
ABSTRACT
Signal Transducers and Activators of Transcription-3 (STAT3), a potent oncogenic transcription factor, is constitutively activated in lung cancer, but mutations in pathway genes are infrequent. Protein Tyrosine Phosphatase Receptor-T (PTPRT) is an endogenous inhibitor of STAT3 and PTPRT loss-of-function represents one potential mechanism of STAT3 hyperactivation as observed in other malignancies. We determined the role of PTPRT promoter methylation and sensitivity to STAT3 pathway inhibitors in non-small cell lung cancer (NSCLC). TCGA and Pittsburgh lung cancer cohort methylation data revealed hypermethylation of PTPRT associated with diminished mRNA expression in a subset of NSCLC patients. We report frequent hypermethylation of the PTPRT promoter which correlates with transcriptional silencing of PTPRT and increased STAT3 phosphorylation (Y705) as determined by methylation-specific PCR (MSP) and real time quantitative reverse transcription (RT)-PCR in NSCLC cell lines. Silencing of PTPRT using siRNA in H520 lung cancer cell line resulted in increased pSTAT3Tyr705 and upregulation of STAT3 target genes such as Cyclin D1 and Bcl-XL expression. We show this association of PRPRT methylation with upregulation of the STAT3 target genes Cyclin D1 and Bcl-XL in patient derived lung tumour samples. We further demonstrate that PTPRT promoter methylation associated with different levels of pSTAT3Ty705 in lung cancer cell lines had selective sensitivity to STAT3 pathway small molecule inhibitors (SID 864,669 and SID 4,248,543). Our data strongly suggest that silencing of PTPRT by promoter hypermethylation is an important mechanism of STAT3 hyperactivation and targeting STAT3 may be an effective approach for the development of new lung cancer therapeutics.
Introduction
Lung cancer is the most common cancer worldwide and the leading cause of cancer-related mortality in the United States [Citation1] and the world. In 2017, an estimated 222,500 new cases of lung cancer are expected to be diagnosed in the US, representing about 14 percent of all cancer diagnoses, but 155,870 deaths are expected, accounting for 27 percent of total cancer deaths [Citation1,Citation2]. Despite therapeutic advances, the five year survival rate for lung cancer patients remains low at only 17% [Citation3,Citation4], much worse than for other common cancers, including breast (89%), prostate (99%) and colon (64%) cancer [Citation5].
The identification of molecular changes that can be targeted has provided one means to improve lung cancer treatment outcomes. Activating mutations in EGFR (epidermal growth factor receptor) and KRAS (a member of the RAS/MAPK pathway) occur in about 10–30% of NSCLC [Citation6]. Although EGFR-Tyrosine kinase inhibitors (TKIs) are an established treatment for patients with a confirmed EGFR mutation [Citation7], most patients develop drug resistance over time [Citation8,Citation9]. Also, patients with a KRAS mutation do not have a targeted therapy available to them [Citation10]. Thus, considering the small subgroup of NSCLC patients who can currently benefit from a targeted therapy, there is a need to identify novel therapeutic strategies that a significant population of NSCLC patients are likely to benefit from.
Emerging studies continue to implicate STAT3 as an oncogene in many different cancers [Citation11], including lung cancer [Citation12–Citation14]. STAT3 belongs to a family of seven latent cytoplasmic transcription factors that are activated in response to various extracellular cytokines, hormones, and growth factors leading to recruitment of STAT3 to the activated receptor via phosphotyrosine-SH2 recognition and subsequent phosphorylation of tyrosine residue Y705 [Citation15]. pSTAT3 undergoes homo (with another pSTAT3) or hetero (with pSTAT1) dimerization through reciprocal phosphotyrosine-SH2 interactions and activated pSTAT3 dimers translocate into the nucleus where they bind to DNA response elements in the promoter regions of targeted genes. There, they mediate gene resulting in cell proliferation, differentiation, apoptosis, angiogenesis, metastasis, inflammation, and immune response [Citation16]. Multiple molecular mechanisms have been identified that influence the persistent activation of STAT3 in different cancers [Citation17]. STAT3 gain-of-function mutations are not frequent in either lung adenocarcinoma or squamous cell lung cancer [Citation18,Citation19]. On the contrary, Grabner et al, elucidate that KRAS mutant lung tumours exhibit reduced STAT3 levels [Citation13], however numerous studies including the most recently published work suggest that STAT3 function as an oncogene in NSCLC [Citation20].
In many human cancers, including lung cancer, STAT3 is constitutively activated by upstream regulators including the tyrosine kinase growth factor receptors EGFR and its ligands, such as EGF (Epidermal Growth Factor), amphiregulin, and TGF-alpha, Src (Proto-oncogene tyrosine-protein kinase) [Citation21]. Of importance is IL-6 (interleukin-6), an upstream STAT3 regulator acting through the IL6R-gp130 receptor complex [Citation14], leading to STAT3 hyperactivation [Citation22]. Further in NSCLC, STAT3 has been shown to be activated by JAK2, and the signalling is independent of known driver oncogenic mutations [Citation23,Citation24]. The importance of disruption of STAT3 regulation includes key STAT3 target genes linked to tumour formation, such as Cyclin D1, which is important in cell cycle control, the antiapoptotic protein Bcl-XL, and many others [Citation14].
In addition to the upstream STAT3 activating ligands associated with initiating STAT3 signalling events, STAT3 is negatively regulated by several downstream regulators, and deregulation of the negative regulators can lead to constitutive activation of STAT3 [Citation23]. PTPRT negatively regulates STAT3 function via dephosphorylating STAT3 tyrosine Y705, the residue known to be critical for activation of STAT3 signal transduction [Citation25]. PTPRT is one of the most frequently inactivated genes across all human cancers [Citation26]. The first study to report somatic mutations of PTPRT was performed by Wang et al in colorectal cancers [Citation27]. A study led by Lee et al., reported that in several types of human cancers, such as colon, gastric, acute leukaemia’s, hepatocellular, breast and NSCLC, the occurrence of a mutation in the PTPRT phosphatase domain is rare [Citation28]. In head and neck cancer, PTPRT has been shown to be mutated in a small subset of tumours [Citation29]. Since the scarcity of PTPRT mutations is inconsistent with the frequency of STAT3 activation in lung cancer, other mechanisms, include the epigenetic alterations of negative regulators of STAT3 may play a role. PTPRT may also promote excessive STAT3 activity, thereby enhancing tumour growth [Citation26]. PTPRT promoter hypermethylation has been studied in only a few solid tumours [Citation26,Citation30]. Therefore, the present study was aimed at assessing the gene promoter methylation status of PTPRT in lung cancer, and through preclinical studies of pathway activation and genetic and pharmacologic inhibition, determining the importance of epigenetic silencing of PTPRT. Our results suggest that targeting STAT3 with newly developed STAT3 pathway inhibitors may demonstrate an effective therapeutic approach to tumours with STAT3 activation from PTPRT gene silencing.
Materials and methods
The cancer genome atlas (TCGA) data analyses
Data analysis was performed using R/Bioconductor software [Citation31,Citation32] with built-in packages and custom routines. Figures were created using built-in R graphics functions. We used Spearman’s correlation coefficient to test association of DNA methylation and gene expression. TCGA DNA methylation was measured on the Illumina Human Methylation 450K platform [Citation33]. Level 3 data were downloaded from TCGA Data Portal. We used the cg03403065 probe to represent PTPRT promoter methylation. The RNA sequencing (RNAseq) data as Level 3 RSEM normalized values were accessed from the Broad Institute TCGA GDAC Firehose (http://ezid.cdlib.org/id/doi:10.7908/C16W9975) and log2-transformed for further analysis. Mutation, amplification, deep deletion and multiple alteration data for STAT3 and PTPRT were downloaded from cBioPortal [Citation34,Citation35].
The University of Pittsburgh lung cancer cohort and analyses
Fresh-frozen banked lung cancer tissue previously characterized for mRNA expression [Citation36] were also analysed for DNA methylation using the Illumina Human Methylation 27K platform. These early stage non-small cell lung cancers are 75% adenocarcinoma and 25% squamous cell carcinoma [Citation36]. DNA and RNA were isolated from frozen tumour tissue and, after passing quality checks, DNA was used to determine methylation status of genes, upon bisulphite conversion, in NSCLC tumours, and RNA was used to assess mRNA expression with the Illumina Human HT-12 v4 BeadChip.
MSP of NSCLC tumour samples
To determine PTPRT methylation status in NSCLC Pittsburgh cohort, lung tumour tissues obtained during biopsy were processed for extraction of DNA using the QIAamp DNA Blood Mini Kit (Qiagen, Hilden, Germany). For MSP, bisulphite conversion of 1 μg of DNA per sample was carried out using the EZ DNA Methylation-DirectTM kit (Zymo Research, Irvine, CA, USA). MSP primers for PTPRT were designed using MethPrimer software [Citation37]. The MSP primers were PTPRT Methylated Sense: 5ʹ-CGTATTTTTTTCGGGGTCGATTAGTTC-3ʹ, PTPRT Methylated Antisense: 5ʹ-GAAAACCCTACCCGACGTATAAATACG-3ʹ, PTPRT Unmethylated Sense: 5ʹ-TTTTTTGTGTATTTTTTTTGGGGTTGATTAGTTTG-3ʹ, PTPRT Unmethylated Antisense: 5ʹ-AAAAAACAAAAACCCTACCCAACATATAAATACA-3ʹ. MSP was performed using the EpiTect® MSP Kit (Qiagen, Hilden, Germany). After reaction completion, products were analysed by gel electrophoresis on an 8% polyacrylamide gel and stained with Gel Red (Biotium, Fremont, CA, USA). β-Actin was used as a normalization control. The β-Actin primers were β-Actin-FP 5ʹ-tagggagtatataggttggggaagtt-3ʹ and β-Actin-RP 5ʹ-aacacacaataacaaacacaaattcac-3ʹ.
Determination of PRPRT and STAT3 target gene expression in NSCLC tumour samples and cell lines by RT-PCR
To measure PTPRT, Bcl-XL and Cyclin D1 expression, RNA was extracted from NSCLC tumour samples and cell lines using the RNeasy mini kit (Qiagen, Hilden, Germany). cDNA was synthesized using the qScript cDNA Synthesis Kit (Quantabio, Beverly, MA, USA). Bcl-XL, Cyclin D1 and β-Actin mRNA expressions were determined by RT-PCR [Citation38], using the MyiQTM Single-Colour Real-Time PCR Detection System (Bio-Rad, Hercules, CA, USA). The PCR primer sequences for PTPRT were: PTPRT-FP 5ʹ-ctcctctgtggtgatgctga-3ʹ, PTPRT-RP 5ʹ atgtctgcggagacgaactc-3ʹ; Bcl-XL were: Bcl-XL-FP 5ʹ-gatccccatggcagcagtaaagcaag-3ʹ, Bcl-XL-RP 5ʹ-ccccatcccggaagagttcattcact-3ʹ; Cyclin D1 were: Cyclin D1 -FP 5ʹctgtgctgcgaagtggaaacc-3ʹ, Cyclin D1-RP 5ʹcaggaagcggtccaggtagtt-3ʹ; and β-Actin were: β-Actin -FP 5ʹ-ctggcacccagcacaatg-3ʹ, β-Actin -RP 5ʹ-gggccggactcgtcatac-3ʹ. Continuous measurement of the PCR product was enabled by incorporation of SYBR-Green® fluorescent dye into the double-stranded PCR products.
Cell culture
Seven lung cancer cell lines, A549, H23, H1435, H1650, H1793, H1703 and H520 were obtained from ATCC (Manassas, VA, USA). Cell lines were authenticated by genotyping with a multiplex STR assay (Genetica). The cell lines were cultured in RPMI-1640 (ATCC, Manassas, VA, USA) containing 10% foetal bovine serum (Flowery Branch, GA, USA).
MSP of NSCLC cell lines
Cell lines were plated at a density of 1 × 106 cells/10 cm dish and after 24h of plating, DNA was isolated using the QIAamp DNA Blood Mini Kit (Qiagen, Hilden, Germany). Bisulphite conversion of 1 μg of DNA per sample was carried out using the EZ DNA Methylation-DirectTM kit (Zymo Research, Irvine, CA, USA). MSP using PTPRT methylated and unmethylated primers as stated previously, were performed using the EpiTect® MSP Kit (Qiagen, Hilden, Germany). After reaction completion, products were analysed by gel electrophoresis on an 8% polyacrylamide gel and stained with Gel Red (Biotium, Fremont, CA, USA).
Determination of pSTAT3Tyr705 expression in lung cancer cell lines
The 7 NSCLC lung cancer cell lines were plated and, after 24h, cells were harvested for protein extraction. To determine pSTAT3Tyr705 protein expression, protein concentration was determined using the Pierce™ BCA Protein Assay Kit (Thermo Fisher Scientific, Pittsburgh PA). Proteins (40 μg/lane) were separated by 10% sodium dodecyl sulphate polyacrylamide gel electrophoresis (SDS-PAGE), probed with rabbit anti-pSTAT3Tyr705 or mouse anti-STAT3 monoclonal antibodies (Cell Signalling Technology, Boston, MA) and developed using the Immobilon Western Chemiluminescent HRP Substrate (EMD Millipore, Massachusetts, MA, USA). Rabbit anti-β-tubulin polyclonal antibody (Abcam Inc, Cambridge, MA, USA), was used as a loading control.
Transient knockdown of PTPRT
siRNA oligonucleotide against PTPRT and scrambled siRNA were obtained from Dharmacon Research, Inc (Lafayette, CO, USA). The siRNA was resuspended in siRNA buffer provided by the manufacturer to the desired final concentration and 1μg of siRNA mixed with OPTIMEM were added per well in a 12 well plate. Cells were replaced with complete media after 48h, and RNA or protein were extracted after 72h or 96h. PTPRT, Bcl-XL, Cyclin D1 and β-Actin mRNA expressions were determined by RT-PCR as described previously. To determine pSTAT3Tyr705, STAT3 and STAT3 targets such as Cyclin D1 and Bcl-XL protein expressions, 40 μg/lane were separated by 10% SDS-PAGE and probed with rabbit anti-pSTAT3Tyr705 or mouse anti-STAT3 monoclonal antibodies (Cell Signalling Technology, Boston, MA), rabbit anti-human Cyclin D1 polyclonal or mouse anti-human Bcl-XL monoclonal antibodies (Santa Cruz Biotechnology, Santa Cruz, CA, USA) and developed using the Immobilon Western Chemiluminescent HRP Substrate (EMD Millipore, Massachusetts, MA, USA). Rabbit anti-β-tubulin polyclonal antibody (Abcam Inc, Cambridge, MA, USA), was used as a loading control.
Small molecule inhibitors
Compounds SID 864,669 (6-(3,4-dichlorophenyl)-3-(1,4,5,6-tetrahydrocyclopenta[c]pyrazol-3-yl)-7H-[1,2,4]triazolo[3,4 b][1,3,4] thiadiazine) and UPCDC-10,205 (3-(4-chlorophenyl)-5-[6-(4-methoxyphenyl)-7-methyl-7H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazin-3-yl]-1H-pyrazole) were prepared as previously reported [Citation39]. The synthesis of SID 4,248,543 (1-[(1-{[2-(4-ethylphenyl)-5-methyl-1,3-oxazol-4-yl] methyl} piperidin-4-yl) carbonyl]-4-phenylpiperazine), and UPCDC-10,381 (1-benzyl-4-[6-(4-methoxyphenyl)-7-methyl-7H- [1,2,4] triazolo[3,4-b] [1,3,4] thiadiazin-3-yl]-1H-1,2,3-triazole) has been included in Supplementary Methods Section.
Proliferation assay
Lung cancer cell lines identified as PTPRT methylated (H23, H1703) or PTPRT unmethylated (H520) were plated at a density of 30,000 cells/well in 1 mL RPMI1640 + 10%FBS. After 24h, cells were treatment with increasing concentrations of the compounds SID 864,669, SID 4,248,543 and UPCDC 10,205 (SID 864,669, SID 4,248,543 were identified from a high content screening campaign [Citation39,Citation40]; UPCDC-10,205 was an optimized analog of SID 864,669) or DMSO alone as a vehicle control in triplicate in the complete media. The compound UPCDC 10,381 that was identified as an inactive compound in the high content STAT3 inhibitor screening assay [Citation40], was tested on the lung cancer cell lines as a negative control. At the end of 72 h, MTT (Thizolyl Blue Tetrazolium Bromide, Sigma-Aldrich, St Louis, MO, USA), assays were performed and the data were analysed quantitatively using GraphPad Prism 5 (GraphPad, La Jolla, CA, USA).
Effect of the STAT3 inhibitor, SID 864669 on mRNAexpression of STAT3 target genes as determined by RT-PCR
Lung cancer cell lines (H23, H1703 and H520) were plated at a density of 8 × 105 cells in a 10cm dish. After 24h of plating, cells were treated with varying concentrations of SID 864,669 or DMSO alone as a vehicle control. At the end of 24h, cells were harvested and RNA extracted and PTPRT, Bcl-XL, Cyclin D1 and β-Actin mRNA expressions were determined by RT-PCR as described earlier.
Effect of transient PTPRT expression with subsequent siRNA treatment on STAT3 expression as determined by western analyses
The PTPRT plasmid, pcDNA4 TO/PTPRT-Full -myc-His was a kind gift from Dr. Zhenghe J. Wang (Case Western Reserve University, Cleveland, Ohio). The NSCLC cell line H1703 was plated and after 24 h, transfected with 2.5 ug of the PTPRT plasmid followed by treatment with siRNA (1 µg, as described previously) targeting PTPRT or scrambled siRNA. At the end of 72h, cells were harvested, and western analyses was done to determine pSTAT3Tyr705, STAT3 and β-Tubulin.
Statistical analyses
For STAT3 inhibitor treatment experiments, to compare two treatment groups in NSCLC cell lines, p values were analysed using an exact two-tailed test and p ≤ 0.05 was considered significant. Statistical analyses were performed using Graph pad prism.
Results
Epigenetic inactivation of PTPRT in NSCLC
In NSCLC, frequent constitutive activation of STAT3 makes it a plausible therapeutic target. However, mutational activation of STAT3 is a rare event. This was verified from the TCGA data analyses in NSCLC tumours where only a very small fraction of somatic mutation frequency in the STAT3 gene was found in lung adenocarcinoma (2%, 95% score confidence interval (CI) 1%-5%) [Citation18] and lung squamous cell carcinoma tumours (1%, 95% CI 0%-3%) ([Citation19] (Supplemental Figure S1). Similarly, amplification and deep deletion of the STAT3 gene was seen in about 0.5% in lung adenocarcinoma and lung squamous cell carcinoma tumours respectively (Supplemental Figure S1). Alternative mechanisms of STAT3 activation include genetic or epigenetic alterations of the negative regulators of STAT3. PTPRT, a negative regulator of STAT3, through mutation, copy number loss/deletion, copy number amplification or promoter hypermethylation may influence STAT3 activation status, as pSTAT3 is a confirmed substrate of PTPRT [Citation25]. To assess this, lung adenocarcinoma and lung squamous cell carcinoma tumours from TCGA were evaluated, and some of the above genetic variations in the PTPRT gene were observed in a very small population such as somatic mutation frequency in 11%, (which was an overestimation of functional alterations since most were not inactivating mutations), copy number amplification in 1% and copy number deletion was seen in less than 0.5% in lung adenocarcinoma (Supplemental Figure S2). In lung squamous cell carcinoma, somatic mutation frequency was seen in 8% (the majority missense), copy number amplification in 1% and copy number deletion as well as multiple alterations were observed in less than 0.5% (Supplemental Figure S2).
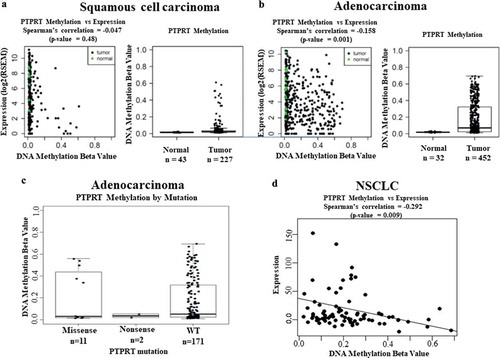
Without evidence of mutational mechanisms for STAT3 activation, we then examined PTPRT methylation in lung adenocarcinoma and lung squamous cell carcinoma using TCGA data obtained with the Illumina HumanMethylation450 platform. We determined aberrant methylation as a fractional methylation level (beta value) and correlated with PTPRT mRNA expression in both lung squamous cell carcinoma ()) and lung adenocarcinoma tumours ()). Using a beta value threshold of > 0.2, as we have previously shown to be associated with cancer specific repression of gene expression [Citation41], TCGA data demonstrated that PTPRT promoter is methylated in 157 out of 452 (35%, 95% CI 30%-39%) of lung adenocarcinomas and 14 out of 227 (6%, 95% CI 4%-10%) lung squamous cell carcinomas. Normal lung tissues were also included in the analyses but did not show methylation in this region. Higher DNA methylation beta values were associated with lower levels of mRNA expression (), p = 0.48 and 1(b), p = 0.001). Further, to determine if negative regulation of STAT3 is mediated by simultaneous occurrence of PTPRT mutation and methylation, lung adenocarcinoma tumours from TCGA were analysed. The analysis showed that most PTPRT methylated tumours had no mutations in PTPRT, with only a few with missense mutations that could be non-functional passenger mutations ()). This observation suggests that loss-of-function of negative regulators of STAT3, like PTPRT promoter hypermethylation, is a possible mechanism of activation of STAT3.
Figure 1. (a&b) Scatter plots of DNA promoter methylation of PTPRT (the cg03403065 probe) and mRNA expression in TCGA squamous cell carcinoma and adenocarcinoma. Tumour samples are black dots, and normal samples are green triangles. The plots demonstrated loss of expression in methylated tumours. Boxplots of DNA promoter methylation of PTPRT in the same samples showed promoter hypermethylation of tumour samples. (c) Boxplots of DNA promoter methylation of PTPRT by PTPRT mutation types. Most of PTPRT hypermethylated samples are PTPRT wild type (WT). Only a small subset of the missense mutated samples showed hypermethylation suggesting that these mutations could be non-functional passenger mutations. (d) Scatter plots of DNA promoter methylation of PTPRT (cg13168820 probe) and mRNA expression in the Pittsburgh Lung Cancer Cohort. As for TCGA, there was a trend for loss of expression in methylated tumours.
We further extended studies to include the Pittsburgh Lung Cancer Cohort, which had previously been studied for genome wide gene expression [Citation36]. Thirty-six of 80 tumours (49%, 95% CI 38%-60%) demonstrated hypermethylation (beta >0.2) of the PTPRT promoter region, and this regional methylation increase was again associated with diminished mRNA expression of the PTPRT (), p = 0.009). We then developed a gene specific assay to assess PTPRT status using MSP to confirm the methylation status of PTPRT promoter region in NSCLC lung tumours ()). Out of the 12 tumour samples, six tumours had detectable promoter region methylation (Patient #s 2, 4, 6, 8, 9 and 11) whereas the remaining 6 tumours demonstrated very low methylation of the PTPRT promoter region (Patient #s 1, 3, 5, 7, 10 and 12), confirming PTPRT promoter methylation can be detected by an independent method and might be a frequent mechanism for STAT3 activation in lung cancer. In future, the real time PCR assay to determine PTPRT methylation can be used in patient samples in the clinical setting.
Figure 2. (a) Methylation of PTPRT promoter region in NSCLC tumours. NSCLC tumours samples from Stage 1A/1B patients were processed to extract DNA followed by bisulphite conversion to determine PTPRT promoter methylation by Real Time PCR. Out of the 12 tumour samples, six tumours had detectable promoter region methylation (Patient #s 2, 4, 6, 8, 9 and 11) whereas the remaining 6 tumours demonstrated very low methylation of the PTPRT promoter region (Patient #s 1, 3, 5, 7, 10 and 12), which compared well with the methylated beta values. (b&c) Association of PTPRT promoter methylation to STAT3 target gene expression in lung tumours. Lung tumours (Patient #s 1, 2 and 3) were assessed for STAT3 target gene expression by RT-PCR. Cyclin D1 (Figure 2(b)) and Bcl-XL (Figure 2(c)) mRNA expression was found to associate with PTPRT methylation status. (d&e) Association of PTPRT promoter methylation and Bcl-XL and Cyclin D1 mRNA expression. Scatter plots of DNA promoter methylation of PTPRT and Bcl-XL and Cyclin D1 mRNA expression in TCGA adenocarcinoma showed an increase in Bcl-XL (Figure 2(d)) and Cyclin D1 (Figure 2(e)) mRNA expression with increase in PTPRT methylation.
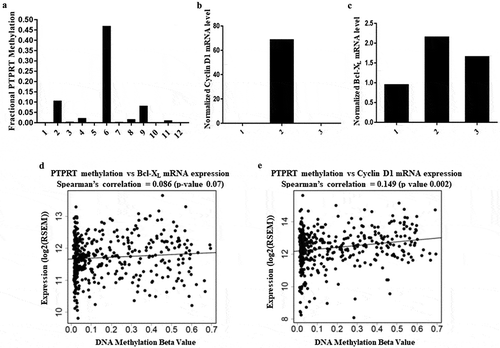
Association of PTPRT methylation status with STAT3 target gene expression in lung tumours
These primary tumours allowed us to determine if PTPRT promoter methylation was associated with upregulation of STAT3 targets. We examined mRNA expression of the STAT3 target genes in three samples with adequate mRNA (Patient #s 1, 2 and 3), finding that Cyclin D1 ()) and Bcl-XL ()) mRNA expression in the tumour with methylated PTPRT promoter region (patient #2) was increased compared to the other two tumours (patient #s 1 and 3) where PTPRT was unmethylated. We extended these analyses to lung adenocarcinoma patients in TCGA. We identified similar higher Bcl-XL ()) and Cyclin D1 ()) mRNA expression with higher PTPRT methylation, again suggesting an association of PTPRT inactivation with STAT3 activation in lung cancer. While these correlations are statistically significant, the imperfect relationship between PTPRT methylation and STAT3 target activation suggest additional factors may modify PTPRT expression and target gene activation.
Hypermethylation of the PTPRT promoter associated with PTPRT and pSTAT3Tyr705 expression in NSCLC cell lines
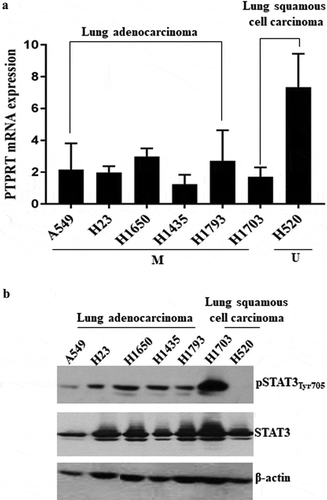
To extend our studies of STAT3 activation, we examined lung cancer cell lines for this epigenetic change. Hypermethylation of the CpG island upstream of the PTPRT promoter was determined using MSP amplification of the bisulphite-treated DNA in seven NSCLC cell lines as done for our primary tumour samples. Out of the seven NSCLC cell lines tested, six (A549, H23, H1435, H1650, H1793 and H1703) demonstrated PTPRT promoter methylation, with H1435, H1650 and H1793 demonstrating partial unmethylation for PTPRT as well. Only in H520 was PTPRT found to be unmethylated. These results indicate that PTPRT could be frequently methylated in lung cancer (Supplemental Figure S3). This PTPRT mRNA expression was tested in the lung cancer cell lines and we observed translational silencing of PTPRT mRNA associated with PTPRT promoter hypermethylation ()). In the NSCLC cell lines such as A549, H23, H1435, H1650, H1793 and H1703, PTPRT mRNA expression was downregulated suggesting that methylation at the CpG islands of PTPRT may be responsible for the reduced PTPRT mRNA expression in the lung cancer cell lines ()). Conversely, in the lung cancer cell line H520, loss of PTPRT promoter methylation is concomitant to gain of PTPRT mRNA expression ()). These results suggest that PTPRT methylation associates with PTPRT mRNA expression in lung cancer cell lines. To test whether PTPRT expression negatively regulates STAT3 phosphorylation since pSTAT3 was identified as an important substrate of PTPRT [Citation25], we determined the phosphorylation level (Y705) of STAT3 in the lung cancer cell lines by western analyses. Based on our hypothesis that PTPRT promoter hypermethylation may lead to increased pSTAT3 levels, the NSCLC cell lines (A549, H23, H1435, H1650, H1793 and H1703) with PTPRT promoter methylation demonstrated increased pSTAT3Tyr705 expression, however in the lung cancer cell line H520 which showed unmethylation of the PTPRT promoter region, reduced expression of pSTAT3Tyr705 was observed ()). This result is consistent with low pSTAT3 expression in H520 cell line as shown in another lung cancer study [Citation42]. Furthermore, this result suggests that promoter hypermethylation of PTPRT leads to loss of its phosphatase function in lung cancer, concomitant with increased activation of STAT3.
Figure 3. (a) PTPRT mRNA expression associates with PTPRT methylation status in lung cancer cell lines as demonstrated by RT-PCR. Downregulation of PTPRT mRNA expression was observed in lung cancer cell lines, A549, H23, H1650, H1435, H1793 and H1703 which demonstrated hypermethylation in PTPRT promoter region whereas the cell line, H520 showed increased PTPRT mRNA expression that associated with unmethylation of the PTPRT promoter region. This experiment was repeated twice with similar results. In this Figure, M = Methylation and U = Unmethylation of the PTPRT gene in the cell lines tested. (b) PTPRT promoter methylation status associates with pSTAT3Tyr705 expression in NSCLC cell lines. Lung cancer cell lines tested for pSTAT3Tyr705 levels demonstrated that methylation of the PTPRT promoter region strongly associated with pSTAT3Tyr705 expression levels as demonstrated by western analyses. Out of the seven lung cancer cell lines, A549, H23, H1650, H1435, H1793 and H1703 which showed PTPRT promoter methylation also showed expression of pSTAT3Tyr705 whereas the expression of pSTAT3Tyr705 was almost negligible in the cell line H520 which associated with the unmethylation status of PTPRT, as demonstrated in our study. This experiment was repeated twice with similar results.
Loss of function of PTPRT causes increased STAT3 target gene expression
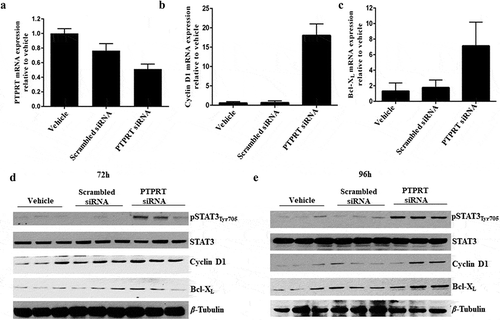
To test whether PTPRT function is directly controlling STAT3 phosphorylation, knockdown of PTPRT by siRNA was performed in the lung cancer cell line H520. STAT3 activates multiple downstream targets including genes involved in cell cycle progression (Cyclin D1, Fos, c-Myc, etc) and anti-apoptotic genes (Survivin, Bcl-2, Bcl-XL and others). In this study, we have focused on cyclin D1 and Bcl-XL as downstream targets of STAT3, as they have been well characterized and studied in lung cancer [Citation43]. Data shown in ) suggest that transfection of siRNA against PTPRT in H520 cells inhibited PTPRT mRNA expression compared to the cells transfected with scrambled siRNA or vehicle at the end of 72h ()). Further, inhibition of PTPRT mRNA expression using siRNA correspond to increased mRNA expression of the STAT3 target genes such as Cyclin D1 and Bcl-XL compared with cells transfected with scrambled siRNA control or vehicle (,c)). In addition, effect of PTPRT knockdown on STAT3 and STAT3 target protein expression showed that in H520 cells transfected with PTPRT siRNA, inhibition of PTPRT results in increased expression of pSTAT3Tyr705 and activation of STAT3 target proteins such as Cyclin D1 and Bcl-XL at the end of 72h ()) and 96h ()), thus providing mechanistic indication that PTPRT methylation is an important epigenetic event that drives upregulation of pSTAT3Tyr705.
Figure 4. (a) Knockdown of PTPRT mRNA expression in H520 cells by PTPRT siRNA. PTPRT knockdown by siRNA in H520 cells which demonstrated PTPRT mRNA expression in our study resulted in decreased expression of PTPRT mRNA. H520 cells were treated with PTPRT siRNA in presence of OPTI-MEM and after 48h, cells were replaced with complete media and at the end of 72h, RNA was extracted. RT-PCR data showed that H520 cells transfected with PTPRT siRNA had significantly lower 48% (p < 0.05) PTPRT mRNA expression compared to cells treated with vehicle. PRPRT mRNA expression in H520 cells transfected with scrambled siRNA did not show any significant decrease when compared to cells treated with vehicle. Results are from three triplicate wells in one experiment. (b & c) Increased expression of Cyclin D1 and Bcl-XL upon PTPRT knockdown in H520 cells. H520 cells which demonstrated unmethylation of the PTPRT promoter region were transfected with PTPRT siRNA. After 72h, the cells showed increased mRNA expression of the STAT3 target genes such as Cyclin D1 and Bcl-XL compared to the cells treated with vehicle or scrambled siRNA as determined by RT-PCR. Results are from three triplicate wells in one experiment. (d & e) Increased pSTAT3Tyr705 along with STAT3 target gene expression upon PTPRT knockdown by siRNA. H520 cells transfected with PTPRT siRNA caused increased pSTAT3Tyr705 along with Cyclin D1 and Bcl-XL protein expression compared to the cells treated with vehicle or scrambled siRNA at the end of 72h and 96h as determined by western analyses. The results are from triplicate wells from one experiment.
PTPRT promoter methylation leads to increased STAT3 activation and sensitivity to STAT3 inhibition
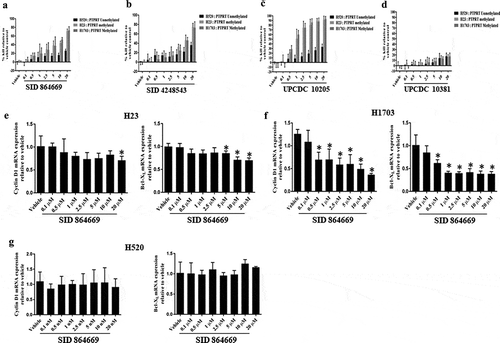
To assess whether PTPRT promoter methylation may increase sensitivity to STAT3 inhibition, lung cancer cell lines which were methylated as well as unmethylated for PTPRT in association with differing levels of pSTAT3Tyr705 expression were chosen. In this study, two of the STAT3 pathway inhibitors, SID 864,669 and SID 4,248,543 were identified from a library of 94,491 compounds from the Molecular Library Screening Centre Network (MLSCN) and have been shown previously to inhibit pSTAT3 activation [Citation40,Citation44,Citation45]. A third STAT3 pathway inhibitor, UPCDC 10,205 was an optimized analog synthesized based on the structure of 669 [Citation39]. The inactive compound UPCDC 10,381 was also synthesized as part of the optimization library. The STAT3 pathway inhibitors which have selectivity for STAT3 vs STAT1 as demonstrated earlier [Citation40,Citation44], were used to assess the sensitivity of lung cancer cell lines with varying pSTAT3 expression levels. These inhibitors have been shown to block IL-6-induced STAT3 activation in head and neck cancer model [Citation40,Citation45]. Data from -) suggest that the lung cancer cell lines H23 and H1703 with PTPRT promoter hypermethylation and STAT3 activation, when exposed to varying concentrations of SID 864,669, SID 4,248,543 and UPCDC 10,205, inhibited cellular proliferation and induced cell death in a dose dependent manner, whereas H520 cells that exhibit unmethylation in the PTPRT promoter region in association with undetectable level of pSTAT3Tyr705 are much less sensitive to SID 864,669, SID 4,248,543 and UPCDC 10,205 at the end of 72h. The inactive compound UPCDC 10,381 did not have any effect on cell death in cell lines at any of the concentrations tested ()), suggesting the specificity of the STAT3 inhibitors. Further we tested the effect of one of the inhibitors, SID 864,669, on the mRNA expression of the STAT3 target genes such as Cyclin D1 and Bcl-XL in the lung cancer cell lines. Our results indicate that SID 864,669 inhibited Cyclin D1 and Bcl-XL mRNA expression in H23, although the effect was modest ()) and H1703 ()) cell lines at all the concentrations tested after 24h. However, in the H520 cell line ()), SID 864,669 did not have any effect on the mRNA expression of the STAT3 target genes, suggesting that pSTAT3Tyr705 expression might predict sensitivity to STAT3 inhibitors. We examined 6 additional lung cancer cell lines for PTPRT methylation to confirm this correlation of PTPRT inactivation with STAT3 activation and sensitivity to STAT3 inhibition. However, these 6 cell lines also had PTPRT methylation, suggesting that in vitro growth conditions may select for this phenotype (since the minority of primary lung cancer have PTPRT inactivation), and thus our ability to causally link these findings.
Figure 5. (a, b, c & d) Increased PTPRT promoter methylation is associated with increased sensitivity to STAT3 inhibition. Lung cancer cell lines which are methylated (H23 and H1703) or unmethylated (H520) in the PTPRT promoter region were treated with increasing concentrations of the three STAT3 inhibitors SID 864,669, SID 4,248,543 and UPCDC 10,205 in complete media. At the end of 72h, MTT assay was done to determine % kill which increased with increasing concentrations of SID 864,669 (Figure 5(a)), SID 4,248,543 (Figure 5(b)) and UPCDC 10,205 (Figure 5(c)) and was more pronounced in H23 and H1703 which are methylated in the PTPRT promoter region in conjunction with high pSTAT3Tyr705 expression. The inactive compound UPCDC 10,381 did not have an effect in any cell lines tested (Figure 5(d)). Results are from four separate experiments. (e, f & g) Increased PTPRT promoter methylation was accompanied with increased sensitivity to STAT3 inhibition as evident from the reduction of STAT3 target gene expression. H23, H1703 and H520 cells were treated with varying concentrations of SID 864,669 and at the end of 24h, cells were harvested for RNA extraction and RT-PCR was performed to determine the mRNA expression of the STAT3 target genes. The STAT3 inhibitor SID 864,669 caused a reduction in Cyclin D1 and Bcl-XL mRNA expression in H23 (Figure 5(e), * indicates p < 0.05) and H1703 (Figure 5(f), * indicates p < 0.05) cells which showed PTPRT promoter methylation with concomitant expression of pSTAT3Tyr705. In H520 cells, there was no effect of SID 864,669 on the mRNA expression of STAT3 target genes (Figure 5(g)).
Discussion
Lung cancer is the leading cause of cancer death worldwide [Citation46,Citation47] . Although progress has been made to understand the pathogenesis of the disease [Citation46], the five year survival rate has not significantly improved over the last few decades [Citation48,Citation49]. The development of targeted agents has been on the rise in the past two decades [Citation49]. The clinical outcome of patients treated with targeted therapy depends on the presence of a variety of factors, such as activating mutations, overexpression of the target protein, gene copy number and others [Citation49]. Tyrosine kinases especially are an intensively pursued drug target because they play an important role in the modulation of growth factor signalling [Citation50]. In lung cancer, response to Erlotinib, an EGFR-tyrosine kinase inhibitor is predicted by the presence of an EGFR mutation more commonly seen in non-smokers [Citation49], as well as overexpression of the EGFR protein, EGFR gene amplification and methylation [Citation51,Citation52]. However, lung cancer patients previously treated with a single agent EGFR-TKI developed secondary resistance through alternative pathways that bypass the inhibited EGFR pathway [Citation53]. In addition, only a few small molecule inhibitors exhibit the highest degree of kinase selectivity [Citation50]. STAT3 represents one important transcription factor, that plays an oncogenic role in several malignancies. In NSCLC, STAT3 is persistently activated in 22%~65% of the patient population [Citation12]. Thus, aberrant activation of STAT3 has prompted the development of different strategies to target STAT3 pathway inhibition. At present, evidence for clinical activity is lacking, perhaps due to insufficient specificity or selectivity [Citation54]. However, the testing of this approach has also been limited by the lack of identification of a sensitive target population.
Alteration of the epigenome resulting from changes in epigenetic regulators is another central mechanism of cancer [Citation55]. Increased hypermethylation of tumour suppressor genes has been associated with the initiation and progression of cancer and are being investigated as potential biomarkers for early detection, prediction of cancer progression and drug sensitivity [Citation56]. PTPRT as has been known to dephosphorylate STAT3, is hypermethylated in cancers, leading to loss of function of these phosphatases with concurrent increased activation of STAT3 [Citation26]. In a study with gastric cancer patients, PTPRT was found to be hypermethylated in a subset of patient tumours [Citation56]. In head and neck squamous cell carcinoma, although frequent hypermethylation has been shown, however the region examined provides an incorrect greater frequency of methylation for cancer, by using an Infinium probe which has prevalent methylation in normal tissue [Citation26]. Our study clearly defines the region and demonstrates a correlation with sensitivity to STAT3 inhibition, which in the clinical setting may be used to identify patients for treatment. In sporadic colorectal cancer, promoter sequence of PTPRT, was found to be hypermethylated compared with healthy colon tissue [Citation30]. In this study, we have identified epigenetic alterations of PTPRT in lung cancer, demonstrating an association with increased STAT3 signalling and increased sensitivity to STAT3 pathway inhibition.
TCGA analyses of lung tumour data revealed PTPRT promoter methylation in association with downregulation of PTPRT mRNA expression. To validate PTPRT promoter methylation in NSCLC human cohort, a subset of tumours showed PTPRT promoter methylation, with a few further tested revealed an association between PTPRT promoter methylation with an increase in the STAT3 target genes such as Cyclin D1 and Bcl-XL mRNA expression. Studies in lung cancer cell lines demonstrate that the PTPRT promoter region is completely or partially methylated and associated with PTPRT mRNA and pSTAT3Tyr705 protein expression status. Subsequent silencing of PTPRT using PTPRT siRNA in a lung cancer model with PTPRT mRNA expression caused induction in the STAT3 target genes such as Cyclin D1 and Bcl-XL mRNA as well as pSTAT3Tyr705, Cyclin D1 and Bcl-XL protein expression. Further, targeting lung cancer cell lines with STAT3 pathway inhibitors demonstrated specificity and selectivity of the STAT3 inhibitors based on their activity as well as the expression of pSTAT3Tyr705 in the cell lines. These cumulative data suggest that epigenetic silencing of PTPRT may play a critical role for STAT3 activation in lung cancer, and that by identifying tumours with this pathway addiction, selection of patients who will be most likely to respond to STAT3 pathway inhibitors may be possible.
Supplemental Material
Download Zip (817.5 KB)Acknowledgments
We would like to thank Sonali C. Joyce for running the western analyses for Supplemental Figure S5.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
Correction Statement
This article has been republished with minor changes. These changes do not impact the academic content of the article.
Additional information
Funding
Related Research Data
References
- Cancer Facts and. Figures. American Cancer Society; 2015.
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66(1):7–30.
- American cancer society cancer facts and figures. Atlanta, GA; 2016.
- David EA, Daly ME, Li CS, et al. Increasing rates of no treatment in advanced-stage non-small cell lung cancer patients: a propensity-matched analysis. J Thorac Oncol. 2017;12:437–445.
- Centers for Disease Control and Prevention National Center for Health Statistics CDC WONDER On-line database, compiled from compressed mortality file 1990-2010 series 20 No. 2P. 2013.
- Vijayalakshmi R, Krishnamurthy A. Targetable “driver” mutations in non small cell lung cancer. Indian J Surg Oncol. 2011;2(3):178–188.
- Isla D, De Castro J, Juan O, et al. Costs of adverse events associated with erlotinib or afatinib in first-line treatment of advanced EGFR-positive non-small cell lung cancer. Clinicoecon Outcomes Res. 2017;9:31–38.
- Fujita K, Kim YH, Yoshizawa A, et al. Concomitant T790M mutation and small-cell lung cancer transformation after acquired resistance to epidermal growth factor receptor-tyrosine kinase inhibitor. Respirol Case Rep. 2017;5(1):e00206.
- Wang W, Song Z, Zhang Y. A Comparison of ddPCR and ARMS for detecting EGFR T790M status in ctDNA from advanced NSCLC patients with acquired EGFR-TKI resistance. Cancer Med. 2017;6(1):154–162.
- Kaufman J, Stinchcombe TE. Treatment of KRAS-mutant non-small cell lung cancer: the end of the beginning for targeted therapies. JAMA. 2017;317(18):1835–1837.
- Bowman T, Garcia R, Turkson J, et al. STATs in oncogenesis. Oncogene. 2000;19(21):2474–2488.
- Harada D, Takigawa N, Kiura K. The role of STAT3 in non-small cell lung cancer. Cancers (Basel). 2014;6(2):708–722.
- Grabner B, Schramek D, Mueller KM, et al. Disruption of STAT3 signalling promotes KRAS-induced lung tumorigenesis. Nat Commun. 2015;6:6285.
- Song L, Turkson J, Karras JG, et al. Activation of Stat3 by receptor tyrosine kinases and cytokines regulates survival in human non-small cell carcinoma cells. Oncogene. 2003;22(27):4150–4165.
- Darnell JE Jr. STATs and gene regulation. Science. 1997;277(5332):1630–1635.
- Liu LJ, Leung KH, Chan DS, et al. Identification of a natural product-like STAT3 dimerization inhibitor by structure-based virtual screening. Cell Death Dis. 2014;5:e1293.
- Al Zaid Siddiquee K, Turkson J. STAT3 as a target for inducing apoptosis in solid and hematological tumors. Cell Res. 2008;18(2):254–267.
- Cancer Genome Atlas Research N. Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014;511(7511):543–550.
- Cancer Genome Atlas Research N. Comprehensive genomic characterization of squamous cell lung cancers. Nature. 2012;489(7417):519–525.
- Njatcha C, Farooqui M, Kornberg A, et al. STAT3 Cyclic Decoy Demonstrates Robust Antitumor Effects in Non-Small Cell Lung Cancer. Mol Cancer Ther. 2018;17(9):1917–1926.
- Wee P, Wang Z. Epidermal growth factor receptor cell proliferation signaling pathways. Cancers (Basel). 2017;9:5.
- Durant L, Watford WT, Ramos HL, et al. Diverse targets of the transcription factor STAT3 contribute to T cell pathogenicity and homeostasis. Immunity. 2010;32(5):605–615.
- Looyenga BD, Hutchings D, Cherni I, et al. STAT3 is activated by JAK2 independent of key oncogenic driver mutations in non-small cell lung carcinoma. PloS One. 2012;7(2):e30820.
- Ward AC, editor. STAT3 inhibitors in cancer. Cancer Drug Discovery and Development;2016.
- Zhang X, Guo A, Yu J, et al. Identification of STAT3 as a substrate of receptor protein tyrosine phosphatase T. Proc Natl Acad Sci U S A. 2007;104(10):4060–4064.
- Peyser ND, Freilino M, Wang L, et al. Frequent promoter hypermethylation of PTPRT increases STAT3 activation and sensitivity to STAT3 inhibition in head and neck cancer. Oncogene. 2016;35(9):1163–1169.
- Wang Z, Shen D, Parsons DW, et al. Mutational analysis of the tyrosine phosphatome in colorectal cancers. Science. 2004;304(5674):1164–1166.
- Lee JW, Jeong EG, Lee SH, et al. Mutational analysis of PTPRT phosphatase domains in common human cancers. APMIS. 2007;115(1):47–51.
- Lui VW, Peyser ND, Ng PK, et al. Frequent mutation of receptor protein tyrosine phosphatases provides a mechanism for STAT3 hyperactivation in head and neck cancer. Proc Natl Acad Sci U S A. 2014;111(3):1114–1119.
- Laczmanska I, Karpinski P, Bebenek M, et al. Protein tyrosine phosphatase receptor-like genes are frequently hypermethylated in sporadic colorectal cancer. J Hum Genet. 2013;58(1):11–15.
- R tR. A language and environment for statistical computing. Vienna, Austria: R foundation for statistical computing; 2004.
- Gentleman RC, Carey VJ, Bates DM, et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5(10):R80.
- Bibikova M, Barnes B, Tsan C, et al. High density DNA methylation array with single CpG site resolution. Genomics. 2011;98(4):288–295.
- Gao J, Aksoy BA, Dogrusoz U, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6(269):pl1.
- Cerami E, Gao J, Dogrusoz U, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2(5):401–404.
- Siegfried JM, Lin Y, Diergaarde B. et al. Expression of PAM50 genes in lung cancer: evidence that interactions between hormone receptors and HER2/HER3 contribute to poor outcome. Neoplasia (New York, NY). 2015;17(11):817–825.
- Li LC, Dahiya R. MethPrimer: designing primers for methylation PCRs. Bioinformatics. 2002;18(11):1427–1431.
- Leeman-Neill R. Prevention and treatment of head and neck cancer with natural compound inhibitors of STAT3. Doctoral Dissertation, University of Pittsburgh. PhD thesis, University of Pittsburgh 2008.
- LaPorte MG, Wang Z, Colombo R, et al. Optimization of pyrazole-containing 1,2,4-triazolo-[3,4-b]thiadiazines, a new class of STAT3 pathway inhibitors. Bioorg Med Chem Lett. 2016;26(15):3581–3585.
- Johnston PA, Sen M, Hua Y, et al. HCS campaign to identify selective inhibitors of IL-6-induced STAT3 pathway activation in head and neck cancer cell lines. Assay Drug Dev Technol. 2015;13(7):356–376.
- Pelosof L, Yerram S, Armstrong T, et al. GPX3 promoter methylation predicts platinum sensitivity in colorectal cancer. Epigenetics. 2017;12(7):540–550.
- Kluge A, Dabir S, Vlassenbroeck I, et al. Protein inhibitor of activated STAT3 expression in lung cancer. Mol Oncol. 2011;5(3):256–264.
- Chen Y, Cao Y, Yang D, et al. Increase of the therapeutic effect on non-small-cell lung cancer cells with combination treatment of shRNA against cyclin D1 and Bcl-xL in vitro. Exp Ther Med. 2012;3(2):255–260.
- Johnston PA, Sen M, Hua Y, et al. High-content pSTAT3/1 imaging assays to screen for selective inhibitors of STAT3 pathway activation in head and neck cancer cell lines. Assay Drug Dev Technol. 2014;12(1):55–79.
- Sen M, Johnston PA, Pollock NI, et al. Mechanism of action of selective inhibitors of IL-6 induced STAT3 pathway in head and neck cancer cell lines. J Chem Biol. 2017;10(3):129–141.
- Chan BA, Hughes BG. Targeted therapy for non-small cell lung cancer: current standards and the promise of the future. Transl Lung Cancer Res. 2015;4(1):36–54.
- Vizoso M, Puig M, Carmona FJ, et al. Aberrant DNA methylation in non-small cell lung cancer-associated fibroblasts. Carcinogenesis. 2015;36(12):1453–1463.
- Koinis F, Kotsakis A, Georgoulias V. Small cell lung cancer (SCLC): no treatment advances in recent years. Transl Lung Cancer Res. 2016;5(1):39–50.
- Rocha-Lima CM, Raez LE. Erlotinib (tarceva) for the treatment of non-small-cell lung cancer and pancreatic cancer. P T. 2009;34(10):554–564.
- Zhang J, Yang PL, Gray NS. Targeting cancer with small molecule kinase inhibitors. Nat Rev. 2009;9(1):28–39.
- Sequist LV, Bell DW, Lynch TJ, et al. Molecular predictors of response to epidermal growth factor receptor antagonists in non-small-cell lung cancer. J Clin Oncol. 2007;25(5):587–595.
- Pan ZY, Jiang ZS, Ouyang HQ. Study of the methylation patterns of the EGFR gene promoter in non-small cell lung cancer. Genet Mol Res. 2015;14(3):9813–9820.
- Brugger W, Thomas M. EGFR-TKI resistant non-small cell lung cancer (NSCLC): new developments and implications for future treatment. Lung Cancer. 2012;77(1):2–8.
- Sen M, Thomas SM, Kim S, et al. First-in-human trial of a STAT3 decoy oligonucleotide in head and neck tumors: implications for cancer therapy. Cancer Discov. 2012;2(8):694–705.
- Timp W, Feinberg AP. Cancer as a dysregulated epigenome allowing cellular growth advantage at the expense of the host. Nat Rev. 2013;13(7):497–510.
- Liu Z, Zhang J, Gao Y, et al. Large-scale characterization of DNA methylation changes in human gastric carcinomas with and without metastasis. Clin Cancer Res off J Am Assoc Cancer Res. 2014;20(17):4598–4612.