophages. Sci Rep. 2016;6. PubMed PMID: 27349785; PubMed Central PMCID: PMCPMC4924096. eng. doi: 10.1038/srep28845
- Li MF, Zhang R, Li TT, et al. High glucose increases the expression of inflammatory cytokine genes in macrophages through H3K9 methyltransferase mechanism. J Interferon Cytokine Res. 2016 Jan;36(1):48–61. PubMed PMID: 26406561; eng.
- Fu X, Wang X, Duan Z, et al. Histone H3k9 and H3k27 acetylation regulates IL-4/STAT6-mediated igepsilon transcription in B lymphocytes. Anat Rec (Hoboken). 2015 Aug;298(8):1431–1439. PubMed PMID: 25952120; eng.
- Xu G, Liu G, Xiong S, et al. The histone methyltransferase Smyd2 is a negative regulator of macrophage activation by suppressing interleukin 6 (IL-6) and tumor necrosis factor α (TNF-α) production. J Biol Chem. 2015;290(9):5414–5423.
- Binda O, LeRoy G, Bua DJ, et al. Trimethylation of histone H3 lysine 4 impairs methylation of histone H3 lysine 9: regulation of lysine methyltransferases by physical interaction with their substrates. Epigenetics. 2010 Nov 16;5(8):767–775. PubMed PMID: 21124070; PubMed Central PMCID: PMCPMC3052887. eng.
- Shi L, Sun L, Li Q, et al. Histone demethylase JMJD2B coordinates H3K4/H3K9 methylation and promotes hormonally responsive breast carcinogenesis. Proceedings of the National Academy of Sciences of the United States of America. 2011 Apr 18;108( 18):7541–7546. PubMed PMID: PMC3088624.
- Janzer A, Lim S, Fronhoffs F, et al. Lysine-specific demethylase 1 (LSD1) and histone deacetylase 1 (HDAC1) synergistically repress proinflammatory cytokines and classical complement pathway components. Biochem Biophys Res Commun. 2012 May 18;421(4):665–670.
- Rai G, Kawamura A, Tumber A, et al. Discovery of ML324, a JMJD2 demethylase inhibitor with demonstrated antiviral activity. Probe Reports from the NIH Molecular Libraries Program: Bethesda (MD): National Center for Biotechnology Information (US); 2012 Dec 17 [ Updated 2013 Sep 16].
- Herbert BA, Steinkamp HM, Gaestel M, et al. Mitogen-activated protein kinase 2 signaling shapes macrophage plasticity in aggregatibacter actinomycetemcomitans-induced bone loss. Infect Immun. 2017 Jan;85(1). PubMed PMID: 27795356; PubMed Central PMCID: PMCPMC5203644. eng. doi: 10.1128/iai.00552-16
- Berghaus LJ, Moore JN, Hurley DJ, et al. Innate immune responses of primary murine macrophage-lineage cells and RAW 264.7 cells to ligands of Toll-like receptors 2, 3, and 4. Comp Immunol Microbiol Infect Dis. 2010 Sep;33(5):443–454. PubMed PMID: 19732955; PubMed Central PMCID: PMCPMC2888975.
- Meng S, Zhang L, Tang Y, et al. BET inhibitor JQ1 Blocks inflammation and bone destruction. J Dent Res. 2014 Jul;93(7):657–662. PubMed PMID: 24799421; PubMed Central PMCID: PMCPMC4107547. eng.
- Herbert BA, Steinkamp HM, Gaestel M, et al. MK2 signaling shapes macrophage plasticity in aggregatibacter actinomycetemcomitans-induced bone loss. Infect Immun. 2016 Oct;17:2016.
- Yates A, Akanni W, Amode MR, et al. Ensembl 2016. Nucleic Acids Research. 2016;44(D1):D710–D716.
- The KDM4B inhibition assay procedure was run by BPS biosciences (San Diego, CA) using AlphaScreen® technology.
- Papadopoulos G, Weinberg EO, Massari P, et al. Macrophage-specific TLR2 signaling mediates pathogen-induced TNF-dependent inflammatory oral bone loss. J Immunol. 2013 Feb 1 2013;190(3):1148–1157.
- Dunmyer J, Herbert B, Li Q, et al. Sustained mitogen-activated protein kinase activation with aggregatibacter actinomycetemcomitans causes inflammatory bone loss. Mol Oral Microbiol. 2012 Oct;27(5):397–407. PubMed PMID: 22958388; eng.
- Zhang X, Hyer JM, Yu H, et al. DUSP1 phosphatase regulates the proinflammatory milieu in head and neck squamous cell carcinoma. Cancer Res. 2014 Dec 15 2014;74(24):7191–7197.
- Herbert BA, Novince CM, Kirkwood KL. Aggregatibacter actinomycetemcomitans, a potent immunoregulator of the periodontal host defense system and alveolar bone homeostasis. Mol Oral Microbiol. 2016;31(3):207–227.
- Madeira MFM, Queiroz-Junior CM, Costa GM, et al. MIF induces osteoclast differentiation and contributes to progression of periodontal disease in mice. Microbes Infect. 2012 Feb;14(2):198–206.
- Ye L, Fan Z, Yu B, et al. Histone demethylases KDM4B and KDM6B promotes osteogenic differentiation of human MSCs. Cell Stem Cell. 2012 Jul 6;11(1):50–61. PubMed PMID: 22770241; PubMed Central PMCID: PMCPMC3392612. eng.
- Valerio MS, Basilakos DS, Kirkpatrick JE, et al. Sex-based differential regulation of bacterial-induced bone resorption. J Periodontal Res. 2017 Jun;52(3):377–387. PubMed PMID: 27509894; PubMed Central PMCID: PMCPMC5303566.
- Whetstine JR, Nottke A, Lan F, et al. Reversal of histone lysine trimethylation by the JMJD2 family of histone demethylases. Cell. 2006 May 5;125(3):467–481.
- Ponnaluri VKC, Vavilala DT, Putty S, et al. Identification of non-histone substrates for JMJD2A–C histone demethylases. Biochem Biophys Res Commun. 2009 Dec 11;390(2):280–284.
- Vazquez D. Inhibitors of protein synthesis. FEBS Letters. 1974 Mar 23;40: S48–S62. .
- Loe H The gingival index, the plaque index and the retention index systems. J Periodontol. 1967 Nov-Dec;38(6):Suppl:610–616. PubMed PMID: 5237684; eng.
- Zhang X, Goncalves R, Mosser DM. The isolation and characterization of murine macrophages. Curr Protoc Immunol. 2008 Nov ; Chapter:Unit-14.11. PubMed PMID: 19016445; PubMed Central PMCID: PMCPmc2834554. eng. doi: 10.1002/0471142735.im1401s83
- Weischenfeldt J, Porse B. Macrophages (BMM): isolation and applications. Cold Spring Harb Protoc. 2008 Dec 1;2008(12):pdb.prot5080.
- Valerio MS, Herbert BA, Basilakos DS, et al. Critical role of MKP-1 in lipopolysaccharide-induced osteoclast formation through CXCL1 and CXCL2. Cytokine. 2015 Jan;71(1):71–80. PubMed PMID: 25261746; PubMed Central PMCID: PMCPMC4254057. eng.