
ABSTRACT
Methylation of histone 3 at lysine 79 (H3K79) is one of the principal mechanisms involved in gene expression. The histone methyltransferase DOT1L, which mono-, di- and trimethylates H3K79 using S-adenosyl-L-methionine as a co-factor, is involved in cell development, cell cycle progression, and DNA damage repair. However, changes in normal expression levels of this enzyme are found in prostate, breast, and ovarian cancer. High levels of H3K79me are also detected in acute myeloid leukaemia patients bearing MLL rearrangements (MLL-r). MLL translocations are found in approximately 80% of paediatric patients, leading to poor prognosis. DOT1L is recruited on DNA and induces hyperexpression of HOXA9 and MEIS1. Based on these findings, selective drugs have been developed to induce apoptosis in MLL-r leukaemia cells by specifically inhibiting DOT1L. The most potent DOT1L inhibitor pinometostat has been investigated in Phase I clinical trials for treatment of paediatric and adult patients with MLL-driven leukaemia, showing promising results.
Introduction
Histone modifications are the principal ‘actors’ in chromatin remodelling, opening and closing chromatin to establish an euchromatin or heterochromatin state. This mechanism induces a continuous modulation of gene expression. The principal histone modifications described to date are divided into five groups: methylation, acetylation, ubiquitination, SUMOylation and phosphorylation. These modifications are reversible and are in fact dynamically added or removed by epigenetic enzymes. Writers add a substrate to specific targets, readers read the substrate, and finally, erasers eliminate it [Citation1]. Histone methylation by histone methyltransferases (HMTs) is one of the most studied epigenetic changes [Citation2]. As well as modulating gene expression, this modification is involved in X-chromosome inactivation [Citation3,Citation4], DNA damage repair [Citation5], and in the determination and differentiation of embryonic stem cells [Citation6,Citation7]. However, it is widely reported that dysregulation of HMT expression and activity is frequently observed in cancer [Citation8–Citation11]. Two main histone residues, arginine and lysine groups, are methylated by HMTs. While arginine undergoes mono- and dimethylation by protein arginine methyltransferases (PRMTs) [Citation12,Citation13], lysine is present in all three methylated states [Citation14]. Depending on methylation state and position, protein methyltransferases (PMTs) induce variations in chromatin rearrangement in the histone core, causing either gene inhibition or activation [Citation15]. For example, H3K4me, H3K4me2, H3K4me3, H3K36me3, H3K79me, H3K79me2, H3K9me, and H3K27me are known to be associated with gene transcription. Differential levels of methylation in the same histone position have different effects, with H3K9me2, H3K9me3, H3K27me2, H3K27me3, and H4K20me linked to gene repression [Citation16].
Lysine methyltransferases and lysine demethylases
Histone methylation is a reversible modification. A methyl group is dynamically added by lysine methyltransferases (KMTs), such as enhancer of zeste homolog 2 (EZH2) and disruptor of telomeric silencing 1-like (DOT1L), and removed by lysine demethylases (KDMs).
KMTs are divided into two main groups depending on their catalytic site. The first group includes EZH2, the most studied epigenetic enzyme, which contains the evolutionarily conserved catalytic Su(var)3–9 Enhancer-of-Zeste and Trithorax (SET) domain [Citation17,Citation18]. This enzyme regulates differentiation and modulates mono-, di- and trimethylation of H3K27, a histone mark associated with transcriptional repression. Mutations of Y641, A677, and A687 residues in the catalytic site of the enzyme induce a variation in substrate specificity with an increase in methylation at H3K27. Increased expression levels of EZH2 are associated with tumour development in prostate and breast cancer, as well as in follicular lymphoma [Citation19–Citation21]. EZH2 inhibitors reducing H3K27me3 levels kill mutant lymphoma cells and were found to be effective in a rhabdoid tumour mouse xenograft model [Citation22–Citation24]. The second KMT group is made up of enzymes that do not contain the SET domain. These enzymes have a catalytic site for methylation homologous to DNA methyltransferases (DNMTs) and PRMT1, using S-adenosyl-L-methionine (SAM) as a cofactor. The enzymes catalyse methylation of histone lysines and non-histone proteins using the SAM methyl group, generating S-adenosyl-L-homocysteine (SAH) as a by-product and methylated lysine residue [Citation25]. One of the most studied enzymes in this group is DOT1L (also known as KMT4) [Citation26]. DOT1L and its homologs are involved in numerous processes, including transcriptional regulation, cell cycle progression, and DNA damage repair, and are implicated in several cancers. High levels of DOT1L were observed in prostate [Citation27], breast [Citation28,Citation29], and ovarian cancer [Citation30], and in acute myeloid leukaemia (AML) with mixed-lineage leukaemia (MLL) translocations (MLL-AF9 and MLL-ENL) [Citation31,Citation32]. To date, 94 MLL fusions have been identified in different leukaemia types, the most frequent translocations being MLL-AF9, MLL-ENL, MLL-AF4, MLL-AF10, MLL-ELL, MLL-AF1Q, MLL-CT45A2, MLL-EPS15, MLL-PTD, and MLL-RABGAP1 [Citation33].
KDMs are also divided into two main groups depending on their mechanism of action. The first demethylase enzyme to be discovered was KDM1 (also known as LSD1). This enzyme is a member of the monoamine oxidase family, which catalyzes mono- or di-demethylation of H3K4 and H4K9 through a redox reaction. Specifically, oxidation of flavin adenine dinucleotide by means of an oxygen molecule allows conversion of H3K4me and H3K4me2 into unmethylated H3K4 [Citation34,Citation35]. The second group of KDM enzymes, which contain the Jumonji C (JmjC) domain, has a different mechanism of action. In this reaction, Fe(II) and α-ketoglutarate are used as cofactors and are indispensable for a redox reaction. Fe(II) is oxidized to Fe(III), producing an unstable hydroxy-amine intermediate, which spontaneously develops a demethylated lysine substrate and produces formaldehyde as a by-product of reaction [Citation36]. Unlike KDM1, KDMs with the JmjC domain are able to act on all three methylated states of lysine.
DOT1L mechanism of action
DOT1 was found for the first time in 1998 by Singer M. et al. in Saccharomyces cerevisiae [Citation37]. By genetic screening, the authors determined which enzymes overexpressed in cells induced disruption of telomeric silencing. These studies led to the isolation of several genes, some of which had not yet been identified, including DOT1, DOT4, DOT5, DOT6, and TLC1 [Citation38]. Homolog genes of DOT1 (DOT1-like or DOT1L) showed 88% similarity at amino acid level [Citation39,Citation40] and were subsequently found in mammals [Citation41], Drosophila [Citation42], and protozoa [Citation43]. As previously mentioned, unlike KMTs such as EZH2, DOT1L does not contain a SET domain, but an AdoMet-binding motif similar to PRMT1 and DNMTs, using SAM as a cofactor. Specifically, DOT1L transfers the S-methyl group of SAM to the amino group of lysine, producing a methylated substrate and S-adenosyl-L-homocysteine (SAH; ). Crystallographic studies investigating the structure of DOT1L showed that the SAM binding site, at position 186, is located close to the lysine substrate binding site, while the catalytic domain is in the C-terminal [Citation39,Citation44–Citation46].
Figure 1. DOT1L mechanism of action. DOT1L transfers the S-methyl group of S-adenosyl-L-methionine to the amino group of H3K79, producing a methylated substrate and S-adenosyl-L-homocysteine.
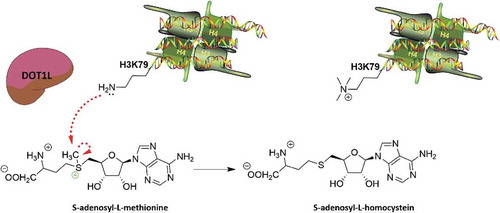
DOT1L is not active on free histones and to date is the only KMT known to mono-, di- and trimethylate H3K79. Although highly conserved and distributed in the eukaryotic genome, this histone target is not found in telomeric loci or in ribosomal DNA. Unlike all amino-terminal tails on which other epigenetic enzymes act, H3K79me is located on the globular domain of H3 [Citation40,Citation47], making H3K79 methylation more influenced by other epigenetic modifications, such as ubiquitination of H2BK120 [Citation48] and acetylation of H4K14 by the histone deacetylase (HDAC) Rpd3 [Citation49], and by silent information regulator (SIR) complex.
DOT1L and ubiquitination
Mono- and dimethylation of H3K79 are directly related to ubiquitination of H2BK120 [Citation50], which is determined by Bre1 and Rad6 (a ubiquitin-conjugating E2 enzyme). Bre1 is a ubiquitin E3 ligase of H2B, which acts on H2BK120 in association with Rad6/UBC2. H2BK120ub is a tag for epigenetic transcriptional activation, elongation by RNA polymerase II, and telomeric silencing [Citation51–Citation53]. Ubiquitination is a fundamental modification for both H3K79 and H3K4me formation. Deletion of these two enzymes blocks methylation of H3K4 and H3K79 [Citation48,Citation54]. Remarkably, mutagenesis of H2BK120 results in the loss of both methylations. These findings indicate that H2BK120 ubiquitination is a prerequisite for H3K4 and H3K79 methylation.
H2BK120 and H3K79 are found on the same nucleosome surface [Citation55]. Several mechanisms were initially proposed to explain the crosstalk between these two modifications, based on hypotheses of either an indirect or direct association between DOT1L and H2BK120ub [Citation56]. Very recently, in vitro experiments confirmed the existence of a direct link [Citation56]. The structure of DOT1L was observed by cryo-electron microscopy, and the enzyme was shown to bind directly H2BK120 on an acid patch of the nucleosome, exploiting the binding of an arginine. In this conformation, ubiquitin interacts with DOT1L inducing a conformational change that makes DOT1L more favourable to H3K79 methylation in normal and leukaemic cells.
DOT1L and SIR complex
In S. cerevisiae, DOT1L was found to compete with Sir3 for the same binding site, creating a dynamic telomeric modulation. Sir1–Sir4 are proteins responsible for transcriptional repression, modulating chromatin structure at silent mating-type loci (HML and HMR) [Citation57–Citation59]. Methylation of H3K79 by DOT1L prevents binding of Sir3, while binding of Sir3 to the unmethylated DNA region prevents association of DOT1L to acetylated H4 and therefore H3K79 methylation, inducing telomeric silencing [Citation60]. Both ubiquitination of H2BK120 and acetylation of H4K14 are thus indispensable for DOT1L activity.
DOT1L and Rpd3
In thymic lymphoma, crosstalk occurs between histone deacetylation and H3K79 methylation, specifically between DOT1L and the HDAC Rpd3, which is associated with transcriptional repression. Increased levels of Rpd3 cause a decrease in methylation levels of H3K79 induced by DOT1L. Rpd3 decreases acetyl levels, inducing transcriptional repression and therefore absence of ubiquitination in H2BK120, fundamental for DOT1L activity. These findings clearly demonstrate the opposite correlation between deacetylation and DOT1L methylation. Indeed, in thymic lymphoma cells with HDAC1 depletion, DOT1L inhibitors induce apoptosis [Citation49]. DOT1L inhibitors are therefore promising candidates for treatments of this cancer subtype.
Role of DOT1L in normal cells
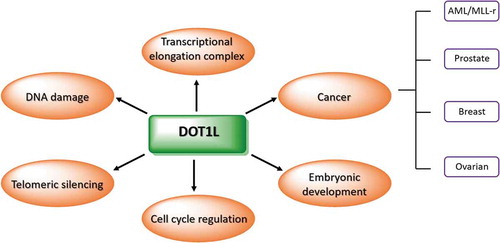
DOT1L is one of the most studied PMT enzymes due to its involvement in normal cellular processes and in tumour development. Through methylation of its target, DOT1L is responsible for telomeric silencing, cell cycle regulation, and DNA repair ().
Figure 2. DOT1L involvement in normal and tumour cells.
Telomeric silencing
As previously mentioned, DOT1L was identified for the first time in 1998 by telomeric screening performed to identify all enzymes whose overexpression determined a block of telomeric silencing [Citation37]. Regulation of telomeric silencing is mediated by SIR protein complex, responsible for H4K14 deacetylation. In S. cerevisiae, telomeres recruit the HDAC Sir2 and the adapter proteins Sir3 and Sir4 from the SIR complex [Citation61,Citation62]. This mechanism promotes a closed chromatin configuration, limiting access to RNA polymerase II and silencing genes in sub-telomeric regions. DOT1L methylation acts on telomeric sequences against H4K14-Sir3 deacetylation, creating a dynamic balance between gene activation and repression [Citation63,Citation64].
The silencing effect on telomeric sequences of DOT1L and POL30, a protein involved in eukaryotic DNA replication, was demonstrated in 2011 using URA3 reporter assay with 5-fluoroorotic acid [Citation65]. Defects in DOT1L expression induced an alteration in ribonucleotide reductase and a promoter of URA3 at telomere VII-L [Citation65,Citation66]. In addition, in normal cells, the role of DOT1L in chromatin formation was only found at telomeric level [Citation66]. Nevertheless, it is clear that methylation of H3K79 is responsible for recruitment of SIR proteins and euchromatin formation.
Cell cycle regulation
Various enzymes such as cyclins and cyclin-dependent kinases are responsible for regulating cell cycle and inducing a block of proliferation. However, in recent years the importance of the methylation status of H2K79 in various phases of cell cycle progression has emerged. Specifically, H3K79 methylation is involved in progression of G1 phase, S phase, mitosis, and meiosis. It is known that different methylation states are present in different phases of the cell cycle, and above all that methylation levels change in tumour compared to normal cells. In normal cells, high levels of H3K79me2 are detected in mitosis phase, with lower levels in G1 phase increasing only in G2/M. H3K79me3 levels are the same in all phases except for M, in which trimethylation is not present. In cancer cells, methylation levels are substantially different. For example, in A549 lung cancer and HeLa cervical cancer cells, overall methylation levels in G1/S transition are high, suggesting that inhibition of DOT1L in tumour cells may lead to an arrest in G1 phase [Citation43,Citation67].
DNA damage repair
Rupture of the double helix of DNA and its subsequent repair is one of the fundamental mechanisms of normal cell growth. All mechanisms used to repair DNA damage prevent the onset of mutations and the formation of tumour cells. When mechanisms are not sufficient for repair, the cell undergoes apoptosis. Major factors responsible for DNA damage include intrinsic factors such as the presence of free oxygen radicals and ultraviolet (UV) radiation. The two main mechanisms that generate breaks in DNA are:
single strand breakage. In this case, the second filament is used as a mould to repair the damage.
rupture of the double helix, also called double-strand break, which is much more complex to repair [Citation5]. In eukaryotes, repair of the double stand requires chromatin remodelling by post-translational modifications [Citation68].
To repair damage induced by radiation, histone modifications recruit tumour suppressor p53 binding protein 1 (53BP1) to sites of DNA damage. 53BP1 is then hyperphosphorylated and produces nuclear foci colocalizing with phosphorylated H2AX (gamma-H2AX) [Citation69]. Methylation of H3K79 or H4K4 is fundamental histone modifications for recruitment of 53BP1 and CAMP response element-binding protein 2 (CREB2, also known as ATF2) on DNA sites. Specifically, an abundance of H3K79 methylation by DOT1L is observed during the repair process, and DOT1L deletion reduces DNA repair. DOT1L and CREB2 therefore play an essential role in DNA damage repair in late G2 phase [Citation70,Citation71].
DNA repair by loci formation of 53BP1 is also promoted by HLA-B-associated transcript 3 (Bat3). Bat3 is involved in dimethylation of H3K4, inducing transcriptional gene regulation and DNA damage-induced apoptosis. Like DOT1L, Bat3 also has a role in embryonic development and cell division [Citation72]. Colocalization of Bat3 and DOT1L was shown to be indispensable for DNA repair. The absence of Bat3 decreases DOT1L interaction with H3 and consequently methylation of K79.
Another mechanism involved in DNA damage repair is DOT1L methylation and H2AX phosphorylation at serine 139. Depletion or inhibition of DOT1L reduces repair of DNA damage with decreased H2AX phosphorylation at serine 139, usually associated with PI3K family members, early markers of DNA damage. In addition, DOT1L inhibition does not increase levels of the phosphorylated protein KAP1, which is linked to DNA repair in heterochromatin regions, supporting the hypothesis that DOT1L induces DNA repair in these regions [Citation73].
Lastly, DOT1L was shown to have a protective effect against UV radiation due to the ability of DNA to protect itself. This mechanism was triggered by methylation of H3K79 and recruitment of xeroderma pigmentosum, complementation group C, a component of the nucleotide excision repair pathway. A decrease in H3K79 methylation levels led to greater sensitivity to UV radiation [Citation74].
DOT1L as a target in MLL-r leukaemia
Leukaemia is characterized by a neoplastic proliferation of haematopoietic stem cells, which induce the growth of a high number of abnormal white blood cells. Different types of leukaemia are grouped according to the speed of development – acute (fast growth) and chronic (slow growth) – and whether the disease starts from myeloid or lymphoid cells. Based on this classification, leukaemia is divided into four main groups: AML, chronic myeloid leukaemia (CML), acute lymphoblastic leukaemia (ALL), and chronic lymphocytic leukaemia [Citation75,Citation76].
AML and CML are cancers of the myeloid cells, which normally develop into mature blood cells such as red blood cells, white blood cells, and platelets. AML is characterized by an increase in the number of myeloid cells in the bone marrow and an arrest in their maturation, frequently resulting in haematopoietic insufficiency (granulocytopenia, thrombocytopenia, or anaemia) with or without leukocytosis [Citation77]. Instead, ALL arises from the malignant transformation and proliferation of lymphoid progenitor cells in the bone marrow and blood.
The American Cancer Society reported that 6,590 new cases of leukaemia were diagnosed in 2016, with over 1,400 deaths, and 80% of ALL occur in children (SEER Cancer Statistics Review [CSR] 1975-–2016). Despite early diagnosis and numerous chemotherapy regimens available, only 30–40% of adult patients with ALL currently achieve long-term remission [Citation78].
In patients diagnosed during infancy and before the ages of 14, the percentage of survival is even lower [Citation79].
ALL is characterized by genetic alterations that induce mutation in the differentiation and proliferation of lymphocytes. In 75% of cases, the main mutation comes from a change in B cells and the remaining percentage from T cells. As in AML, ALL is also caused by the chromosomal translocations [ETV6-RUNX1], [TCF3-PBXI], [BCR-ABL], and MLL rearrangements (MLL-r). The MLL chromosomal translocation (11q23) is found in adult AML and ALL (5–10% [Citation80] and 5% [Citation81], respectively). Incidence is higher in paediatric patients (70% for AML [Citation82] and 35–50% for ALL [Citation83]), determining an aggressive clinical course with intermediate prognosis [Citation83]. The greater percentage of MLL translocation in paediatric patients (<2 years of age) compared to adults is caused by low latency and prenatal origin [Citation84]. However, age and cytogenic effects are independent prognostic parameters [Citation85].
MLL translocation
The MLL1 gene, today named histone-lysine N-methyltransferase 2A (KMT2A), encodes for the HMT MLL1 protein, a positive regulator of gene transcription. Located at chromosome 11q23, MLL1 is often found rearranged with different genes [Citation86,Citation87]. These gene fusions induce the formation of chimeric proteins characterized by interaction of the amino-terminal portion of MLL with the C-terminal portion of more than 90 different proteins. Ten of these proteins, -AF9, -ENL, -AF4, -AF10, -ELL, -AF1Q, -CT45A2, -EPS15, -PTD, and -RABGAP1, were shown to be involved in tumorigenesis [Citation33,Citation88,Citation89]. Some of these proteins are nuclear proteins that, by interacting with pTEFb complex and phosphorylating Pol II, regulate transcription and recruit DOT1L [Citation90,Citation91].
ALF family proteins and ENL are the most common protein partners of MLL. The AF4 gene (ALL1-fused gene from chromosome 4), t(4,11)(q21,q23) translocation, found predominantly in ALLs [Citation86,Citation92], is responsible for infant ALL with MLL1 rearrangement in 50% of cases in children and 75% in adults [Citation93]. t(9;11)(p22;q23) translocation is instead principally associated with AML, resulting in chimeric MLL-AF9 protein [Citation94]. Like AF4, AF9 (ALL1-fused gene from chromosome 9), which is associated with myeloid leukaemia, and ENL, associated with lymphoid and myeloid leukaemia [Citation93]. AF9 and ENL belong to the family of YEATS domain proteins, identified as a super elongation complex comprising AF4, ENL, pTEFb, and DotCom complex. These proteins exhibit homology with the hydrophobic C-terminal ANC1 homology domain, responsible for recruitment of numerous proteins. AF9 and ENL interact with AF4, which contains complexes for pTEFb and DOT1L binding. In AML, this protein cooperation (MLL-AF10, MLL-AF4, MLL-AF9, and MLL-ENL) binds DNA and recruits DOT1L to promoters of MLL target genes. DOT1L recruitment leads to an increase in levels of H3K79 methylation in promoter genes and subsequent hyperexpression of proteins, such as HOXA9 and its cofactor MEIS1 [Citation95–Citation98]. HOXA is the most overexpressed gene cluster (HOXA7, HOXA9, and HOXA10) in MLL-r leukaemia, and is responsible for segmentation and haematopoiesis [Citation80]. MEIS1 is necessary for leukaemia growth and is associated with oncogenic factors [Citation99]. Dysregulated expression of HOXA and MEIS1 is therefore a critical event in leukaemia development, contributing to growth and survival factors that promote their oncogenic potential.
Preferentially, DOT1L directly interacts with ENL [Citation100], AF10 (also known as MLLT10), and AF17 (also known as MLLT6). The interaction is mediated by an octapeptide motif and a leucine zipper region of AF10 required for leukaemic transformation by MLL-AF10. This association increases recruitment of DOT1L on DNA and therefore HMT activity. These complexes have different functions in leukaemogenesis. MLL-AF10 transforms haematopoietic progenitors via the DOT1L interaction domain, while MLL-ENL/AF9 does so through the ANC1 homology domain, which is responsible for its association with both AF4 and DOT1L [Citation101].
DOT1L and epigenetic therapy in MLL-r Patients
Most chimeric proteins involved in MLL-r are reported as being responsible for DOT1L recruitment [Citation102]. This mechanism induces an increase in the expression of genes involved in leukaemogenesis. DOT1L is therefore one of the most effective pharmacological targets used to treat AML harbouring an MLL translocation.
Preclinical studies in a mouse xenograft model showed that the use of DOT1L inhibitors, such as EPZ004777 and EPZ5676 (also known as pinometostat), reduces HOXA9 gene expression with subsequent differentiation and death of AML cells [Citation103,Citation104]. Pinometostat is the most potent DOT1L inhibitor and was investigated in Phase I clinical trials in patients with MLL-r leukaemia [Citation105]. Pinometostat shows good anticancer activity in vitro and in mice models, reducing leukaemia cell growth [Citation104]. However, in adult [Citation106] and paediatric patients [Citation107], pinometostat administration as monotherapy is not effective while combination with other drugs results active in both younger and older patients.
In the following subsections, we will provide an overview of known DOT1L inhibitors and their mechanisms of action. Given the close correlation between DOT1L activity and some epigenetic modifications, we will also discuss the possibility of using combination therapy to improve the antileukemic effect of these inhibitors.
DOT1L inhibitors
DOT1L is a KMT with an AdoMet binding motif that catalyzes H3K79 methylation using SAM as a cofactor. Based on this mechanism of action, efforts of the scientific community have focused on synthetizing new molecules able to occupy the enzymatic pocket of SAM, displacing it from its binding site and thus inhibiting the methyltransferase mechanism of DOT1L. Most DOT1L inhibitors are therefore mimetics of the SAM molecule. Some of these compounds (EPZ004777 [3], EPZ5676 [5], and SGC0946 [4]; ) down-regulate MLL fusion target genes, such as HOXA9 and MEIS1, inhibiting proliferation of MLL-r leukaemia cells. However, DOT1L is also involved in maintenance of adult haematopoiesis [Citation108,Citation109]. Therefore, although these molecules displayed high potency against leukaemia, total DOT1L inhibition induced numerous side effects. For example, in mice treated for 14 days with EPZ004777, an increase in white blood cells, with high levels of neutrophils, monocytes and lymphocytes, was observed [Citation103].
Figure 3. Structure of S-adenosyl-L-methionine (SAM) and DOT1L inhibitors.
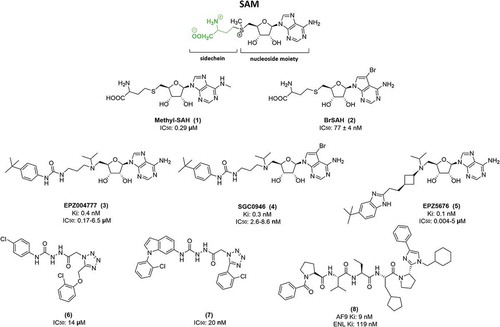
In recent years, new compounds with different mechanisms of action have been developed. Unlike the first class of DOT1L inhibitors, mimetic peptide molecules such as compound (8), shown in , do not determine total inhibition of DOT1L activity, but bind AF9/ENL, thus blocking recruitment of DOT1L to MLL target genes and reducing levels of HOAX9 [Citation110].
Based on SAH structure, several compounds were synthetized in which more hydrophobic groups were added. Specific inhibitors of DOT1L, direct SAH analogs, were identified by alkylating the primary amine at the adenine ring of SAH (methyl-SAH [1]) and through substitution at the homocysteine [Citation111]. Another molecule was synthetized with a similar SAH structure but with bromide on the adenosine ring (BrSAH [3]), which dramatically decreased DOT1L activity and selectivity compared to other KMTs [Citation112]. A high-throughput screening approach identified a new inhibitor (6) with an IC50 of 14 μM [Citation113], and from this structure a new compound (7) with nM activity (IC50 = 20 nM) was developed (). However, all these molecules were very hydrophilic and did not display any cellular activity. The SAM competitor EPZ004777, belonging to the first class of DOT1L inhibitors, was the first molecule able to strongly inhibit DOT1L, displaying antiproliferative activity in MLL-r leukaemic cells both in vitro and in vivo. Characterized for the first time in 2011 [Citation103], this compound has an IC50 of 0.4 nM with very high DOT1L selectivity compared to other PMTs. In vitro studies showed that the compound inhibits PRMT5 with IC50 >500 nM and other PMTs with IC50 >50 µM. EPZ004777 decreases expression of HOXA9 and MES1 by acting on MLL-AF9/AF10/AF6 complexes. This compound also exerts an apoptotic effect not only in MLL-r, as in RS4;11 (MLL-AF4; IC50 = 6.47 µM), SEM (MLL-AF4; IC50 = 1.72 µM), MV4-11 (MLL-AF4; IC50 = 0.17 µM), MOLM-13 (MLL-AF9; IC50 = 0.72 µM), but also non-MLL-r leukaemic cell lines, inducing cell death in Kasumi-1 cells (IC50 = 32.99 µM) [Citation103]. In in vivo studies, EPZ004777 dramatically reduced cancer cell proliferation, with an apoptotic effect, activating the caspase cascade, and led to an increase in the percentage of survival of MLL-r leukaemia xenograft mice. Experimental procedures performed for 12 days also demonstrated the ability of EPZ004777 to induce cell differentiation before the apoptotic event [Citation103]. Further, these in vivo studies showed that EPZ004777 is well tolerated at 50, 100 and 150 mg/ml concentrations. However, this compound is a mimetic SAM inhibitor and, as previously mentioned, completely blocks the activity of DOT1L, inducing some side effects [Citation103].
Despite its strong inhibitory activity in vitro (400 pM) and moderate activity in cellula (<3 µM), EPZ004777 has very low pharmacological properties, which precluded the use of normal oral administration and prevented its adoption in therapy.
EPZ004777 requires subcutaneous administration with a continuous infusion pump for 7 days at 50 mg/ml. The low half-life of the drug, resulting from the introduction of a ribose group [Citation114], leads to the lowering of plasma levels necessary for a good pharmacological effect; However, the continuous administration of the drug for 14 days to maintain high plasma levels causes a significant increase in neutrophils, monocytes, and lymphocytes [Citation103]. Mice treated with EPZ004777 show a significant increase in survival, but with a modest duration.
In a cell-free assay, SGC0946, an analog of EPZ004777 with a bromide on the adenosine ring, showed an IC50 of 0.3 nM, and an IC50 of 2.6 nM and 8.8 nM in A431 and MCF10a cells, respectively [Citation115]. SGC0946 results more active than its precursor.
In 2013, Daigle S.R. et al. characterized the molecule EPZ5676 [(2R,3R,4S,5R)-2-(6-amino-9H-purin-9-yl)-5-((((1r,3S)-3-(2-(5-(tert-butyl)-1H-benzo[d]imidazol-2-yl)ethyl)cyclobutyl)(isopropyl) amino)methyl)tetrahydrofuran-3,4-diol], a derivative of EPZ004777. This compound, also known as pinometostat, showed a Ki ≤80 pM with 37,000-fold selectivity compared to other PMTs. Crystallographic studies demonstrated that the compound occupies the SAM site on DOT1L, inducing a conformational change by opening the hydrophobic pocket [Citation104]. This change is able to increase affinity between DOT1L and the molecule. In in vitro and in vivo preclinical studies, the compound inhibited H3K79 methylation and HOXA9 and MEIS1 MLL fusion target gene expression. Pinometostat selectively induces cell death in cell lines with MLL-AF9, MLL-AF4, and MLL-ENL fusions. Its substrate specificity and high potency led to the drug being tested in animal models.
Although pinometostat strongly inhibits DOT1L in enzymatic assays (Ki ≤80 pM) and in cells with nanomolar concentration activity, the molecule has low oral and high intraperitoneal bioavailability [Citation116], necessitating intraperitoneal administration.
Pinometostat also has some pharmacokinetic limitations. The half-life of the drug ranges from one to 5 hours (with a greater t ½ found in rats compared to mice and dogs), and administering a single dose at 20 mg/ml reach the plasma concentration equal to 5 µM, which, however decreases after 2 hours, reaching concentrations lower than the dose necessary to block tumour proliferation. In addition, intermittent administration of the drug for eight consecutive hours was less effective than continuous administration, again due to the decrease in plasma drug levels.
It was also shown that the effects are visible only after more than 7 days of treatment. Mice subjected to administration for only 7 days showed no significant improvement as a result of therapy. The administration protocol used in xenograft models therefore provides for continuous intraperitoneal administration by osmotic pump, from 21 to 28 days at a concentration of 70 mg/ml [Citation104].
However, administration of the compound in MV4-11 xenografts for 21 days induced tumour regression and was tolerated up to 70 mg/kg/day. In addition, little or no tumour regrowth was observed for over 30 days beyond cessation of treatment with the compound. In some cases, resistance developed after 14 days of treatment. AML resistance is usually caused by N676, F691, D839, and Y843 mutations in the FMS-like tyrosine kinase 3 site [Citation117,Citation118]. However, resistance to pinometostat occurs in both MLL-ENL and MLL-AF9 due to an increase in efflux of the drug by activation of PI3K/AKT and RAS/RAF/MEK/ERK pathways, without any mutation in DOT1L. ABCB1 and ABCG2, proteins belonging to ABC-superfamily xenobiotic transporters, were reported to induce drug resistance in MLL-ENL cells. Treatment with ABCB1 inhibitors restores pinometostat sensitivity [Citation119].
Pinometostat was also investigated in Phase I clinical trials against relapsed/refractory acute MLL-r in children (NCT02141828) and adults (NCT01684150). The final report, from 2016, of the Phase I study in children shows that continuous intravenous administration has an acceptable safety profile up to 70 mg/m2/day for 23 days of treatment [Citation120]. In adults, the safe dose is 90 mg/m2/day after 21 or 28 days of treatment, with >15% of patients experiencing mild adverse effects, such as nausea, constipation, vomiting, diarrhoea, hypocalcaemia, hypokalemia, hypomagnesaemia, leukocytosis, and anaemia [Citation107]. However, a clinical trial performed in 2017 (NCT01684150) showed that out of 51 patients treated with pinometostat, only two achieved total remission at 25 mg/ml of drug administration [Citation106].
These data suggest that the effect of pinometostat in mice and humans is completely different. In the clinic, the drug is not sufficient when administered as monotherapy to exert a strong therapeutic effect. Combination with other chemotherapeutic agents such as Ara-C or azacitidine has shown an improvement in cell killing in vitro and better tolerability in vivo. Two different clinical trials are now recruiting to evaluate the combination of pinometostat with cytarabine, daunorubicin, daunorubicin hydrochloride, and azacitidine (NCT03724084; NCT03701295).
Cooperation between DOT1L and epigenetic enzymes in MLL-r treatment
Due to its location on the globular domain of H3 [Citation40,Citation47], H3K79me modification is tightly associated with other epigenetic modifications, such as H2BK120 ubiquitination [Citation48], Rpd3 acetylation [Citation49], and with SIR complex (). DOT1L activity is also closely linked to the expression and activity of epigenetic enzymes, such as BRD4 and DNMT3A. These mechanisms suggested the use of DOT1L inhibitors in combination with small molecules selective for a different target in order to increase their anticancer activity in MLL-r leukaemia [Citation121].
Figure 4. Schematic representation showing histone colocalization of DOT1L and other epigenetic enzymes. DOT1L methylation is affected by several histone modifications and DNA methylation.
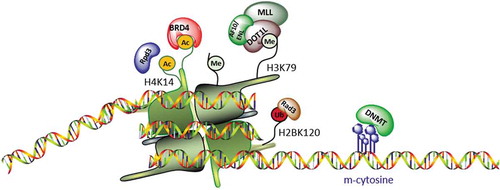
Although part of two distinct complexes, BRD4 and DOT1L cooperate in MLL-r leukaemia. DOT1L acts by methylating H3K79 and recruits p300, which regulates the binding of BRD4 to chromatin. Specifically, the increase in H3K79me2 levels leads to chromatin opening by inhibiting localization of the transcriptional repressors SIRT1 and SUV39H1, and facilitating binding of CREB1 with p300. The increase in p300 on chromatin increases acetylation levels and therefore localization of BRD4. Co-administration of the BRD4 inhibitor I-BET and SGC0946 had a synergistic effect in MLL-r mouse and human leukaemia at lower doses than those used in monotherapy. However, further studies showed that the dual inhibition of these enzymes also decreases proliferation in other leukaemia types [Citation122].
The DNMT3A mutation occurs in 20% of adult AML patients, determining poor prognosis, and DOT1L and DNMT3A are associated with MLL-r leukaemia. In haematopoietic cell systems in which DNMT3A was deleted, an increase in H3K79 methylation was observed. Specifically, the decrease in DNMT3A led to an increase in DNA methylation, including in regions responsible for transcription of genes deregulated in leukaemia. These findings suggest the use of DOT1L inhibitors in the treatment of AML patients with mutated DNMT3A [Citation121].
Several studies were also performed to evaluate the use of pinometostat in combination with standard-of-care agents in acute leukaemias. MOLM-13 (MLL-AF9), MVA-11 (MLL-AF4), and non-MLL-r cell lines were challenged with EPZ5676 in combination with cytarabine and daunorubicin, standard-of-care agents used in the clinic for MLL-r treatment, and showed a high percentage of cell death. These findings suggest the ability of these compounds to block MLL-r leukaemia growth both alone and above all in combination with other chemotherapeutics [Citation123].
Conclusions
MLL rearrangements in acute leukaemias induce complex biological changes that are still not fully explained. The MLL-r translocation, associated with the most aggressive leukaemic subtypes, is present in nearly 80% of paediatric leukaemias, leading to poor prognosis. Many clinical trials in Europe, America, and Japan are investigating chemotherapeutic agents for the treatment of MLL-r leukaemia. However, relapse rates are high and effective treatment times are very long. In recent years, several research groups have focused on studying the mechanisms responsible for leukaemogenesis, highlighting the involvement of the epigenetic target DOT1L. Epigenetic modifications are fundamental processes in chromatin remodelling, and HMTs play a key role in gene activation and silencing. The HMT DOT1L, known to be involved in cell development, cell cycle progression, and DNA damage repair, methylates H3K79. Studies show that an increase in levels of H3K79me is a major cause of leukaemogenesis in AML patients bearing MLL-r. MLL-AF10 and MLL-ENL recruit DOT1L on DNA, inducing hypermethylation of H3K79 in MLL-r target genes such as HOXA and MEIS1.
To overcome the drawbacks of conventional chemotherapeutic drugs, novel compounds have recently been developed to inhibit DOT1L and thus counteract leukaemic growth. Pinometostat is currently being investigated in Phase I clinical trials for paediatric and adult patients with acute leukaemia harbouring MLL-r and appears to yield good results in inhibiting MLL-r leukaemia progression with good tolerability. Other clinical trials are starting to evaluate the tolerability and percentage of remission in co-administration of pinometostat with daunorubicin hydrochloride and cytarabine (NCT03724084), and with 5-azacitidine (NCT03701295).
Acknowledgments
We thank C. Fisher for linguistic editing.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Biswas S, Rao CM. Epigenetic tools (The writers, the readers and the erasers) and their implications in cancer therapy. Eur J Pharmacol. 2018 Oct;15(837):8–24.
- Trievel RC. Structure and function of histone methyltransferases. Crit Rev Eukaryot Gene Expr. 2004;14(3):147–169.
- Greciano PG, Goday C. Methylation of histone H3 at Lys4 differs between paternal and maternal chromosomes in Sciara ocellaris germline development. J Cell Sci. 2006 Nov 15;119(Pt 22):4667–4677.
- Brinkman AB, Roelofsen T, Pennings SW, et al. Histone modification patterns associated with the human X chromosome. EMBO Rep. 2006 Jun;7(6):628–634.
- Kari V, Raul SK, Henck JM, et al. The histone methyltransferase DOT1L is required for proper DNA damage response, DNA repair, and modulates chemotherapy responsiveness. Clin Epigenetics. 2019 Jan 7;11(1):4.
- Eom GH, Kim KB, Kim JH, et al. Histone methyltransferase SETD3 regulates muscle differentiation. J Biol Chem. 2011 Oct 7;286(40):34733–34742.
- Chen X, Cao G, Wu J, et al. The histone methyltransferase EZH2 primes the early differentiation of follicular helper T cells during acute viral infection. Cell Mol Immunol. 2019. doi:10.1038/s41423-019-0219-z.
- Chen Y, Liu X, Li Y, et al. Lung cancer therapy targeting histone methylation: opportunities and challenges. Comput Struct Biotechnol J. 2018;16:211–223.
- Tran TQ, Lowman XH, Kong M. Molecular pathways: metabolic control of histone methylation and gene expression in cancer. Clin Cancer Res. 2017 Aug 1;23(15):4004–4009.
- Wilson S, Fan L, Sahgal N, et al. The histone demethylase KDM3A regulates the transcriptional program of the androgen receptor in prostate cancer cells. Oncotarget. 2017 May 2;8(18):30328–30343.
- Li T, Chen H, Li W, et al. Promoter histone H3K27 methylation in the control of IGF2 imprinting in human tumor cell lines. Hum Mol Genet. 2014 Jan 1;23(1):117–128.
- Shen NY, Ng SY, Toepp SL, et al. Protein arginine methyltransferase expression and activity during myogenesis. Biosci Rep. 2018 Feb 28;38(1):1–17.
- Wei H, Mundade R, Lange KC, et al. Protein arginine methylation of non-histone proteins and its role in diseases. Cell Cycle. 2014;13(1):32–41.
- Black JC, Van Rechem C, Whetstine JR. Histone lysine methylation dynamics: establishment, regulation, and biological impact. Mol Cell. 2012 Nov 30;48(4):491–507.
- Rea S, Eisenhaber F, O’Carroll D, et al. Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature. 2000 Aug 10;406(6796):593–599.
- Shilatifard A. Molecular implementation and physiological roles for histone H3 lysine 4 (H3K4) methylation. Curr Opin Cell Biol. 2008 Jun;20(3):341–348.
- Dillon SC, Zhang X, Trievel RC, et al. The SET-domain protein superfamily: protein lysine methyltransferases. Genome Biol. 2005;6(8):227.
- Wu H, Zeng H, Dong A, et al. Structure of the catalytic domain of EZH2 reveals conformational plasticity in cofactor and substrate binding sites and explains oncogenic mutations. PLoS One. 2013;8(12):e83737.
- Kleer CG, Cao Q, Varambally S, et al. EZH2 is a marker of aggressive breast cancer and promotes neoplastic transformation of breast epithelial cells. Proc Natl Acad Sci U S A. 2003 Sep 30;100(20):11606–11611.
- Takawa M, Masuda K, Kunizaki M, et al. Validation of the histone methyltransferase EZH2 as a therapeutic target for various types of human cancer and as a prognostic marker. Cancer Sci. 2011 Jul;102(7):1298–1305.
- Varambally S, Cao Q, Mani RS, et al. Genomic loss of microRNA-101 leads to overexpression of histone methyltransferase EZH2 in cancer. Science. 2008 Dec 12;322(5908):1695–1699.
- Knutson SK, Wigle TJ, Warholic NM, et al. A selective inhibitor of EZH2 blocks H3K27 methylation and kills mutant lymphoma cells. Nat Chem Biol. 2012 Nov;8(11):890–896.
- Konze KD, Ma A, Li F, et al. An orally bioavailable chemical probe of the lysine methyltransferases EZH2 and EZH1. ACS Chem Biol. 2013;8(6):1324–1334.
- Qi W, Chan H, Teng L, et al. Selective inhibition of Ezh2 by a small molecule inhibitor blocks tumor cells proliferation. Proc Natl Acad Sci U S A. 2012 Dec 26;109(52):21360–21365.
- Struck AW, Thompson ML, Wong LS, et al. S-adenosyl-methionine-dependent methyltransferases: highly versatile enzymes in biocatalysis, biosynthesis and other biotechnological applications. Chembiochem. 2012 Dec 21;13(18):2642–2655.
- Steger DJ, Lefterova MI, Ying L, et al. DOT1L/KMT4 recruitment and H3K79 methylation are ubiquitously coupled with gene transcription in mammalian cells. Mol Cell Biol. 2008 Apr;28(8):2825–2839.
- Annala M, Kivinummi K, Leinonen K, et al. DOT1L-HES6 fusion drives androgen independent growth in prostate cancer. EMBO Mol Med. 2014 Sep;6(9):1121–1123.
- Nassa G, Salvati A, Tarallo R, et al. Inhibition of histone methyltransferase DOT1L silences ERalpha gene and blocks proliferation of antiestrogen-resistant breast cancer cells. Sci Adv. 2019 Feb;5(2):eaav5590.
- Lee JY, Kong G. DOT1L: a new therapeutic target for aggressive breast cancer. Oncotarget. 2015 Oct 13;6(31):30451–30452.
- Wang X, Wang H, Xu B, et al. Depletion of H3K79 methyltransferase Dot1L promotes cell invasion and cancer stem-like cell property in ovarian cancer. Am J Transl Res. 2019;11(2):1145–1153.
- Okuda H, Stanojevic B, Kanai A, et al. Cooperative gene activation by AF4 and DOT1L drives MLL-rearranged leukemia. J Clin Invest. 2017 May 1;127(5):1918–1931.
- Liu W, Deng L, Song Y, et al. DOT1L inhibition sensitizes MLL-rearranged AML to chemotherapy. PLoS One. 2014;9(5):e98270.
- Meyer C, Burmeister T, Groger D, et al. The MLL recombinome of acute leukemias in 2017. Leukemia. 2018 Feb;32(2):273–284.
- Kong X, Ouyang S, Liang Z, et al. Catalytic mechanism investigation of lysine-specific demethylase 1 (LSD1): a computational study. PLoS One. 2011;6(9):e25444.
- Hayward D, Cole PA. LSD1 histone demethylase assays and inhibition. Methods Enzymol. 2016;573:261–278.
- Accari SL, Fisher PR. Emerging roles of JmjC domain-containing proteins. Int Rev Cell Mol Biol. 2015;319:165–220.
- Singer MS, Kahana A, Wolf AJ, et al. Identification of high-copy disruptors of telomeric silencing in Saccharomyces cerevisiae. Genetics. 1998 Oct;150(2):613–632.
- Chen S, Li L, Chen Y, et al. Identification of novel disruptor of telomeric silencing 1-like (DOT1L) inhibitors through structure-based virtual screening and biological assays. J Chem Inf Model. 2016 Mar 28;56(3):527–534.
- Min J, Feng Q, Li Z, et al. Structure of the catalytic domain of human DOT1L, a non-SET domain nucleosomal histone methyltransferase. Cell. 2003 Mar 7;112(5):711–723.
- Feng Q, Wang H, Ng HH, et al. Methylation of H3-lysine 79 is mediated by a new family of HMTases without a SET domain. Curr Biol. 2002 Jun 25;12(12):1052–1058.
- Schubeler D, MacAlpine DM, Scalzo D, et al. The histone modification pattern of active genes revealed through genome-wide chromatin analysis of a higher eukaryote. Genes Dev. 2004 Jun 1;18(11):1263–1271.
- List O, Togawa T, Tsuda M, et al. Overexpression of grappa encoding a histone methyltransferase enhances stress resistance in Drosophila. Hereditas. 2009 Feb;146(1):19–28.
- Janzen CJ, Hake SB, Lowell JE, et al. Selective di- or trimethylation of histone H3 lysine 76 by two DOT1 homologs is important for cell cycle regulation in Trypanosoma brucei. Mol Cell. 2006 Aug;23(4):497–507.
- Frederiks F, Tzouros M, Oudgenoeg G, et al. Nonprocessive methylation by Dot1 leads to functional redundancy of histone H3K79 methylation states. Nat Struct Mol Biol. 2008 Jun;15(6):550–557.
- van Leeuwen F, Gafken PR, Gottschling DE. Dot1p modulates silencing in yeast by methylation of the nucleosome core. Cell. 2002 Jun 14;109(6):745–756.
- Jones B, Su H, Bhat A, et al. The histone H3K79 methyltransferase Dot1L is essential for mammalian development and heterochromatin structure. PLoS Genet. 2008 Sep 12;4(9):e1000190.
- Lacoste N, Utley RT, Hunter JM, et al. Disruptor of telomeric silencing-1 is a chromatin-specific histone H3 methyltransferase. J Biol Chem. 2002 Aug 23;277(34):30421–30424.
- Ng HH, Xu RM, Zhang Y, et al. Ubiquitination of histone H2B by Rad6 is required for efficient Dot1-mediated methylation of histone H3 lysine 79. J Biol Chem. 2002 Sep 20;277(38):34655–34657.
- Vlaming H, McLean CM, Korthout T, et al. Conserved crosstalk between histone deacetylation and H3K79 methylation generates DOT1L-dose dependency in HDAC1-deficient thymic lymphoma. Embo J. 2019 Jun 17;38:1–14.
- Shahbazian MD, Zhang K, Grunstein M. Histone H2B ubiquitylation controls processive methylation but not monomethylation by Dot1 and Set1. Mol Cell. 2005 Jul 22;19(2):271–277.
- Hwang WW, Venkatasubrahmanyam S, Ianculescu AG, et al. A conserved RING finger protein required for histone H2B monoubiquitination and cell size control. Mol Cell. 2003 Jan;11(1):261–266.
- Wood A, Krogan NJ, Dover J, et al. Bre1, an E3 ubiquitin ligase required for recruitment and substrate selection of Rad6 at a promoter. Mol Cell. 2003 Jan;11(1):267–274.
- Xiao T, Kao CF, Krogan NJ, et al. Histone H2B ubiquitylation is associated with elongating RNA polymerase II. Mol Cell Biol. 2005 Jan;25(2):637–651.
- Sun ZW, Allis CD. Ubiquitination of histone H2B regulates H3 methylation and gene silencing in yeast. Nature. 2002 Jul 4;418(6893):104–108.
- Luger K, Mader AW, Richmond RK, et al. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature. 1997 Sep 18;389(6648):251–260.
- McGinty RK, Kim J, Chatterjee C, et al. Chemically ubiquitylated histone H2B stimulates hDot1L-mediated intranucleosomal methylation. Nature. 2008 Jun 5;453(7196):812–816.
- Manning BJ, Peterson CL. Direct interactions promote eviction of the Sir3 heterochromatin protein by the SWI/SNF chromatin remodeling enzyme. Proc Natl Acad Sci U S A. 2014 Dec 16;111(50):17827–17832.
- Grunstein M, Gasser SM. Epigenetics in Saccharomyces cerevisiae. Cold Spring Harb Perspect Biol. 2013 Jul 1;5(7):a017491-a017491.
- Bupp JM, Martin AE, Stensrud ES, et al. Telomere anchoring at the nuclear periphery requires the budding yeast Sad1-UNC-84 domain protein Mps3. J Cell Biol. 2007 Dec 3;179(5):845–854.
- Anderson CJ, Baird MR, Hsu A, et al. Structural basis for recognition of ubiquitylated nucleosome by Dot1L methyltransferase. Cell Rep. 2019 Feb 12;26(7):1681–1690 e5.
- Moretti P, Freeman K, Coodly L, et al. Evidence that a complex of SIR proteins interacts with the silencer and telomere-binding protein RAP1. Genes Dev. 1994 Oct 1;8(19):2257–2269.
- Imai S, Armstrong CM, Kaeberlein M, et al. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature. 2000 Feb 17;403(6771):795–800.
- Palladino F, Laroche T, Gilson E, et al. SIR3 and SIR4 proteins are required for the positioning and integrity of yeast telomeres. Cell. 1993 Nov 5;75(3):543–555.
- Smith JS, Boeke JD. An unusual form of transcriptional silencing in yeast ribosomal DNA. Genes Dev. 1997 Jan 15;11(2):241–254.
- Rossmann MP, Luo W, Tsaponina O, et al. A common telomeric gene silencing assay is affected by nucleotide metabolism. Mol Cell. 2011 Apr 8;42(1):127–136.
- Takahashi YH, Schulze JM, Jackson J, et al. Dot1 and histone H3K79 methylation in natural telomeric and HM silencing. Mol Cell. 2011 Apr 8;42(1):118–126.
- Kim W, Kim R, Park G, et al. Deficiency of H3K79 histone methyltransferase Dot1-like protein (DOT1L) inhibits cell proliferation. J Biol Chem. 2012 Feb 17;287(8):5588–5599.
- Price BD, D’Andrea AD. Chromatin remodeling at DNA double-strand breaks. Cell. 2013 Mar 14;152(6):1344–1354.
- Rappold I, Iwabuchi K, Date T, et al. Tumor suppressor p53 binding protein 1 (53BP1) is involved in DNA damage-signaling pathways. J Cell Biol. 2001 Apr 30;153(3):613–620.
- Jorgensen S, Schotta G, Sorensen CS. Histone H4 lysine 20 methylation: key player in epigenetic regulation of genomic integrity. Nucleic Acids Res. 2013 Mar 1;41(5):2797–2806.
- Lazzaro F, Sapountzi V, Granata M, et al. Histone methyltransferase Dot1 and Rad9 inhibit single-stranded DNA accumulation at DSBs and uncapped telomeres. Embo J. 2008 May 21;27(10):1502–1512.
- Wakeman TP, Wang Q, Feng J, et al. Bat3 facilitates H3K79 dimethylation by DOT1L and promotes DNA damage-induced 53BP1 foci at G1/G2 cell-cycle phases. Embo J. 2012 May 2;31(9):2169–2181.
- Geuting V, Reul C, Lobrich M. ATM release at resected double-strand breaks provides heterochromatin reconstitution to facilitate homologous recombination. PLoS Genet. 2013;9(8):e1003667.
- Zhu B, Chen S, Wang H, et al. The protective role of DOT1L in UV-induced melanomagenesis. Nat Commun. 2018 Jan 17;9(1):259.
- De Kouchkovsky I, Abdul-Hay M. Acute myeloid leukemia: a comprehensive review and 2016 update. Blood Cancer J. 2016 Jul 1;6(7):e441.
- Jamieson CH. Chronic myeloid leukemia stem cells. Hematology Am Soc Hematol Educ Program. 2008;2008:436–442.
- Lowenberg B, Downing JR, Burnett A. Acute myeloid leukemia. N Engl J Med. 1999 Sep 30;341(14):1051–1062.
- Oriol A, Vives S, Hernandez-Rivas JM, et al. Outcome after relapse of acute lymphoblastic leukemia in adult patients included in four consecutive risk-adapted trials by the PETHEMA Study Group. Haematologica. 2010 Apr;95(4):589–596.
- Pulte D, Jansen L, Gondos A, et al. Survival of adults with acute lymphoblastic leukemia in Germany and the United States. PLoS One. 2014;9(1):e85554.
- Chen CW, Armstrong SA. Targeting DOT1L and HOX gene expression in MLL-rearranged leukemia and beyond. Exp Hematol. 2015 Aug;43(8):673–684.
- Marchesi F, Girardi K, Avvisati G. Pathogenetic, clinical, and prognostic features of adult t(4;11)(q21;q23)/MLL-AF4 positive B-cell acute lymphoblastic leukemia. Adv Hematol. 2011;2011:621627.
- Heerema NA, Sather HN, Ge J, et al. Cytogenetic studies of infant acute lymphoblastic leukemia: poor prognosis of infants with t(4;11) - a report of the Children’s Cancer Group. Leukemia. 1999 May;13(5):679–686.
- Balgobind BV, Zwaan CM, Pieters R, et al. The heterogeneity of pediatric MLL-rearranged acute myeloid leukemia. Leukemia. 2011 Aug;25(8):1239–1248.
- Raimondi SC, Chang MN, Ravindranath Y, et al. Chromosomal abnormalities in 478 children with acute myeloid leukemia: clinical characteristics and treatment outcome in a cooperative pediatric oncology group study-POG 8821. Blood. 1999 Dec 1;94(11):3707–3716.
- Schoch C, Schnittger S, Klaus M, et al. AML with 11q23/MLL abnormalities as defined by the WHO classification: incidence, partner chromosomes, FAB subtype, age distribution, and prognostic impact in an unselected series of 1897 cytogenetically analyzed AML cases. Blood. 2003 Oct 1;102(7):2395–2402.
- Gu Y, Nakamura T, Alder H, et al. The t(4;11) chromosome translocation of human acute leukemias fuses the ALL-1 gene, related to Drosophila trithorax, to the AF-4 gene. Cell. 1992 Nov 13;71(4):701–708.
- Tkachuk DC, Kohler S, Cleary ML. Involvement of a homolog of Drosophila trithorax by 11q23 chromosomal translocations in acute leukemias. Cell. 1992 Nov 13;71(4):691–700.
- Bursen A, Schwabe K, Ruster B, et al. The AF4.MLL fusion protein is capable of inducing ALL in mice without requirement of MLL.AF4. Blood. 2010 Apr 29;115(17):3570–3579.
- Wachter K, Kowarz E, Marschalek R. Functional characterisation of different MLL fusion proteins by using inducible sleeping beauty vectors. Cancer Lett. 2014 Oct 1;352(2):196–202.
- Ma C, Staudt LM. LAF-4 encodes a lymphoid nuclear protein with transactivation potential that is homologous to AF-4, the gene fused to MLL in t(4;11) leukemias. Blood. 1996 Jan 15;87(2):734–745.
- Li Y, Wen H, Xi Y, et al. AF9 YEATS domain links histone acetylation to DOT1L-mediated H3K79 methylation. Cell. 2014 Oct 23;159(3):558–571.
- Domer PH, Fakharzadeh SS, Chen CS, et al. Acute mixed-lineage leukemia t(4;11)(q21;q23) generates an MLL-AF4 fusion product. Proc Natl Acad Sci U S A. 1993 Aug 15;90(16):7884–7888.
- Meyer C, Hofmann J, Burmeister T, et al. The MLL recombinome of acute leukemias in 2013. Leukemia. 2013 Nov;27(11):2165–2176.
- Iida S, Seto M, Yamamoto K, et al. MLLT3 gene on 9p22 involved in t(9;11) leukemia encodes a serine/proline rich protein homologous to MLLT1 on 19p13. Oncogene. 1993 Nov;8(11):3085–3092.
- Okada Y, Feng Q, Lin Y, et al. hDOT1L links histone methylation to leukemogenesis. Cell. 2005 Apr 22;121(2):167–178.
- Milne TA, Martin ME, Brock HW, et al. Leukemogenic MLL fusion proteins bind across a broad region of the Hox a9 locus, promoting transcription and multiple histone modifications. Cancer Res. 2005 Dec 15;65(24):11367–11374.
- Guenther MG, Lawton LN, Rozovskaia T, et al. Aberrant chromatin at genes encoding stem cell regulators in human mixed-lineage leukemia. Genes Dev. 2008 Dec 15;22(24):3403–3408.
- Thiel AT, Blessington P, Zou T, et al. MLL-AF9-induced leukemogenesis requires coexpression of the wild-type Mll allele. Cancer Cell. 2010 Feb 17;17(2):148–159.
- Wong P, Iwasaki M, Somervaille TC, et al. Meis1 is an essential and rate-limiting regulator of MLL leukemia stem cell potential. Genes Dev. 2007 Nov 1;21(21):2762–2774.
- Mueller D, Bach C, Zeisig D, et al. A role for the MLL fusion partner ENL in transcriptional elongation and chromatin modification. Blood. 2007 Dec 15;110(13):4445–4454.
- Zhang H, Zhou B, Qin S, et al. Structural and functional analysis of the DOT1L-AF10 complex reveals mechanistic insights into MLL-AF10-associated leukemogenesis. Genes Dev. 2018 Mar 1;32(5–6):341–346.
- Nguyen AT, Taranova O, He J, et al. DOT1L, the H3K79 methyltransferase, is required for MLL-AF9-mediated leukemogenesis. Blood. 2011 Jun 23;117(25):6912–6922.
- Daigle SR, Olhava EJ, Therkelsen CA, et al. Selective killing of mixed lineage leukemia cells by a potent small-molecule DOT1L inhibitor. Cancer Cell. 2011 Jul 12;20(1):53–65.
- Daigle SR, Olhava EJ, Therkelsen CA, et al. Potent inhibition of DOT1L as treatment of MLL-fusion leukemia. Blood. 2013 Aug 8;122(6):1017–1025.
- Waters NJ, Daigle SR, Rehlaender BN, et al. Exploring drug delivery for the DOT1L inhibitor pinometostat (EPZ-5676): subcutaneous administration as an alternative to continuous IV infusion, in the pursuit of an epigenetic target. J Control Release. 2015 Dec 28;220(Pt B):758–765.
- Stein EM, Garcia-Manero G, Rizzieri DA, et al. The DOT1L inhibitor pinometostat reduces H3K79 methylation and has modest clinical activity in adult acute leukemia. Blood. 2018 Jun 14;131(24):2661–2669.
- Neerav Shukla M, CW MD PhD, MMOB MD, et al. Final report of phase 1 study of the DOT1L inhibitor, pinometostat (EPZ-5676), in children with relapsed or refractory MLL-r acute leukemia. blood. 2016 december 2;128(22):2175–2178.
- Nguyen AT, He J, Taranova O, et al. Essential role of DOT1L in maintaining normal adult hematopoiesis. Cell Res. 2011 Sep;21(9):1370–1373.
- Jo SY, Granowicz EM, Maillard I, et al. Requirement for Dot1l in murine postnatal hematopoiesis and leukemogenesis by MLL translocation. Blood. 2011 May 5;117(18):4759–4768.
- Du L, Grigsby SM, Yao A, et al. Peptidomimetics for targeting protein-protein interactions between DOT1L and MLL oncofusion proteins AF9 and ENL. ACS Med Chem Lett. 2018 Sep 13;9(9):895–900.
- Yao Y, Chen P, Diao J, et al. Selective inhibitors of histone methyltransferase DOT1L: design, synthesis, and crystallographic studies. J Am Chem Soc. 2011 Oct 26;133(42):16746–16749.
- Yu W, Smil D, Li F, et al. Bromo-deaza-SAH: a potent and selective DOT1L inhibitor. Bioorg Med Chem. 2013 Apr 1;21(7):1787–1794.
- Chen C, Zhu H, Stauffer F, et al. Discovery of novel Dot1L inhibitors through a structure-based fragmentation approach. ACS Med Chem Lett. 2016 Aug 11;7(8):735–740.
- Anglin JL, Song Y. A medicinal chemistry perspective for targeting histone H3 lysine-79 methyltransferase DOT1L. J Med Chem. 2013 Nov 27;56(22):8972–8983.
- Yu W, Chory EJ, Wernimont AK, et al. Catalytic site remodelling of the DOT1L methyltransferase by selective inhibitors. Nat Commun. 2012;3:1288.
- Basavapathruni A, Olhava EJ, Daigle SR, et al. Nonclinical pharmacokinetics and metabolism of EPZ-5676, a novel DOT1L histone methyltransferase inhibitor. Biopharm Drug Dispos. 2014 May;35(4):237–252.
- Williams AB, Nguyen B, Li L, et al. Mutations of FLT3/ITD confer resistance to multiple tyrosine kinase inhibitors. Leukemia. 2013 Jan;27(1):48–55.
- Pauwels D, Sweron B, Cools J. The N676D and G697R mutations in the kinase domain of FLT3 confer resistance to the inhibitor AC220. Haematologica. 2012 Nov;97(11):1773–1774.
- Campbell CT, Haladyna JN, Drubin DA, et al. Mechanisms of pinometostat (EPZ-5676) treatment-emergent resistance in MLL-rearranged leukemia. Mol Cancer Ther. 2017 Aug;16(8):1669–1679.
- Stein EM, Garcia-Manero G, Rizzieri DA, et al. A phase 1 study of the DOT1L inhibitor, pinometostat (EPZ-5676), in adults with relapsed or refractory leukemia: safety, clinical activity, exposure and target inhibition. blood. 2015 december 3;126(23):2547.
- Rau RE, Rodriguez BA, Luo M, et al. DOT1L as a therapeutic target for the treatment of DNMT3A-mutant acute myeloid leukemia. Blood. 2016 Aug 18;128(7):971–981.
- Gilan O, Lam EY, Becher I, et al. Functional interdependence of BRD4 and DOT1L in MLL leukemia. Nat Struct Mol Biol. 2016 Jul;23(7):673–681.
- Klaus CR, Iwanowicz D, Johnston D, et al. DOT1L inhibitor EPZ-5676 displays synergistic antiproliferative activity in combination with standard of care drugs and hypomethylating agents in MLL-rearranged leukemia cells. J Pharmacol Exp Ther. 2014 Sep;350(3):646–656.