
ABSTRACT
Human papillomavirus-related oropharyngeal squamous cell carcinoma (HPV+ OPSCC) represents a unique disease entity within head and neck cancer with rising incidence. Previous work has shown that alternative splicing events (ASEs) are prevalent in HPV+ OPSCC, but further validation is needed to understand the regulation of this process and its role in these tumours. In this study, eleven ASEs (GIT2, CTNNB1, MKNK2, MRPL33, SIPA1L3, SNHG6, SYCP2, TPRG1, ZHX2, ZNF331, and ELOVL1) were selected for validation from 109 previously published candidate ASEs to elucidate the post-transcriptional mechanisms of oncogenesis in HPV+ disease. In vitro qRT-PCR confirmed differential expression of 9 of 11 ASE candidates, and in silico analysis within the TCGA cohort confirmed 8 of 11 candidates. Six ASEs (MRPL33, SIPA1L3, SNHG6, TPRG1, ZHX2, and ELOVL1) showed significant differential expression across both methods. Further evaluation of chromatin modification revealed that ASEs strongly correlated with cancer-specific distribution of acetylated lysine 27 of histone 3 (H3K27ac). Subsequent epigenetic treatment of HPV+ HNSCC cell lines (UM-SCC-047 and UPCI-SCC-090) with JQ1 not only induced downregulation of cancer-specific ASE isoforms, but also growth inhibition in both cell lines. The UPCI-SCC-090 cell line, with greater ASE expression, also showed more significant growth inhibition after JQ1 treatment. This study confirms several novel cancer-specific ASEs in HPV+OPSCC and provides evidence for the role of chromatin modifications in regulation of alternative splicing in HPV+OPSCC. This highlights the role of epigenetic changes in the oncogenesis of HPV+OPSCC, which represents a unique, unexplored target for therapeutics that can alter the global post-transcriptional landscape.
Introduction
Head and neck squamous cell carcinoma (HNSCC) is the sixth leading cause of cancer deaths worldwide. While these tumours have been traditionally associated with heavy tobacco and alcohol use, the risk factors are changing, and there has been a significant increase in oropharynx tumours related to human papillomavirus (HPV). The incidence of HPV-positive (HPV+) oropharyngeal squamous cell carcinoma (OPSCC) has more than doubled over the past decades making it one of the most rapidly growing cancer populations in the United States, while the incidence of HPV-negative (HPV−) OPSCC has decreased [Citation1]. These trends are likely attributable to decreasing tobacco use as well as rising prevalence of HPV within the population [Citation2]. Notably, within TCGA, HPV+ tumours show relatively few genetic alterations in cancer drivers at the exome level compared to HPV− tumours [Citation3]. This may be related to fewer carcinogenic exposures to cause DNA alterations, but it may also suggest that changes in post-transcriptional regulation or epigenetic changes may take precedence in HPV-related head and neck carcinogenesis. Indeed, post-transcriptional changes have been shown to have an important role in head and neck cancer in mediating oncogenesis, tumour progression and drug resistance [Citation4–Citation10]. Furthermore, the epigenetic landscape (on the level of both DNA methylation and histone modification) of HPV+ HNSCC samples correlates with the expression of cancer-related genes and cancer prognosis [Citation10–Citation13]
Alternative splicing is a post-transcriptional process that allows for diversity in protein production in the absence of genetic alterations. Previous literature has shown that variations in the splicing of a wild-type (WT) isoform of a gene can contribute to carcinogenesis. For example, alternatively spliced forms of genes BCL2L1 and TNR6 have been shown to be either pro or anti-apoptotic in human cancers, based on exon inclusion and expression [Citation14]. Within head and neck cancer, multiple genes, such as LAMA3, DST, and TP63 are alternatively spliced [Citation8]. Recent work has also revealed functionally active alternatively spliced variants of AKT3 and GSN in HPV+ OPSCC [Citation9,Citation15]. In addition, over 100 additional splicing events were identified in a previous publication, but their function and regulation remain unstudied [Citation9].
Epigenetic DNA methylation has been well established as a method of gene expression regulation in tumours including in the head and neck. In HNSCC, epigenetic silencing of tumour suppressors have been shown to play a role in tumorigenesis [Citation5–Citation7], and histone modifications may mediate drug resistance [Citation4]. Moreover, a growing body of literature also has implicated epigenetic changes in splicing regulation. In other tumour types, it has been observed that DNA methylation is higher in exons and intergenic regions compared to introns, suggesting that DNA methylation may regulate alternative splicing [Citation16,Citation17]. It has also been proposed that the generation of aberrant isoforms with alternative starts is a potential mechanism by which cancer cells circumvent suppression of oncogene transcription by hypermethylation [Citation9,Citation18,Citation19].
Prior literature has established the interplay between chromatin state and gene splicing. Histone methylation marks are significantly enriched at exons, and studies have shown H3K4me3 to recruit spliceosomal factors through adaptor proteins [Citation20]. In addition, histone acetyltransferases such as the SAGA protein complex interact with and recruit splicing machinery, and facilitate recognition of intron branchpoints [Citation21]. In addition, these histone modifications associated with open chromatin, including H3K27-acetylation, are associated with enhancers which regulate the transcriptional activity of multiple neighbouring gene targets, up to 1–1.5 megabases away, through the formation of chromatin loops [Citation22,Citation23]. These super-enhancers (SEs) are defined by recruitment of tissue-specific transcription factors, H3K27 acetylation, BRD4 and MED1 proteins [Citation24].
Herein, we propose that epigenetic modification in HPV-related OPSCC, specifically regions of chromatin, enriched by H3K27ac, regulate expression of alternative splicing variants which contribute to oncogenesis in these tumours. To evaluate this hypothesis, we first performed validation of 11 alternatively spliced genes from a list of previously identified candidate 109 ASEs [Citation9] to confirm both their presence in OPSCC primary tumours and the validity of ASEs identified in silico. We then utilize these ASEs to evaluate the correlation between the epigenetic landscape and ASE expression. HPV+ head and neck cell lines were subsequently treated with JQ1, the inhibitor of BRD4, which recognizes the H3K27ac histone mark. JQ1 treatment resulted in alteration of splice variant balance as well as growth inhibition in multiple head and neck cell lines. This is the first study to demonstrate the role of H3K27ac landscape and SE positioning in the epigenetic regulation of ASEs in HPV+ HNSCC.
Results
Validation of 11 candidate ASEs by qRT-PCR and with TCGA
A total of 11 ASEs were selected from 109 previously published ASEs [Citation9] for qRT-PCR validation (). The ASEs selected for validation were among the most prevalent ASEs identified in the tumour cohort, and which also showed correlation with DNA methylation and chromatin state, as previously published [Citation9]. The 11 ASEs included 8 alternative start events, 2 deletion events, and one insertion event. All ASEs except ELOVL1 exhibited outlier over-expression in the tumour group relative to normal tissues. Of the 11 ASEs, only 4 were canonical (CTNNB1, MKNK2, SYCP2, and ZNF331). shows the sashimi plots of 2 representative HPV+ OPSCC and 2 non-cancer samples for the ZHX2 ()) and MKNK2 ()) ASEs.
Table 1. Eleven alternative splicing events selected for validation .
Figure 1. Two validated splice variants ZHX2 and MKNK2 ASEs.
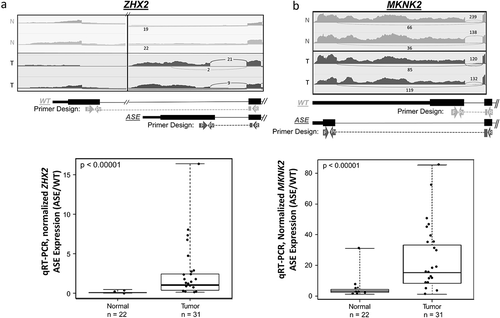
In vitro validation by qRT-PCR revealed 9 out of the 11 genes to have significantly higher rates of outlier ASE expression (FDR < 0.05) in tumour samples compared to normal samples (). TPRG1 ASE had the highest prevalence of outlier overexpression in tumour samples at 94% (n = 29 out of 31), followed by MRPL33 at 90% (n = 28 out of 31), and MKNK2 at 77% (n = 24 out of 31, ), FDR<0.00001). Notably, for most ASEs investigated, qRT-PCR showed higher sensitivity for detection of ASEs with a higher prevalence of outlier expression compared to RNA-Seq within this cohort ().
Table 2. Validation of 11 ASEs in vitro (qRT-PCR) and in silico (TCGA).
Cross-validation was then performed in silico on an independent cohort (TCGA) of 16 normal samples and 44 HPV+ OPSCC. Within the TCGA data set, 8 out of the 11 ASEs had significant differential outlier ASE expression (FDR < 0.05) between the tumour and normal samples (). The ASEs with the highest prevalence in the tumour group in the TCGA cohort were ELOVL1 at 91% (n = 40 out of 44), followed by SYCP2 at 80% (n = 35 out of 44). ZNF331 ASE was not identified as an outlier in any of the tumour samples. In addition, when comparing ASE expression among all three platforms, outlier detection in normal samples remained consistently at a frequency below 5% (1 sample or less, ). All 11 candidate ASEs were validated either in vitro or in silico. Consistent differential expression was found in six genes (MRPL33, SIPA1L3, SNHG6, TPRG1, ZHX2, and ELOVL1).
Spatial correlation between chromatin modifications and ASE
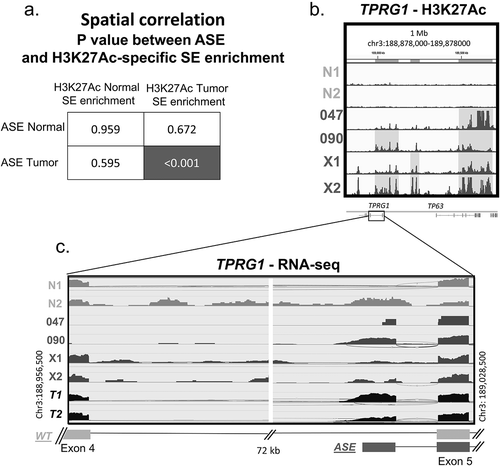
Next, GenometriCorr (projection statistics) analysis was performed to evaluate the potential relationship between ASE expression and chromatin modification. This analysis showed that the spatial distribution of the 109 ASEs significantly correlated with tumour-specific H3K27ac super-enhancer (SE) regions as defined by ROSE analysis (), p < 0.001) but not with normal-specific H3K27ac SEs. Interestingly, GenometriCorr analysis also -showed that tumour-specific SEs were significantly correlated with the location of alternative start sites (p = 0.021), as well as both canonical (p = 0.011) and non-canonical alternatively spliced tumour-related gene isoforms (p = 0.005), while no correlation was seen with normal tissue super-enhancers (p > 0.2) (Supplemental Table 2). One example of H3K27ac enrichment in relationship with a splicing event is shown in the TPRG1 gene (). ROSE analysis identified nearby SE regions, enriched with H3K27ac, within the 1 million bp region surrounding TPRG1 in PDX and cell lines, but not normal tissue (highlighted in )). Within this region, the presence of the TPRG1 ASE expression is confirmed by RNA-Seq analysis only in samples (SCC090 & X2) that demonstrated H3K27ac enrichment with absence in normal tissue. Notably, both cell lines had relatively low expression of canonical WT TPRG1.
Figure 2. Spatial correlation of chromatin reorganization and alternative splicing events.
In addition to H3K27ac, three other chromatin marks (H3K4me3, H3K9ac and H3K9me3) were studied in relationship to alternative splicing expression. These data show that active marks H3K27ac and H3K4me3 both show consistent spatial enrichment in regions of tumour-related ASEs, only in tumour-specific marks. H3K9ac showed spatial correlation that was not specific to normal or tumour tissue, an effect that was observed also when relating to gene expression in previously published work [Citation10]. H3K9me3 is a repressive mark, and as expected did not show any significant relationship to ASE expression in either normal or tumour-specific marks.
Next, we evaluated the spatial relationship of enrichment of these histone marks to specific ASE types (Supplemental Table 2). Interestingly, in all active histone marks, there was a significant spatial correlation between histone marks and non-canonical ASEs. Next, alternative start site ASE isoforms were evaluated and all three histone marks showed tumour-specific spatial correlation with alternative start site splicing events. As previously published work has shown, the active histone marks (H3K27ac, H3k4me3, and H3K9 c) demonstrate enrichment in transcription start site regions which may relate to these alternative start site isoforms. Analysis of relationship of these chromatin marks for insertion and deletion events was deferred, given the lower prevalence of these splicing events.
Epigenetic-driven changes in ASE landscape decrease cell proliferation
Two HPV+ cell lines (UM-SCC-047 and UPCI-SCC-090) were then treated with the bromodomain inhibitor, JQ1, which is expected to downregulate the activity of SEs [Citation25]. Out of 109 ASE previously detected for HPV+ OPSCC, 26 ASEs were detected in UM-SCC-047 and 60 ASEs in UPCI-SCC-090 using RNA-Seq analysis. When compared to the number of ASEs in tumours, UM-SCC-047 was within the lower quartile and UPCI-SCC-090 was in the upper quartile ()). JQ1 suppressed proliferation in both UPCI-SCC-090 and UM-SCC-047 (p < 0.0001 and p < 0.005, respectively, ). However, the growth suppression effect of JQ1 was more pronounced in ASE-rich cell line, UPCI-SCC-090.
Figure 3. JQ1 treatment of HPV+ HNSCC cell lines.
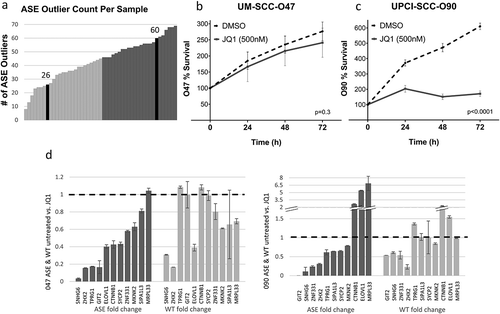
JQ1 treatment resulted in changes in the splicing balance of several of the 11 genes studied, producing varying effects on the expression of the ASE and WT isoforms. In the UM-SCC-047 cell line, nearly all genes showed a decrease in ASE expression after JQ1 treatment, while wild-type gene expression was not significantly changed in five genes, and decreased in six of those studied. In UPCI-SCC-090, again a majority of genes (8 of 11) showed decrease in splice variant expression after JQ1 treatment. In this cell line, JQ1 had more variable impact on wild-type gene expression, with four genes showing decreased expression, and the remainder with stable or increased expression. In addition, the splice variant ELOVL1, which was suppressed in tumours, showed enhanced expression after JQ1 treatment in this cell line. It is also noted that the splice variant of MRPL33 showed enhanced expression after JQ1 treatment in both cell lines.
RNA-Seq analysis of both UM-SCC-047 and UPCI-SCC-090 cell lines was conducted comparing JQ1 treatment to control (DMSO). Among 109 ASE candidates [Citation1], 75 splice junctions were captured by RNA-seq with adequate coverage involving 68 unique genes for which overall gene expression was available. The UM-SCC-047 cell line had relatively low overall expression of ASEs, with 24 (32%) ASEs showing no expression in both DMSO or JQ1 treated cells. There were 12 junctions with significant differential expression after JQ1 treatment, and 83% (n = 10) showed decreased expression after JQ1 treatment (Supplemental Table 3). However, all but one of these (ASE in BAI2) were accompanied by significant decreases on overall gene expression after JQ1 treatment. In contrast, after JQ1 treatment of UPCI-SCC-090, 91% (n = 10 of 11) of junctions with significant differential expression showed decreased expression. Half of these (n = 5) were accompanied by either no change or increase in overall gene expression, showing splicing-specific changes induced by JQ1 treatment (Supplemental Table 4).
To better understand the specificity of JQ1 treatment, we performed JQ1 treatment in an additional HPV+ HNSCC cell line (93-VU-147 T), as well as immortalized oral keratinocyte cell lines (OKF6 & NOKSI). Significant growth inhibition was seen in all cell lines treated with JQ1. As in UM-SCC-047 and UPCI-SCC-090, JQ1 also significantly decreased a majority of ASEs (9 of 11) in 93-VU-147 T and wild-type expression remained primarily unchanged or increased (7 of 11 genes, Supplemental Figure 1). In immortalized normal cell lines (Supplemental Figure 2), greater off-target effects were seen in response to JQ1 treatment. There was greater inhibition of wild-type gene expression in immortalized oral keratinocytes, with 72–81% of genes of interest showing decrease in wild-type expression with JQ1 treatment in the non-cancer cell lines. Notably, JQ1 did decrease expression of ASEs as well; however, only about half of the studied ASEs were able to be detected in the normal cell lines, even in untreated controls. Therefore, growth inhibition in the normal cell lines may be more related to off-target impact on gene expression.
Evaluation of specificity to HPV+ disease
To understand whether validated ASEs were specific to the HPV+ population of the disease, additional analysis was performed on an HPV− HNSCC cohort from TCGA RNA-sequencing analysis. Normalized junction expression for the 11 identified ASEs was compared between 411 tumours and 34 normal samples. This analysis revealed that about half of the ASEs (n = 6) were also significantly differentially expressed in HPV− tumours (CTNNB1, MKNK2, MRPL33, SNHG6, SYCP2, and ZHX2, FDR<0.05, Supplemental Table 5), suggesting that not all ASEs are specific to the HPV+ sub-type. However, five ASEs did not have a significant differential expression in HPV− tumours and may be more specific to HPV-related tumours (GIT2, SIPA1L3, TPRG1, ZNF331, and ELOVL1).
Furthermore, we sought to understand splicing events in the context of HPV viral integration. Of tumours with detectable HPV16 genome, 16 tumours showed reliably detectable viral integration, 19 did not show evidence of integration and 10 were equivocal with limited HPV−human fusion detected. We then compared the rate of alternative splice variant expression, utilizing the previously published 109 candidates [Citation9], in tumours with integrated HPV viral genome (n = 16) and non-integrated tumours (n = 19). The number of splice variants expressed in integrated tumours was significantly higher than non-integrated tumours (mean 55.1 vs. 41.3 splicing events, Student’s T-test, p = 0.001, Supplemental Figure 3).
Discussion
The scarcity of mutational alterations in HPV+ OPSCC has limited our understanding of the underlying biology of this disease and the detection of oncogenic targets. This study therefore seeks to elucidate the post-transcriptional process of alternative splicing in HPV+ OPSCC through the characterization of cancer-specific ASEs and investigation of their epigenetic control. In other disease processes, alternative splicing of the WT isoform of a gene has been shown to contribute to carcinogenesis [Citation18–Citation20]. For example, alternatively spliced forms of genes BCL2L1 and TNR6 are known to be either pro or anti-apoptotic in human cancers, based on exon inclusion and expression [Citation12]. Similarly, we have recently demonstrated that cancer-specific splicing in AKT3 and GSN leads to oncogenic splice variants in head and neck SCC which promote growth and invasion [Citation9,Citation15].
Alternative splicing variants allow for variant protein forms with oncogenic potential to be expressed in tumours, even in the absence of DNA mutations. However, therapeutic targeting of individual splice variants may be challenging unless the mechanisms allowing for their expression in tumours is better understood. What is the process by which the balance between splice variants is determined; and what alterations in tumours allow for expression of tumour-specific splicing events? The data from this study suggest that chromatin modifications may contribute to the regulation of aberrant cancer-specific splice variants.
In order to study the impact of chromatin modifications on ASE expression, we selected 11 ASEs from a list of 109 previously published ASE candidates for additional validation. These selected candidates offer a representative sample of 10% of the ASEs identified by previously published algorithm, with high prevalence and correlation with DNA methylation [Citation9]. Within validation, the use of outlier statistic methods with multiple testing correction for detection of aberrant splicing events is extremely stringent, only counting those samples with outlier expression at least two standard deviations above average of any normal samples. Despite this stringency, we identified six ASEs (MRPL33, SIPA1L3, SNHG6, TPRG1, ZHX2, and ELOVL1, as having significant tumour-specific expression in both biological evaluation and within TCGA.
These validated splice variants occur in genes of potential biological interest. Upregulation of SNHG6 expression predicts poor prognosis in colorectal cancer [Citation26] and poor clinical features in gastric cancer [Citation27]. Similarly, ZHX2 expression correlates with worse prognosis in renal cell carcinoma [Citation28]. These publications suggest the potential biological role of ASEs in HPV+ OPSCC requiring further in-depth functional analysis that is outside of the scope of the current manuscript. However, multiple recent studies have shown the functional role of tumour-specific splicing variants that contribute to oncogenesis within head and neck cancer. In prior work, a functionally active splice variant of AKT3 was identified, in which its impact on growth in HNSCC was isoform specific [Citation9]. Other splicing variants have recently been reported showing isoform-specific functional activity in head and neck cancer, including an isoform of GSN which promotes migration [Citation15] and an LOXL2 isoform which promotes growth and progression [Citation29].
Interestingly, some of the genes have also been studied in the context of HPV integration. A study by Parfenov et al. found HPV integration to occur on chromosome 3, and that it is associated with chromosomal rearrangements leading to the amplification of TPRG1 along with TP63 [Citation30]. TP63 has been a frequent site of HPV integration in both oropharynx and cervical cancers and play a potential role in SE regulation in those tissues, and the ASE in TPRG1 was found to be specific to HPV-positive tumours [Citation10,Citation31,Citation32]. The MRPL33 ASE is particularly of interest because HPV integration has been noted to occur in the vicinity of MRPL33 – within a 3 million base pair region surrounding the gene; however, the MRPL33 ASE was also seen in HPV− tumours [Citation33], and so may not be exclusively driven by HPV integration. While not all ASEs were specific to HPV+ tumours, we found that tumours with integration of the viral HPV genome exhibited significantly more ASEs.
While epigenetics, such as chromatin modifications and DNA methylation, are widely known to contribute to oncogenesis, mounting evidence over recent years has pointed to an interrelation between chromatin and epigenetic modifications with regulation of alternative splicing in disease. The histone acetyltransferase Gcn5 was demonstrated to interact with two U2 snRNP proteins and its HAT activity is required for their recruitment to pre-mRNA branch points [Citation21]. Chromatin compaction is known to have a kinetic effect on differential splicing in which weaker splice sites can be detected [Citation34]. Likewise, the subunit Brm of the SWI/SNF chromatin remodeller regulates splicing by pausing transcription, thereby promoting inclusion of exons with weaker splice sites [Citation35]. The epigenetic modification of H3K4me3 regulates splicing through binding protein CHD1 recruiting of spliceosome component U2 snRNP. CHD1 knockdown reduced splicing efficiency and combined knockdown with H3K4me4 further reduced U2 snRNP recruitment [Citation20].
In this study, H3K27ac modifications were found to significantly spatially correlate with ASEs, suggesting a stronger link between alternative splicing and histone acetylation than previously understood. H3K27-acetylation is associated with super-enhancers, that can regulate and activate gene expression in large groups of genes through changes in chromatin loop structures and recruitment of tissue-specific transcription factors to significantly change intracellular biology [Citation22,Citation23]. In prior work, tumour-specific gene expression changes were correlated with both active histone marks H3K27ac as well as H3K4me3 [Citation10]. Similarly in this study, these chromatin marks showed tumour-specific enrichment related to alternative splicing events not seen in normal tissue. These relationships were strongest when relating to ASEs which were non-canonical, and ASEs relating to alternative start sites, which may be due to enrichment in transcriptional start sites which has been previously demonstrated [Citation10].
A variety of evidence implicates bromodomain containing protein 4 (BRD4), an acetylated histone-binding protein, in splicing regulation [Citation36]. BRD4 has been associated with U2AF65 activity and the production of mature spliced transcripts, and its mutant form leads to a reduction in U1 snRNP recruitment at intron-containing genes. In the context of this study, assays of the ASEs and their associated WT junction revealed a clear change in splice variant balance after treatment with bromodomain inhibitor JQ1. Both cell lines showed that a majority of the tumour-specific ASEs were downregulated after JQ1 treatment to a greater degree than wild-type isoforms, including SNHG6, ZHX2, TPRG1 and SIPA1L3 which were validated across all methods.
Treatment with JQ1 significant decreased cell growth of both HPV+ head and neck cancer cell lines UM-SCC- 047 and UPCI-SCC-090. There are known off-target growth effects of JQ1 which may be related to JQ1’s suppression of MYC [Citation37]. It is notable that growth suppression was twice as great in UPCI-SCC-090 compared to UM-SCC-047, and UPCI-SCC-090 had a greater degree of aberrant ASE expression. In addition, JQ1 had a more specific suppression of aberrant ASE expression compared to wild-type gene in UPCI-SCC-090 in both PCR and RNA-Seq.
Previous studies [Citation9,Citation15] have shown that ASEs within HPV+ OPSCC (AKT3 and GSN) are functionally relevant and confer oncogenic properties. Given the changes in ASE expression seen after JQ1 treatment, some of the greater growth inhibition seen in the ASE-rich UPCI-SCC-090 cell line may be due to the alteration of its oncogenic ASE expression. Super-enhancers represent exciting therapeutic targets in cancer, with small molecule inhibitors such as JQ1 with the potential to affect global tumour oncogene expression [Citation38]. Chromatin-regulating small molecules such as histone deacetylase inhibitors (vorinostat) and hypomethylating agents (azacitidine and decitabine) have already been FDA approved for treatment of haematologic malignancies [Citation39,Citation40], and recent evidence suggests a role for super-enhancer in the oncogenesis of head and neck cancer [Citation10,Citation41].
One limitation with JQ1 treatment is that it demonstrated greater off-target effects in immortalized oral keratinocytes, compared to treated tumour cell lines, with alteration in wild-type genes. In comparison, in tumour cell lines, expression alteration was more specific to alternative splice site. Indeed, prior work has shown that these histone modifications may also modulate disease-specific gene expression in additional to splicing variants [Citation10]. Therefore, the growth suppression seen from JQ1 treatment may be related to both ASE changes and off-target effects.
We acknowledge the additional limitation of this study in methodology and scope. In the PCR validation of splice variants, most PCR primers were designed de novo, so non-specific annealing of primers may impact reliability and sensitivity of detection of specific splicing events. In addition, there was observed to be variation between the different validation approaches in the prevalence of tumour outliers. These variations may be explained by differences in cohorts and/or techniques. Within PCR validation, several ASEs showed higher prevalence than in the RNA-Seq data, which may relate to its ability to detect specific splice junctions in an unbiased manner dictated by the depth of sequencing. With regard to JQ1 treatment experiments, as discussed previously, off-target effects with impact on gene expression may also impact growth effects observed, particularly in non-tumour models. The changes in ASE expression and balance were observed in only a subset of ASEs consisting of the selected 11 ASEs and changes to the full ASE landscape were not interrogated in all cell lines.
In conclusion, eleven splicing variants were validated in HPV+ OPSCC in vitro, and in silico on an independent TCGA cohort. Six ASEs were confirmed as differentially expressed between normal and tumour samples across methodologies, involving genes which have evidence of potential oncologic impact based on prior literature. In addition, HPV+ head and neck cancer cell lines treated with JQ1 demonstrate how epigenetic-driven changes can both alter the splice variant landscape of these tumours, and potentially inhibit tumour growth, which was more prominent in an ASE-rich cell line. Overall, this study provides novel evidence that ASEs are one mechanism by which the activity of relevant proteins is altered in HPV+ OPSCC and are under regulation by epigenetic changes like histone acetylation and SE machineries influenced by HPV activity. Understanding the ASE landscape of HPV+ OPSCC provides an opportunity for the discovery of novel molecular targets which alter the epigenetic landscape which may result in changes in ASE expression and subsequent tumour inhibition.
Patients and methods/materials
Study cohorts and patient-derived xenografts
The initial discovery cohort was comprised of data from a recently published study of 47 primary tumour tissue samples from patients with HPV+ OPSCC and 25 normal mucosal tissue from uvulopalatopharyngoplasty (UPPP) surgical samples in non-cancer-affected patients [Citation9]. All tissue samples were collected under an institutional review board-approved protocol (#NA_00036235). Due to limited sample RNA quantity, a partial subset of this discovery cohort (n = 22 UPPP non-cancer controls, and n = 31 HPV+ OPSCC samples) was used for the biological validation. Two patient-derived xenografts (PDX) models were generated from two primary tumour samples from this discovery cohort [Citation10]. From The Cancer Genome Atlas (TCGA) [Citation42], additional data for a separate cohort of 44 HPV+ samples (with 16 corresponding normal oropharyngeal tissues) and 411 HPV− samples (with 34 corresponding normal tissues) was obtained for in silico validation. Junction quantification and RSEM gene expression from RNA-sequencing data were obtained from Broad GDAC firebrowse (gdac.broadinstitute.org) utilizing mRNASeq from Illumina HiSeq platform for head and neck squamous cell carcinoma.
Cell culture and drug treatment
Three HPV+ head and neck cell lines were utilized: UM-SCC-047, UPCI-SCC-090 [Citation43], and 93-VU-147 T provided by Dr Thomas Carey (University of Michigan) and Dr Susanne Gollin (University of Pittsburgh). Cell lines were authenticated using the Short Tandem Repeat (STR) Profiling Service by the Johns Hopkins School of Medicine Genetic Resources Core Facility DNA Services. Cells were grown on high-glucose DMEM media (Clontech, Mountain View, CA), supplemented by 10% fetal bovine serum (FBS) and 1% Penicillin-Streptomycin at 37°C in 5% CO2 [Citation10]. Additionally, immortalized oral keratinocyte cell lines (OKF6 and NOKSI) were cultured and drug treated in a similar manner.
HPV+HNSCC and immortalized oral keratinocyte cell lines were grown in 6 well tissue culture dishes and were treated with 500 nM JQ1 (bromodomain inhibitor, Selleck Chemicals, Houston, TX, USA) or with 0.1% of dimethyl sulphoxide (DMSO), as a control, added to high-glucose DMEM media for 72 h at 37°C. The cellular growth-monitoring experiments were performed in pentaplicates using 4-h incubation with 1:10 diluted Alamar Blue (Bio-Rad) in high-glucose DMEM Media measured every 24 h.
RNA isolation, sequencing and qRT-PCR
RNA from treated and non-treated cell lines, as well as primary samples, was extracted using mirVana miRNA Isolation Kit (Ambion, Forster City, CA). RNA-Seq data for this cohort were previously published and analysed (GSE112027) [Citation9,Citation10,Citation44]. MapSplice2 version 2.0.1.9 was used to quantify read counts during the alignment [Citation44,Citation45]. Gene expression values were quantified from RNA-sequencing data using RNA-Seq by Expectation-Maximization (RSEM) version 1.2.9 [Citation46].
Due to limited sample RNA quantity, a partial subset of this discovery cohort (n = 22 UPPP non-cancer controls, and n = 31 HPV+ OPSCC samples) was used for qRT-PCR validation. All RNA samples were reverse transcribed into cDNA using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Forster City, CA). QRT-PCR primers and probes of the ASEs and their WT controls were designed using Integrated DNA Technologies PrimerQuest tool, with the probe spanning across the ASE junction or its associated WT junction (Supplemental Table 1). These were validated by touchdown PCR (data not shown). All qRT-PCR experiments were normalized to GAPDH expression and performed on an Applied Biosystems 7900 Real-Time PCR system in triplicates.
H3K27ac chromatin immunoprecipitation (ChIP), ChIP-Seq analysis, and the detection of H3K27ac super-enhancers
H3K27ac-specific ChIP-Seq data for 2 PDX samples (PDX1 and PDX2) and 2 UPPP samples (UPPP1 and UPPP2) from the cohort described above, and the UM-SCC-047 and UPCI-SCC-090 cell lines, were previously published and analysed (GSE112027) [Citation10]. Chromatin was extracted from these six samples, as previously described [Citation10]. The H3K27ac (8173, CST) antibody was used to isolate DNA segments bound by this histone modification, using SimpleChIP Plus Enzymatic Chromatin IP Kit (9005, CST). MACS (Model-based Analysis of ChIP-Seq algorithm, version 1.4.2) called ChIP-Seq peaks for each sample using the input DNA in that sample as a control [Citation47]. MACS peaks for H3K27ac were further ranked with the ChIP-Seq signal intensity and stitched if necessary by the Ranking Of Super Enhancers (ROSE) analysis software [Citation23] for each chromatin mark and for each sample. The resulting set of merged peaks with heavy ChIP-Seq abundance is referred to as enhancers. The top ranking of these were classified as super-enahcers (SE).
Detection of heavy ChIP-Seq abundance for H3K27ac, H3K4me2, H3K9ac, and H3K9me3
In order to evaluate whether other histone marks have spatial correlation with ASE expression, we completed similar wet-lab and dry lab analysis for additional active histone marks (H3K4me3, and H3K9ac), and repressive mark (H3K9me3). Just as in the case of H3K27ac, we performed chromatin immunoprecipitation of H3K4me3 (9751, CST), H3K9ac (9649, CST), and H9K9me3 (13,969, CST) using the same kit (9005, CST). Similarly, high-throughput sequencing and MACS analysis were completed for these additional marks. To recapitulate H3K27ac analysis, we employed the same ROSE algorithm to call regions of heavy ChIP-Seq abundance peaks, which are normally called ‘super-enhancers’ and ‘typical enhancers’ by ROSE in the case of H3K27ac. However, we intentionally added ‘typical enhancer’ calls for non-enhancer marks H3K4me3, H3K9ac, and H3K9me3, as these would not strictly be referred to as ‘super-enhancers.’ This analysis was repeated for all four marks including H3K27ac in parallel. The ROSE analysis was completed for each mark and each sample.
GenometriCorr spatial correlation analysis
The R package GenometriCorr [Citation48] was used to compare the positional distribution of the 109 ASEs with signal-enriched peaks for 4 chromatin marks (H3K27ac, H3K4me3, H3K9me3, and H3K9ac). The peaks were detected by ROSE software, and the term ‘super-enhancers’ was defined based on prior publication methods for all four chromatin marks.(REF) The package is intended to test the positional correlations (co-localization) of pairs of genome-wide annotations, which are represented as the sets of genomic intervals. For each of the four marks, super-enhancer datasets were categorized into normal-specific and tumour-specific events, via removal of ROSE-called peaks detected in both tumour and normal samples, and the analysis was performed separately for normal-specific and tumour-specific SEs. P-values were adjusted using the Bonferroni correction based on the number of tests performed.
Outlier and median statistical analyses
Differential ASE expression between tumour and normal groups was evaluated with outlier analysis using the OGSA Bioconductor package [Citation49], on ASE expression normalized to gene expression. Samples were categorized as outliers based on expression distribution in normal samples, and presence of outliers was compared between groups using Fisher’s exact test, and FDR adjusted p-values utilizing the Benjamini–Hochberg method [Citation9]. qRT-PCR data were performed in triplicate with two-tailed Mann–Whitney test [Citation50] at a significance level of 0.05
HPV integration
HPV integration status of tumours was determined based on RNA-sequencing analysis using MapSplice, as previously published [Citation10]. In short, to detect integration, a reference genome was created as a chimera including the human genome and HPV16 genome and tumours were mapped to this chimera genome. In this way, viral integration was detected as a fusion site between the human and HPV16 genome. We considered the viral genome integrated if there were at least three discordant pairs (one paired-end read mapped to the viral genome, and its mate pair mapped to the human genome) and one split read (read spanned the human-viral junction). These seven total reads support integration at the same locus [Citation10,Citation30].
Supplemental Material
Download Zip (268.9 KB)Disclosure statement
No potential conflict of interest was reported by the authors.
Supplemental Materials
Supplemental data for this article can be accessed here
Additional information
Funding
References
- Chaturvedi AK, Engels EA, Pfeiffer RM, et al. Human papillomavirus and rising oropharyngeal cancer incidence in the United States. J Clin Oncol. 2011;29(32):4294–4301.
- Rettig EM, D’Souza G. Epidemiology of head and neck cancer. Surg Oncol Clin N Am. 2015;24:379–396.
- Agrawal N, Frederick MJ, Pickering CR, et al. Exome sequencing of head and neck squamous cell carcinoma reveals inactivating mutations in NOTCH1. Science. 2011;333(6046):1154–1157.
- Almeida LO, Abrahao AC, Rosselli-Murai LK, et al. NFkappaB mediates cisplatin resistance through histone modifications in head and neck squamous cell carcinoma (HNSCC). FEBS Open Bio. 2014;4(1):96–104.
- Paluszczak J, Wisniewska D, Kostrzewska-Poczekaj M, et al. Prognostic significance of the methylation of Wnt pathway antagonists-CXXC4, DACT2, and the inhibitors of sonic hedgehog signaling-ZIC1, ZIC4, and HHIP in head and neck squamous cell carcinomas. Clin Oral Investig. 2017;21:1777–1788.
- Worsham MJ, Chen KM, Meduri V, et al. Epigenetic events of disease progression in head and neck squamous cell carcinoma. Arch Otolaryngol Head Neck Surg. 2006;132:668–677.
- Dong SM, Sun DI, Benoit NE, et al. Epigenetic inactivation of RASSF1A in head and neck cancer. Clin Cancer Res. 2003;9(10 Pt 1):3635–3640.
- Li R, Ochs MF, Ahn SM, et al. Expression microarray analysis reveals alternative splicing of LAMA3 and DST genes in head and neck squamous cell carcinoma. PloS One. 2014;9(3):e91263.
- Guo T, Sakai A, Afsari B, et al. A novel functional splice variant of AKT3 defined by analysis of alternative splice expression in HPV-Positive oropharyngeal cancers. Cancer Res. 2017;77(19):5248–5258.
- Kelley DZ, Flam EL, Izumchenko E, et al. Integrated analysis of whole-genome ChIP-Seq and RNA-Seq data of primary head and neck tumor samples associates HPV integration sites with open chromatin marks. Cancer Res. 2017;77(23):6538–6550.
- Degli Esposti D, Sklias A, Lima SC, et al. Unique DNA methylation signature in HPV-positive head and neck squamous cell carcinomas. Genome Med. 2017;9:33.
- Anayannis NV, Schlecht NF, Belbin TJ. Epigenetic mechanisms of human papillomavirus-associated head and neck cancer. Arch Pathol Lab Med. 2015;139(11):1373–1378.
- Fertig EJ, Markovic A, Danilova LV, et al. Preferential activation of the hedgehog pathway by epigenetic modulations in HPV negative HNSCC identified with meta-pathway analysis. PLoS One. 2013;8(11):e78127.
- Chen J, Weiss WA. Alternative splicing in cancer: implications for biology and therapy. Oncogene. 2015;34(1):1–14.
- Kelley DZ, Flam EL, Guo T, et al. Functional characterization of alternatively spliced GSN in head and neck squamous cell carcinoma. Transl Res. 2018;202:109–119.
- Maunakea AK, Chepelev I, Cui K, et al. Intragenic DNA methylation modulates alternative splicing by recruiting MeCP2 to promote exon recognition. Cell Res. 2013;23(11):1256–1269.
- Narayanan SP, Singh S, Shukla S. A saga of cancer epigenetics: linking epigenetics to alternative splicing. Biochem J. 2017;474(6):885–896.
- Hoivik EA, Witsoe SL, Bergheim IR, et al. DNA methylation of alternative promoters directs tissue specific expression of Epac2 isoforms. PLoS One. 2013;8(7):e67925.
- Maunakea AK, Nagarajan RP, Bilenky M, et al. Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature. 2010;466(7303):253–257.
- Sims RJ 3rd, Millhouse S, Chen CF, et al. Recognition of trimethylated histone H3 lysine 4 facilitates the recruitment of transcription postinitiation factors and pre-mRNA splicing. Mol Cell. 2007;28(4):665–676.
- Gunderson FQ, Johnson TL, Madhani HD. Acetylation by the transcriptional coactivator Gcn5 plays a novel role in co-transcriptional spliceosome assembly. PLoS Genet. 2009;5:e1000682.
- Heintzman ND, Hon GC, Hawkins RD, et al. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature. 2009;459(7243):108–112.
- Whyte WA, Orlando DA, Hnisz D, et al. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell. 2013;153(2):307–319.
- Hnisz D, Abraham BJ, Lee TI, et al. Super-enhancers in the control of cell identity and disease. Cell. 2013;155(4):934–947.
- Sengupta D, Kannan A, Kern M, et al. Disruption of BRD4 at H3K27Ac-enriched enhancer region correlates with decreased c-Myc expression in Merkel cell carcinoma. Epigenetics. 2015;10(6):460–466.
- Li M, Bian Z, Yao S, et al. Up-regulated expression of SNHG6 predicts poor prognosis in colorectal cancer. Pathol Res Pract. 2018;214(5):784–789.
- Yan K, Tian J, Shi W, et al. LncRNA SNHG6 is Associated with poor prognosis of gastric cancer and promotes cell proliferation and EMT through epigenetically silencing p27 and sponging miR-101-3p. Cell Physiol Biochem. 2017;42(3):999–1012.
- Kwon RJ, Kim YH, Jeong DC, et al. Expression and prognostic significance of zinc fingers and homeoboxes family members in renal cell carcinoma. PLoS One. 2017;12(2):e0171036.
- Liu C, Guo T, Saki A, et al. A novel splice variant of LOXL2 promotes progression of HPV negative head and neck squamous cell carcinoma. Cancer. 2020;126(4):737–748.
- Parfenov M, Pedamallu CS, Gehlenborg N, Cancer Genome Atlas N. et al. Characterization of HPV and host genome interactions in primary head and neck cancers. Proc Natl Acad Sci U S A. 2014;111(43):15544–15549.
- Rusan M, Li YY, Hammerman PS. Genomic landscape of human papillomavirus-associated cancers. Clin Cancer Res. 2015;21(9):2009–2019.
- Corces MR, Granja JM, Shams S, et al. The chromatin accessibility landscape of primary human cancers. Science. 2018;362 (6413).
- Warburton A, Redmond CJ, Dooley KE, et al. HPV integration hijacks and multimerizes a cellular enhancer to generate a viral-cellular super-enhancer that drives high viral oncogene expression. PLoS Genet. 2018;14(1):e1007179.
- Allo M, Schor IE, Munoz MJ, et al. Chromatin and alternative splicing. Cold Spring Harb Symp Quant Biol. 2010;75:103–111.
- Pacini C, Koziol MJ. Bioinformatics challenges and perspectives when studying the effect of epigenetic modifications on alternative splicing. Philos Trans R Soc Lond B Biol Sci. 2018;373 (1748).
- Hussong M, Kaehler C, Kerick M, et al. The bromodomain protein BRD4 regulates splicing during heat shock. Nucleic Acids Res. 2017;45:382–394.
- Enomoto K, Zhu X, Park S, et al. Targeting MYC as a therapeutic intervention for anaplastic thyroid cancer. J Clin Endocrinol Metab. 2017;102(7):2268–2280.
- Loven J, Hoke HA, Lin CY, et al. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell. 2013;153(2):320–334.
- Issa JP, Kantarjian HM. Targeting DNA methylation. Clin Cancer Res. 2009;15(12):3938–3946.
- Lane AA, Chabner BA. Histone deacetylase inhibitors in cancer therapy. J Clin Oncol. 2009;27(32):5459–5468.
- He L, Gao L, Shay C, et al. Histone deacetylase inhibitors suppress aggressiveness of head and neck squamous cell carcinoma via histone acetylation-independent blockade of the EGFR-Arf1 axis. J Exp Clin Cancer Res. 2019;38(1):84.
- Cancer Genome Atlas N. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature. 2015;517(7536):576–582. .
- Hatakeyama H, Cheng H, Wirth P, et al. Regulation of heparin-binding EGF-like growth factor by miR-212 and acquired cetuximab-resistance in head and neck squamous cell carcinoma. PloS One. 2010;5(9):e12702.
- Guo T, Gaykalova DA, Considine M, et al. Characterization of functionally active gene fusions in human papillomavirus related oropharyngeal squamous cell carcinoma. Int J Cancer J Inter Du Cancer. 2016;139(2):373–382.
- Wang K, Singh D, Zeng Z, et al. MapSplice: accurate mapping of RNA-seq reads for splice junction discovery. Nucleic Acids Res. 2010;38(18):e178.
- Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12(1):323.
- Feng J, Liu T, Qin B, et al. Identifying ChIP-seq enrichment using MACS. Nat Protoc. 2012;7(9):1728–1740.
- Favorov A, Mularoni L, Cope LM, et al. Exploring massive, genome scale datasets with the GenometriCorr package. PLoS Comput Biol. 2012;8(5):e1002529.
- Ochs MF, Farrar JE, Considine M, et al. Outlier analysis and top scoring pair for integrated data analysis and biomarker discovery. IEEE/ACM Trans Comput Biol Bioinform. 2014;11(3):520–532.
- Mann HB, Whitney DR. On a test of whether one of 2 random variables is stochastically larger than the other. Ann Math Stat. 1947;18(1):50–60. .