
ABSTRACT
Alternative protein-coding transcripts of the RASSF1 gene have been associated with dual functions in human cancer: while RASSF1C isoform has oncogenic properties, RASSF1A is a tumour suppressor frequently silenced by hypermethylation. Recently, the antisense long non-coding RNA RASSF1 (ANRASSF1) was implicated in a locus-specific mechanism for the RASSF1A epigenetic repression mediated by PRC2 (Polycomb Repressive Complex 2). Here, we evaluated the methylation patterns of the promoter regions of RASSF1A and RASSF1C and the expression levels of these RASSF1 transcripts in breast cancer and breast cancer cell lines. As expected, RASSF1C remained unmethylated and RASSF1A was hypermethylated at high frequencies in 75 primary breast cancers, and also in a panel of three mammary epithelial cells (MEC) and 10 breast cancer cell lines (BCC). Although RASSF1C was expressed in all cell lines, only two of them expressed the transcript RASSF1A. ANRASSF1 expression levels were increased in six BCCs. In vitro induced demethylation with 5-Aza-2ʹ-deoxicytydine (5-Aza-dC) resulted in up-regulation of RASSF1A and an inverse correlation with ANRASSF1 relative abundance in BCCs. However, increased levels of both transcripts were observed in two MECs (184A1 and MCF10A) after treatment with 5-Aza-dC. Overall, these findings indicate that ANRASSF1 is differentially expressed in MECs and BCCs. The lncRNA ANRASSF1 provides new perspectives as a therapeutic target for locus-specific regulation of RASSF1A.
Introduction
Tumour suppressor function has been demonstrated for several proteins of the Ras-Association Domain (RASSF) family, which comprises 10 members (RASSF1 to RASSF10). These proteins have been implicated in a broad range of cellular processes such as cell cycle control, mitosis, apoptosis, microtubule stabilization, cell adhesion and polarization [Citation1]. RASSF1 was one of the first genes identified as a member of this family [Citation2]. This gene is located within the 120Kb critical deletion region on 3p21.3, which is now recognized as a locus that harbour a tumour suppressor cluster [Citation3–Citation5]. Multiple isoforms termed RASSF1A to RASSF1G are originated from alternative splicing and by differential usage of two distinct promoters, both associated with specific CpG-islands. The well-studied variants are RASSF1A and RASSF1C, transcribed from the upstream and downstream promoters, respectively [Citation6].
RASSF1A and RASSF1C variants are ubiquitously expressed in non-tumour tissues and are also recognized by their cancer-related functions [Citation7]. Whilst RASSF1A can be inactivated by deletion or rarely by point mutations, the transcriptional silencing by promoter hypermethylation is the major mechanism leading to loss or suppression of its function in cancer cells. Aberrant RASSF1A methylation is described as an early and frequent event in human cancers [Citation8,Citation9]. In breast cancer, RASSF1A methylation is observed at high frequencies, and this aberrant epigenetic mark has also been detected in the serum from breast cancer patients in association with metastasis occurrence, large tumour size, and low response to adjuvant therapy [Citation10]. Thus, it has been suggested that hypermethylation of RASSFIA could be used as a potential diagnostic and/or prognostic biomarker in breast cancer.
Contrary to RASSF1A, RASSF1C has been associated to oncogenic activities [1Citation1–Citation13], and no hypermethylation is described in its promoter in human cancer. Given these opposite effects in cancer cells, it has been proposed that RASSF1 has a dual function. In normal tissues, the RASSF1A protein presents a dominant action; thus, a high expression ratio of RASSF1A/RASSF1C could suppress the oncogenic effects of RASSF1C. In cancer cells, the effects on cell growth and apoptosis mediated by RASSF1C isoform could accompany RASSF1A loss or inactivation [Citation14].
In our previous study, we described that RASSF1A isoform is epigenetically silenced in the majority of breast cancer cell lines (15 out of 17), while RASSF1C expression was maintained in all of them [Citation15]. Additionally, we also demonstrated that induced demethylation by using 5-Aza-2ʹ-deoxycytidine (5-Aza-dC), isolated or in combination with the histone deacetylase inhibitor Trichostatin A (TSA), simultaneously up-regulated RASSF1A and its flanking genes while no significant changes were observed in RASSF1C expression levels [Citation15]. These findings suggested the existence of a fine-tuned and locus-specific epigenetic control of the RASSF1 alternative transcripts.
Recently, a new nuclear long non-coding RNA (lncRNA), RASSF1-AS1 (also termed Antisense Intronic Noncoding RASSF1 or ANRASSF1) was characterized by Beckedorff et al. [Citation16]. The authors proposed a model in which this lncRNA has a cis function in the epigenetic silencing of RASSF1A. Accordingly, as ANRASSF1 is transcribed by RNA polymerase II, it remains tethered to its transcriptional site and recruit histone modifying complexes. Functional assays using HeLa cells overexpressing ANRASSF1 and RNA immunoprecipitation with antibody specific to members of PRC2 complex (i.e. SUZ12 and EZH2) indicated an enrichment of the lncRNA ANRASSF1. The occupancy of PRC2 at the RASSF1A promoter was associated with the trimethylation of the lysine 27 of H3 histone (H3K27me3). This repressive histone mark was neither observed in the promoter regions of RASSF1C nor in the four neighbouring genes [Citation16].
The protein EZH2, catalytic subunit of PRC2 complex showing histone methyltransferase activity [Citation17], is also able to interact with DNA methyltransferases (DNMTs) [Citation18]. Thus, the cis-acting function of ANRASSF1 mediated by PRC2 provides a possible link between histone modifications (H3K27me) and de novo locus-specific methylation. ANRASSF1 could indirectly reinforce RASSF1A long-term epigenetic silencing via DNA methylation. In this context, the proposed mechanism of action of ANRASSF1 would explain why the promoter-associated CpG island of RASSF1A is frequently methylated in several human solid tumours while the closest CpG island associated to the RASSF1C promoter region remains unmethylated. Most studies have only focused on the DNA methylation analysis of RASSF1A variant and few reports described simultaneously the methylation patterns of both CpG islands of the gene RASSF1 in the same series of tumour tissue samples.
In this study, we first determined the methylation patterns of RASSF1A and RASSF1C in primary breast tumours and breast cancer cell lines. Then, we correlated the promoter methylation patterns of both variants with the expression levels of the ANRASSF1 non-coding RNA and the protein-coding RASSF1A, RASSF1C, and EZH2 transcripts in breast cancer cells lines and discussed the potential of ANRASSF1 as a new breast cancer biomarker and therapeutic target.
Results
RASSF1A and RASSF1C methylation in breast cancer and breast cancer cell lines
Initially, the methylation patterns of both RASSF1A and RASSF1C promoter regions were evaluated in a set of 75 ductal breast carcinomas by conventional MS-PCR (Methylation-Specific Polymerase Chain Reaction). As expected, a high frequency of RASSF1A methylation was observed (85% of the tissue samples), whereas all samples showed exclusively non-methylated alleles at RASSF1C promoter-region. shows the significant associations between clinical parameters and disease-specific survival rates in this cohort. However, no significant differences were found between clinical and histopathological parameters and RASSF1A methylation (). Also, RASSF1A methylation was not associated with disease-free survival (p = 0.1293, Kaplan-Meier estimation).
Table 1. Disease-specific survival analysis in 75 breast cancer patients.
Table 2. RASSF1A methylation according to clinical, histopathological, and immunohistochemical markers in 75 invasive ductal breast carcinomas.
The RASSF1A and RASSF1C methylation patterns were also evaluated in 10 breast cancer cell lines (BCCs: BT-483, BT-549, Hs578T, MCF7, MDA-MB-134, MDA-MB-231, MDA-MB-415, MDA-MB-453, MDA-MB-468, and SK-BR-3) and in three mammary epithelial cell lines (MECs: 184A1 and 184B5 derived from normal breast epithelium and MCF10A from benign fibrocystic disease). ,) illustrates respectively the physical position of the two CpG islands of the RASSF1 gene and the MS-PCR results. Only MDA-MB-415 cells showed exclusively unmethylated alleles of RASSF1A promoter region. This region was fully methylated in eight breast cancer cell lines. A hemimethylated pattern (presence of both methylated and unmethylated alleles) was detected in Hs578T cancer cells and in three MECs used as normal references. As observed in the primary breast cancer tissue samples, the RASSF1A promoter region was commonly methylated while the promoter-specific CpG island of RASSF1C remained unmethylated in all of them.
Figure 1. (a) Illustrative physical map of RASSF1 locus showing the overlapping of associated coding and non-coding RNAs, as well as the two promoter-associated CpG islands 130 and 83 of the alternative transcripts RASSF1C and RASSF1A, respectively [modified from UCSC Genome Browser on Human Dec. 2013 (GRCh38/hg38) Assembly, http://genome.ucsc.edu)]. (b-d) Expression levels of RASSF1A, ANRASSF1 and RASSF1C, respectively, in three mammary epithelial cells (MECs) and ten breast cancer cell lines (BCCs). The expression of each target gene was normalized with the GAPDH gene expression levels. (e) DNA methylation status of RASSF1A and RASSF1C in MECs and BCCs (M = methylated and U = unmethylated). (f) The colour red represents a gradient heatmap of methylation intensities of the two promoter regions of RASSF1 gene. These data were retrieved from the public database Cancer Methylome System (http://cbbiweb.uthscsa.edu/KMethylomes/), obtained upon genome-wide MBDCap-sequencing (Methyl-CpG binding domain-based capture and sequencing) (p-value was determined by the Mann–Whitney test).
![Figure 1. (a) Illustrative physical map of RASSF1 locus showing the overlapping of associated coding and non-coding RNAs, as well as the two promoter-associated CpG islands 130 and 83 of the alternative transcripts RASSF1C and RASSF1A, respectively [modified from UCSC Genome Browser on Human Dec. 2013 (GRCh38/hg38) Assembly, http://genome.ucsc.edu)]. (b-d) Expression levels of RASSF1A, ANRASSF1 and RASSF1C, respectively, in three mammary epithelial cells (MECs) and ten breast cancer cell lines (BCCs). The expression of each target gene was normalized with the GAPDH gene expression levels. (e) DNA methylation status of RASSF1A and RASSF1C in MECs and BCCs (M = methylated and U = unmethylated). (f) The colour red represents a gradient heatmap of methylation intensities of the two promoter regions of RASSF1 gene. These data were retrieved from the public database Cancer Methylome System (http://cbbiweb.uthscsa.edu/KMethylomes/), obtained upon genome-wide MBDCap-sequencing (Methyl-CpG binding domain-based capture and sequencing) (p-value was determined by the Mann–Whitney test).](/cms/asset/17373416-7363-42b8-995e-3f2d3548f808/kepi_a_1615355_f0001_oc.jpg)
Expression levels of the lncRNA ANRASSF1 and protein-coding transcripts in breast cancer cell lines
The data of DNA methylation analysis were correlated with the expression of RASSF1 transcripts by RT-qPCR: RASSF1C was unmethylated and expressed in all MEC and BCC cells, while RASSF1A was hypermethylated and silenced in most of BCCs (,)). The RASSF1A transcript was detected only in the MDA-MB-415 and Hs578T cells, which exhibit unmethylated and hemimethylated patterns, respectively. Although expressed at very low levels in one MEC (184B5), the lncRNA ANRASSF1 was detected in six BCCs (BT-483, MCF7, MDA-MB-134, MDA-MB-415, MDA-MB-453, MDA-MB-468) ()). Furthermore, the BCCs Hs578T (hemimethylated) and BT-549, MDA-MB-231, and SK-BR-3 (fully methylated) showed results similar to MECs. The gene EZH2 was equally expressed among MECs and BCCs, with exception of MDA-MB-468 cells which expressed higher levels (Figure S1A).
Effect of 5-Aza-dC treatment in the transcriptional expression levels
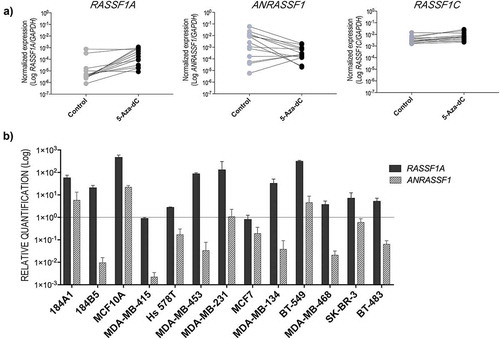
Next, we compared the expression levels of the lncRNA ANRASSF1 and RASSF1A transcripts after 5-Aza-dC treatment. Increased RASSF1A expression levels (p = 0.0020) were observed after induced demethylation, indicating its re-expression in both MEC and BCC groups. As expected, minor changes were observed for RASSF1C expression levels (p = 0.0081). Contrarily, we observed a trend to lower levels of ANRASSF1 expression under this experimental condition (p = 0.0105) ()). ) shows the relative abundance of RASSF1A and ANRASSF1 transcripts in each cell line: 10 of 13 cell lines showed an increase of at least four times in the RASSF1A expression levels. In addition, the relative expression levels of ANRASSF1 were lower compared to untreated cells in 1 MEC and 8 BCCs. Interestingly, ANRASSF1 levels were increased in two MECs (184A1 and MCF10A). In addition, after the treatment with 5-Aza-dC in MDA-MB-415 cells, although no alteration in the relative expression of RASSF1A was found, ANRASSF1A showed the most negative effect compared to the other cell lines. No effects in EZH2 gene expression were observed after 5-Aza-dC treatment (Figure S1B).
Figure 2. (a) Effect of induced demethylation on the expression levels of RASSF1 transcripts after in vitro treatment with the DNMT inhibitor 5-Aza-2-deoxycytidine (5-Aza-dC) in MECs and BCCs. These data show the average of two independent experiments and technical triplicates for each cell line. (b) Abundance of the RASSF1A and ANRASSF1 transcripts after 5-Aza-dC treatment in each cell line relative to the respective paired untreated control (p-value was determined by the Wilcoxon matched-pairs signed rank test).
Discussion
The results of the present study reinforce that while the promoter-associated CpG island of RASSF1A is hypermethylated in high frequency in breast carcinomas, the downstream CpG island associated with the alternative RASSF1C promoter remains unmethylated. These DNA methylation data are in accordance with those available in the public database such as Cancer Methylome System [Citation19] ()) and The Cancer Genome Atlas (TCGA) [Citation20] (Figure S2). RASSF1A methylation is a common epigenetic alteration in a broad spectrum of human solid tumours [Citation8–Citation10], commonly associated with poor prognosis [Citation21–Citation23]. Epigenetic inactivation of RASSF1A has also been associated with specific molecular subtypes in breast cancer [Citation24], although we did not find significant associations probably due to the small sample size.
A panel of three MECs and 10 BCCs was used to evaluate the effect of DNA methylation of the promoter-associated CpG islands of the RASSF1 gene in the expression levels of its protein-coding transcripts (RASSF1A and RASSF1C) and in the recently characterized lncRNA ANRASSF1. In 2004, Reis et al. [Citation25] reported the first evidence of an intronic and antisense transcript associated with the RASSF1 gene which was also correlated with tumour differentiation in prostate cancer. Subsequently, the same research group effectively characterized this new transcript as ANRASSF1 [Citation16]. The authors described higher ANRASSF1 expression levels in breast (MDA-MB-231 and MCF7) and prostate (LNCap and DU145) cancer cell lines when compared with non-tumour cell lines (MCF10A and RWPE-1, respectively). An inverse correlation between ANRASSF1 and RASSF1A expression was also found [Citation16].
Only two other studies have investigated the ANRASSF1 expression levels in primary human cancers [Citation26,Citation27]. Iranpour et al. [Citation26] reported that, in comparison to matched adjacent noncancerous tissues, ANRASSF1 is overexpressed in breast cancer especially of the triple-negative subtype [Citation26]. A more recent study showed that ANRASSF1 was also frequently overexpressed in gastric cancer in comparison with adjacent tissues [Citation27]. These findings suggest a relevant role of ANRASSF1 in cancer progression.
The importance of non-coding RNA (ncRNA) is increasingly being recognized as key regulators of physiological programs during development and in human diseases, including cancer [Citation28,Citation29]. Few lncRNAs have been completely characterized. To date, ANRASSF1 is described as an endogenous unspliced lncRNA, which is transcribed through RNA polymerase II, it is capped and polyadenylated, exhibits nuclear localization and ability to bind to PRC2. Despite its shorter half-life compared to other lncRNAs that bind to PRC2, it has been proposed that the interaction ANRASSF1/PRC2 could act directly in the epigenetic silencing of the RASSF1A isoform [Citation16]. Further, the importance of antisense transcripts at this locus was also demonstrated through an analogue system described in plants and based on small interfering RNA (siRNA)-directed CpG methylation. Castanotto et al. [Citation30] demonstrated that short hairpin RNAs (shRNAs) complementary to the RASSF1A promoter can direct lower levels of de novo DNA methylation and partial gene silencing in HeLa cells.
Herein, we described that ANRASSF1 abundance is lower in three MECs when compared with a panel of 10 BCCs. In addition, the lncRNA was detected in cell lines methylated as well as in a cell line with exclusively unmethylated RASSF1A alleles (MDA-MB-415). These changes in the DNA methylation pattern were not associated with the expression of EZH2. Notably, very low abundance of ANRASSF1 was detected in four hemimethylated cell lines (3 MECs and in Hs578T breast cancer cell line) as well as in three fully methylated BCCs (BT-549, MDA- MB-231, and SK-BR-3). The detection of ANRASSF1 in partially methylated normal mammary epithelial cells and in a cancer cell line unmethylated suggest that this lncRNA may have other functions than those related with RASSF1A epigenetic silencing.
We observed a heterogeneous effect of 5-Aza-dC treatment in the relative abundance of ANRASSF1 and RASSF1A transcripts. While the expression levels of ANRASSF1 clearly decreased in six BCCs (MDA-MB-415 was unmethylated at RASSF1A promoter, and BT-483, MCF7, MDA-MB-134, MDA-MB-453, and MDA-MB-468 were fully methylated), two of three MECs (184A1 and MCF10A) and BT-549 breast cancer cells showed an increase in its relative abundance. Several factors should contribute to this heterogeneous profile, including the differential sensitivity to the DNMT inhibitor and effects of gene dosage.
Mayor et al.[Citation31] showed that although the in vitro treatment with 5-Aza-dC was able to restore the expression of hypermethylated genes in HCT116 colon cancer cells, the remethylation of the associated promoter regions occurred 16 days after 5-Aza-dC withdrawal. These authors also described that the retained levels of chromatin marks at these chromosomal regions accompanied the restitution of the DNA methylation. According to the proposed model for the epigenetic silencing of the RASSF1A driven by ANRASSF1 [Citation16], one could speculate that ANRASSF1 could antagonize the effects of drug-induced demethylation in the promoter region of RASSF1A, contributing for its remethylation after a short-term period in the absence of the DNA methylation inhibitors. In this context, ANRASSF1 could be considered as a promisor molecular target to epigenetic therapy.
The characterization of the molecular mechanisms involved in RASSF1A epigenetic silencing could lead to a deeper understanding of tumour development and allow the identification of new treatment strategies. Commonly, the expression levels of lncRNAs are lower and tend to show tissue-specific patterns in non-tumour tissues, but are overexpressed in cancer cells [Citation32,Citation33]. Overall, these biological features support the use of lncRNAs as diagnostic and prognostic markers, as well as therapeutic targets in cancer [Citation34,Citation35]. Currently, approaches to target lncRNAs have been emerging as an innovative therapeutic strategy to induce the up-regulation of locus-specific endogenous genes [Citation35]. In this context, ANRASSF1 is located in the nucleus and has been implicated in the epigenetic repression of the tumour suppressor RASSF1A. Therapeutic target of ANRASSF1 have potential to revert the repressive chromatin modifications introduced by PRC2 at RASSF1A promoter, potentially leading to RASSF1A up-regulation in breast cancer.
Material and methods
Patients
Breast cancer samples were obtained from Amaral Carvalho Hospital, Jaú, São Paulo, Brazil, under patient-informed consent and Institutional Ethics Committee approval. The DNA methylation patterns of both promoter-associated CpG islands of the RASSF1 gene (RASSF1A and RASSF1C) were analysed in a set of 75 invasive ductal breast carcinoma samples from unrelated patients. The samples were obtained from patients naïve of radio and/or chemotherapy prior to surgery. The mean age of the patients was 58.9 ± 15.7 years and the mean follow-up period was 89.2 ± 35.12 months, ranging from 6 to 153 months.
Methylation specific-polymerase chain reaction (MS-PCR)
Genomic DNA was isolated by standard SDS/proteinase K digestion followed by phenol and chloroform extraction and ethanol precipitation. Approximately 1μg of genomic DNA was subjected to bisulfite modification using the EpiTect Bisulfite Modification Kit (Qiagen, Hilden, Germany), according to the manufacturer’s instructions, and eluted in a final volume of 40μL. In order to evaluate the conversion efficiency, 5μL of modified DNA were amplified by PCR using a primer set that recognize the bisulfite-modified template (control region) but do not discriminate between methylated and unmethylated alleles. The DNA methylation patterns of RASSF1A and RASSF1C promoter regions were determined by conventional MS-PCR. Bisulfite-converted CpG methylated HeLa genomic DNA (New England Biolabs, Ipswich, MA, USA) and the unmethylated EpiTect control DNA (Qiagen, Hilden, Germany) were used to evaluate the specificity of each primer set for methylated and unmethylated alleles, respectively (supplementary material, Table S1). The amplification reactions were performed at a final volume of 25μL containing 0.25mM of each primer, 200mM of each dNTP, 15mM Tris-HCl (pH 8.0), 50mM KCl, 2.5mM MgCl2 and 1U of AmpliTaq Gold DNA polymerase (Applied Biosystems, Foster City, CA, USA). The PCR conditions were as follows: 1 cycle at 95°C for 5 minutes; 31 cycles at 95°C for 45 seconds; 56°C for 30 seconds and 72°C for 30 seconds, and one cycle at 72°C for 4 minutes. The amplified products were visualized after electrophoresis in 6% polyacrylamide gels and stained with silver nitrate.
Cell lines and 5-Aza-dC treatment
Three mammary epithelial cell lines (MECs: 184A1 and 184B5, both chemically transformed and MCF10A derived from benign fibrocystic disease) and 10 breast cancer cell lines (BCCs: BT-483, BT-549, Hs578T, MCF7, MDA-MB-134, MDA-MB-231, MDA-MB-415, MDA-MB-453, MDA-MB-468, and SK-BR-3) were obtained from the Tissue Culture Shared Resource (TCSR) at the Lombardi Comprehensive Cancer Center, Georgetown University, Washington DC. All cell lines were fingerprinted using the Cell ID™ System (Promega, Madison, WI, USA) for genomic authentication by the TCSR of Georgetown University, prior to the experiments. The cultures were maintained at 37°C in a humidified environment containing 5% CO2 and supplemented medium as previously described [Citation15].
Total RNA extraction and mRNA expression analysis
Total RNA was extracted using the RNeasy Mini Kit (Qiagen, Hilden, Germany), purified with DNase I amplification grade (Invitrogen, Carlsbad, CA, USA), and quantified on a NanoDrop ND-1000 Spectrophotometer (Thermo Fisher, Waltham, MA, USA). Total RNA integrity was assessed using an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA). The complementary DNA (cDNA) was obtained with the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA, USA), using random primers in a final volume of 20μL, according to the manufacturer’s instructions. Relative expression levels of the RASSF1A, RASSF1C, and EZH2 alternative transcripts were quantified by RT-qPCR (quantitative real-time polymerase chain reaction) as previously reported [Citation15].
Strand-specific cDNA synthesis and expression analysis of ANRASSF1 by reverse transcriptase-polymerase chain reaction (RT-PCR)
The experimental strategy used for the ANRASSF1 expression analysis based on qualitative and quantitative RT-PCR is detailed in the supplementary Figure S3. Orientation-specific RT-PCR was performed with 1.5 μg of total RNA, reverse transcribed using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA, USA). cDNA synthesis used primer complementary to the antisense or sense strand of ANRASSF1 in a final volume of 20μL. The occurrence of self-priming and DNA contamination was verified in cDNA reactions with no primer or transcriptase reverse, respectively, as additional controls. The ANRASSF1 was detected as previously described using a primer set that generated an amplicon of 516bp [Citation16]. Direct Sanger sequencing confirmed the identity of this PCR product using the DNA automated sequencer ABI 3500 (Applied Biosystems, Foster City, CA, USA) and the CLC Main Workbench software 6.0 (CLC Bio, Aarhus, Denmark) (supplementary material, Table S1 and Figure S1). For the quantitative RT-PCR analysis, the reverse transcription reaction was diluted 1:10 with sterile ultra pure water. Two μL of diluted cDNA were used in a 15μL reaction using the GoTaq qPCR master mix (Promega, Madison, WI, USA) following the conditions: 1 cycle at 95°C for 5 minutes followed by 35 cycles of two steps: denaturation at 95°C for 10 seconds and annealing/extension at 60°C for 30 seconds. The reactions were performed in triplicate using the StepOne Real-time PCR System (Applied Biosystems, Foster City, CA, USA). The results were calculated using the ΔΔCt-method normalized with glyceraldehyde-3-phosphate dehydrogenase (GAPDH) expression levels, which was experimentally selected in a previous study of our group [Citation15].
Immunohistochemical analysis
Estrogen (ER), Progesterone (PgR), Epidermal Growth Factor II receptor (HER2), p53 and Ki-67 antigen were analysed in formalin-fixed and paraffin-embedded (FFPE) tissues by immunohistochemistry analysis, as previously described by Caldeira et al. [Citation36]. ER and PgR scoring were reviewed according to American Society of Clinical Oncology/College of American Pathologists Guideline Recommendations for Immunohistochemical Testing of Estrogen and Progesterone Receptors in Breast Cancer [Citation37].
Statistical analysis
Pairwise associations between RASSF1A methylation patterns and clinical, histopathological and immunohistological variables were assessed using Fisher’s exact test. The survival data, obtained from clinical records, were analysed using the non-parametric Kaplan–Meier method, and the differences were evaluated by log-rank test. Multivariate statistical analysis was performed using the Cox model to evaluate the combined effects of these variables in the risk of death. Comparisons of gene expression levels were based on the average and standard deviation of two independent experiments and three technical replicates. The significance level of 5% was applied for all statistical analysis using the statistical software package SAS/STAT, version 6.0 (SAS Institute, Inc., Cary, NC, USA) or GraphPad Prism 6 (GraphPad Software, Inc., La Jolla, CA, USA).
Supplemental Material
Download MS Word (1.9 MB)Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplementary data can be accessed here.
Additional information
Funding
Related Research Data
References
- Richter AM, Pfeifer GP, Dammann RH. The RASSF proteins in cancer; from epigenetic silencing to functional characterization. Biochim Biophys Acta. 2009 Dec;1796:114–128.
- Pfeifer GP, Dammann R, Li C, et al. Epigenetic inactivation of a RAS association domain family protein from the lung tumour suppressor locus 3p21.3. Nat Genet. 2000 Jul;25:315–319.
- Lerman MI, Minna JD. The 630-kb lung cancer homozygous deletion region on human chromosome 3p21.3: identification and evaluation of the resident candidate tumor suppressor genes. The international lung cancer chromosome 3p21.3 tumor suppressor gene consortium. Cancer Res. 2000 Nov;60:6116–6133.
- Ji L, Minna JD, Roth JA. 3p21.3 tumor suppressor cluster: prospects for translational applications. Future Oncol. 2005 Feb 1;1:79–92.
- Hesson LB, Cooper WN, Latif F. Evaluation of the 3p21.3 tumour-suppressor gene cluster. Oncogene. 2007;26:7283–7301.
- Dammann R, Schagdarsurengin U, Seidel C, et al. The tumor suppressor RASSF1A in human carcinogenesis: an update. Histol Histopathol. 2005;20:645–663.
- Volodko N, Gordon M, Salla M, et al. RASSF tumor suppressor gene family: biological functions and regulation. FEBS Lett. 2014;588:2671–2684.
- Pfeifer GP, Dammann R. Methylation of the tumor suppressor gene RASSF1A in human tumors. Biochem (Mosc). 2005 May;70:576–583.
- Donninger H, Vos MD, Clark GJ. The RASSF1A tumor suppressor. J Cell Sci. 2007 Sep;120:3163–3172.
- Dworkin AM, Huang TH-M, Toland AE. Epigenetic alterations in the breast: implications for breast cancer detection, prognosis and treatment. Semin Cancer Biol. 2009 Jun;19:165–171.
- Amaar YG, Minera MG, Hatran LK, et al. Ras association domain family 1C protein stimulates human lung cancer cell proliferation. Am J Physiol Lung Cell Mol Physiol. 2006;291:L1185–L1190.
- Estrabaud E, Lassot I, Blot G, et al. RASSF1C, an isoform of the tumor suppressor RASSF1A, promotes the accumulation of β-catenin by interacting with βTrCP. Cancer Res. 2007;67:1054–1061.
- Reeves ME, Baldwin SW, Baldwin ML, et al. Ras-association domain family 1C protein promotes breast cancer cell migration and attenuates apoptosis. BMC Cancer. 2010;Oct;10:562.
- Reeves ME, Firek M, Chen S-T, et al. The RASSF1 gene and the opposing effects of the RASSF1A and RASSF1C isoforms on cell proliferation and apoptosis. Mol Biol Int. 2013;2013:145096.
- da Costa Prando E, Cavalli LR, Rainho C. Evidence of epigenetic regulation of the tumor suppressor gene cluster flanking RASSF1 in breast cancer cell lines. Epigenetics. 2011;6:1413–1424.
- Beckedorff FC, Ayupe AC, Crocci-Souza R, et al. The intronic long noncoding RNA ANRASSF1 recruits PRC2 to the RASSF1A promoter, reducing the expression of RASSF1A and increasing cell proliferation. PLoS Genet. 2013;9:e1003705.
- Margueron R, Reinberg D. The polycomb complex PRC2 and its mark in life. Nature. 2011 Jan;469:343–349.
- Viré E, Brenner C, Deplus R, et al. The polycomb group protein EZH2 directly controls DNA methylation. Nature. 2006;439:871–874.
- Gu F, Doderer MS, Huang Y-W, et al. CMS: a web-based system for visualization and analysis of genome-wide methylation data of human cancers. Chuang EY, editor. PLoS One. 2013;8:e60980.
- Díez-Villanueva A, Mallona I, Peinado MA. Wanderer, an interactive viewer to explore DNA methylation and gene expression data in human cancer. Epigenet Chromatin. 2015 Jun;23(8):22.
- Grawenda AM, O’Neill E. Clinical utility of RASSF1A methylation in human malignancies. Br J Cancer. 2015 Jul;113:372–381.
- Xu J, Shetty PB, Feng W, et al. Methylation of HIN-1, RASSF1A, RIL and CDH13 in breast cancer is associated with clinical characteristics, but only RASSF1A methylation is associated with outcome. BMC Cancer. 2012;12:243.
- Han Z-H, Xu C-S, Han H, et al. Value of the level of methylation of RASSF1A and WIF-1 in tissue and serum in neoadjuvant chemotherapeutic assessment for advanced breast cancer. Oncol Lett. 2017;14:4499–4504.
- Hagrass HA, Pasha HF, Shaheen MA, et al. Methylation status and protein expression of RASSF1A in breast cancer patients. Mol Biol Rep. 2014;41:57–65.
- Reis EM, Nakaya HI, Louro R, et al. Antisense intronic non-coding RNA levels correlate to the degree of tumor differentiation in prostate cancer. Oncogene. 2004;23:6684–6692.
- Iranpour M, Soudyab M, Geranpayeh L, et al. Expression analysis of four long noncoding RNAs in breast cancer. Tumor Biol. 2016;37:2933–2940.
- Kangarlouei R, Irani S, Noormohammadi Z, et al. ANRIL and ANRASSF1 long noncoding RNAs are upregulated in gastric cancer. J Cell Biochem. 2019 Mar 4:1–5. Epub ahead of print. DOI: 10.1002/jcb.28520
- Rasool M, Malik A, Zahid S, et al. Non-coding RNAs in cancer diagnosis and therapy. Non-Coding RNA Res. 2016;1:69–76.
- Derrien T, Johnson R, Bussotti G, et al. The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res. 2012;22:1775–1789.
- Castanotto D, Tommasi S, Li M, et al. Short hairpin RNA-directed cytosine (CpG) methylation of the RASSF1A gene promoter in HeLa cells. Mol Ther. 2005;12:179–183.
- Mayor R, Muñoz M, Coolen MW, et al. Dynamics of bivalent chromation domains upon drug induced reactivation and resilencing in cancer cells. Epigenetics. 2011;6(9):1138–1148.
- Djebali S, Davis CA, Merkel A, et al. Landscape of transcription in human cells. Nature. 2012;489:101–108.
- Malih S, Saidijam M, Malih N. A brief review on long noncoding RNAs: a new paradigm in breast cancer pathogenesis, diagnosis and therapy. Tumor Biol. 2016 Feb;37:1479–1485.
- Lavorgna G, Vago R, Sarmini M, et al. Long non-coding RNAs as novel therapeutic targets in cancer. Pharmacol Res. 2016;110:131–138.
- Slaby O, Laga R, Sedlacek O. Therapeutic targeting of non-coding RNAs in cancer. Biochem J. 2017 Dec;474:4219–4251.
- Caldeira JRF, Prando EC, Quevedo FC, et al. CDH1 promoter hypermethylation and E-cadherin protein expression in infiltrating breast cancer. BMC Cancer. 2006;6:48.
- Hammond ME, Hayes DF, Wolff AC, et al. American Society of Clinical Oncology/College of American Pathologists guideline recommendations for immunohistochemical testing of estrogen and progesterone receptors in breast cancer. J Oncol Pract. 2010 Jul;6:195–197.