
ABSTRACT
In eukaryotes, nucleosomes form a barrier to DNA templated reactions and must be dynamically disrupted to provide access to the genome. During nucleosome (re)assembly, histones can be replaced by new histones, erasing post-translational modifications. Measuring histone turnover in mammalian cells has mostly relied on inducible overexpression of histones, which may influence and distort natural histone deposition rates. We have previously used recombination-induced tag exchange (RITE) to study histone dynamics in budding yeast. RITE is a method to follow protein turnover by genetic switching of epitope tags using Cre recombinase and does not rely on inducible overexpression. Here, we applied RITE to study the dynamics of the replication-independent histone variant H3.3 in human cells. Epitope tag-switching could be readily detected upon induction of Cre-recombinase, enabling the monitoring old and new H3.3 in the same pool of cells. However, the rate of tag-switching was lower than in yeast cells. Analysis of histone H3.3 incorporation by chromatin immunoprecipitation did not recapitulate previously reported aspects of H3.3 dynamics such as high turnover rates in active promoters and enhancers. We hypothesize that asynchronous Cre-mediated DNA recombination in the cell population leads to a low time resolution of the H3.3-RITE system in human cells. We conclude that RITE enables the detection of old and new proteins in human cells and that the time-scale of tag-switching prevents the capture of high turnover events in a population of cells. Instead, RITE might be more suited for tracking long-lived histone proteins in human cells.
Introduction
Chromatin in eukaryotic cells is a repeated array of nucleosomes, which consist of DNA packaged around an octamer of histone proteins. Nucleosomes form barriers for DNA templated reactions such as replication, transcription and repair [Citation1–Citation4]. To allow access to the DNA, nucleosomes can be temporarily disrupted by the actions of histone chaperones and chromatin remodelling complexes [Citation5]. Conversely, nucleosomes are generally reassembled rapidly after disruption to safeguard chromatin integrity [Citation6–Citation9]. This reassembly process can involve either recycling of resident histones or the deposition of newly synthesized histones [Citation10].
During S-phase, the duplicated DNA daughter strands are packaged by newly synthesized histones. In mammalian cells, deposition of new histones during DNA replication involves canonical histones, whose expression peaks in S-phase [Citation11]. The canonical form of histone H3 is represented by two proteins, H3.1 and H3.2, which differ by only one amino acid and are both encoded by multiple genes [Citation12]. H3.1 an H3.2 are deposited at replication forks by the CAF1 (chromatin assembly factor 1) complex [Citation13]. CAF1 directly binds to PCNA [Citation14] and as a result canonical H3 deposition is strictly coupled to DNA synthesis [Citation15,Citation16]. In contrast, the histone variant H3.3, which is encoded by two genes in humans (H3F3A and H3F3B) is expressed throughout the cell cycle and can be deposited in both S-phase and interphase [Citation17–Citation21]. H3.3 differs four and five amino acids from H3.1 and H3.2, respectively, which enables specific chaperones such as HIRA and DAXX/ATRX to preferentially interact with H3.3 (in complex with histone H4) and assemble nucleosomes independently of DNA replication [Citation22–Citation24,Citation25].
H3.3 accumulates in non-dividing cells [Citation26,Citation27], which is consistent with both H3.3’s role as a replacement histone and the finding that nucleosomes are disrupted and reassembled using newly synthesized histones even in the absence of DNA replication [Citation28,Citation29,Citation30] Studies on genome-wide replication-independent histone dynamics in mammalian cells indicate that new H3.3 is rapidly incorporated at active genes, particularly at enhancers and promoters, and at regions of transcription termination [Citation32–Citation34]. Interestingly, despite the enrichment of H3.3 in transcribed regions, depletion of H3.3 or its chaperone HIRA leads to only mild gene expression changes in dividing cells [Citation35,Citation36]. In contrast, in non-dividing cells H3.3 is critical for gene regulation and preserving genome integrity as H3.3 prevents the accumulation of nucleosome-free DNA [Citation26,Citation37]. In addition, while H3.3 seems dispensable in dividing cells with a stable gene expression programme, H3.3 plays a prominent role in transcription regulation during processes in which expression programmes are altered, such as development, differentiation, reprogramming and stress responses [Citation36,Citation38–Citation46, reviewed by Citation47]. This indicates that histone turnover in interphase maintains an epigenetic landscape permissive for expression changes in order to facilitate shifts in cellular states.
It is currently unknown how histone replacement is involved in transcription regulation, but addressing this issue requires methods to distinguish between old and new histones [Citation48]. Most studies on genome-wide histone turnover have relied on inducible overexpression of epitope tagged histones [Citation33,Citation34], or conversely on inducible transcription shutdown of tagged histones [Citation49] using the Tet-on and Tet-off system, respectively. A drawback of these methods is that they rely on artificial expression regulation of histones, potentially leading to altered histone deposition and eviction rates. CATCH-IT (covalent attachment of tags to capture histones and identify exchange) is a complementary, metabolic pulse-labelling technique which does not rely on histone overexpression [Citation50]. CATCH-IT provides a genome-wide overview of nucleosome turnover but because labelling occurs in all proteins, no distinction can be made between different histone variants. Recently, another approach called time-ChIP was developed which combines SNAP-tagging of histones with biotin-based pull-down to map old existing histones [Citation51]. Importantly, these methods only allow for following either new or old histones but not both old and new histones simultaneously in the same pool of cells. Specifically, CATCH-IT cannot be used to track the fate of stable parental histone, while time-ChIP requires an extreme sequencing depth to track the absence of highly dynamic new histones.
Previously, we developed a method called recombination-induced tag exchange (RITE) to study histone dynamics in budding yeast [Citation52,Citation53]. In RITE, epitope tags that are integrated at an endogenous gene can be genetically swapped by conditional activation of Cre-mediated site-specific recombination. RITE can be used to endogenously tag histone genes, preserving natural expression regulation, and allows for following old and new histones simultaneously. Using this technique, it was found that replication-independent histone H3 turnover is linked to active transcription in budding yeast [Citation53] and promoted by the NuB4/HAT-B histone acetyltransferase complex [Citation54]. RITE has also been applied to link histone turnover to other epigenetic processes in budding yeast and to measure histone turnover in other organisms [Citation56–Citation64]. An advantage of RITE is that it can also be used to follow old histones, even over multiple cell divisions, as the tag switching is a permanent genetic event. In this way, it has been determined that in replicating yeast cells old H3 preferentially accumulates at the 5ʹ ends of long lowly-expressed genes [Citation63].
Here, we developed RITE in human cells and applied it to study the dynamics of the replication-independent histone variant H3.3. After conditional activation of Cre recombinase, both old and new histones could be monitored simultaneously by Western blot and chromatin immunoprecipitation (ChIP). However, low time resolution due to asynchronous Cre-mediated recombination in human cells limited the ability of RITE to resolve the dynamics of H3.3 in short time scales. Therefore, the concept of RITE can be used to measure protein dynamics in mammalian cells but in the current setup RITE is not suited for measuring the dynamics of proteins with fast turnover.
Methods
Cell culture and transfection
Human non-transformed retinal pigment epithelial cells transduced with the human telomerase gene (hTERT RPE1; designated RPE1 from here on) from ATCC (CRL-4000) were grown in DMEM/F12 (Gibco) supplemented with 10% foetal calf serum (FCS). K562 chronic myeloid cells from ATCC (CCL-243) were grown in RPMI (Gibco) supplemented with 10% FCS. Cells were maintained at 37°C, 5% CO2 in a humidified incubator.
The RITE system consists of two components: a RITE cassette that is integrated in-frame with an endogenous gene and an inducible Cre recombinase. For C-terminal tagging of H3.3 with a RITE cassette, a CRISPR-Cas9 plasmid (pX330, Cong et al. 2013[Citation64]; Addgene #42230) with guide RNA (gRNA) 5ʹ-CCCGAGTGGGATTAATAGTG targeting the last intron of H3F3B was co-transfected (1:1) with a repair plasmid for homologous directed repair (HDR). This intron targeting gRNA was chosen in order to prevent off-target cleavage at homologous histone H3 genes. The HDR plasmid consists of ~1.5 kb homology arms flanking the 3ʹ part of H3F3B with a RITE cassette placed in-frame with H3.3. The gRNA protospacer adjacent motif (PAM) located in the HDR plasmid was mutated (CGG > CCA) using the Quickchange II site-directed mutagenesis kit (Agilent) to prevent plasmid DNA cleavage.
For co-transfection into RPE1 cells, approximately one million cells were transfected with 3 µg plasmid DNA using FugeneHD reagent (Promega) using a DNA:FugeneHD ratio of 1:4. For K562, one million cells were nucleofected with 3 µg plasmid DNA using an Amaxa Nucleofector II device (Lonza) following manufacturers’ protocol. Selection with puromycin (10 μg/mL for RPE1, 2 µg/mL for K562; Invivogen) was started 72 h after transfection. Cells were selected with puromycin for one week.
A 4-hydrotamoxifen (4OHT) inducible version of Cre (Cre-ERT2) was introduced at the AAVS1 safe-harbour locus using CRISPR-Cas9 in polyclonal H3F3B-RITE RPE1 and K562 cells. Using identical transfection methods as described above, pX330 with gRNA 5ʹ-GGGGCCACTAGGGACAGGAT [Citation65] targeting the AAVS1 locus was co-transfected with an AAVS1 HDR plasmid (derived from AAVS1 hPGK-PuroR-pA donor, Hockemeyer et al. 2009 [Citation66]; Addgene #22072) containing a human PGK1 promoter driven Cre-ERT2-IRES-NeoR. Cells were selected with G418 (500 µg/mL for RPE1, 800 µg/mL for K562; Invivogen) for two weeks after which monoclonal cell lines were derived. Cre-ERT2 activity was induced by treating cells with 4OHT (100 nM; Sigma T176) dissolved in 100% ethanol.
Genomic DNA extraction and PCR
For genotyping H3F3B-RITE cell lines and determining DNA recombination efficiency, genomic DNA (gDNA) was isolated from approximately 2 × 106 cells using the ISOLATE II Genomic DNA Kit (BioLine) according to the manufacturer instructions. PCR was performed with 200 ng of gDNA and MyTaq polymerase (BioLine) according to the manufacturer’s protocol. Quantitative PCR of gDNA was done with FastStart SYBRgreen Master Mix (Roche) on a LightCycler 480 II (Roche) instrument. Primers for (q)PCR are listed in .
Table 1. Primers used in this study.
Western blot
Approximately 1 × 107 cells were washed twice with ice-cold phosphate-buffered saline (PBS). Proteins were isolated by adding SDS lysis buffer (50 mM Tris-HCl pH 6.8, 2% SDS, 10% glycerol) supplemented with protease inhibitor cocktail (PIC; Roche). DNA was sheared by sonication for 10 min at high settings (30 s on, 30 s off) using a Bioruptor Pico sonicator (Diagenode) to reduce sample viscosity. Protein concentration was determined with the DC protein assay (Bio-Rad) according to manufactures manual. An amount of 10 μg protein was separated on a 16% SDS-PAGE gel and blotted on 0.2 μm nitrocellulose membranes at 1 ampere for 1 h. Afterwards membranes were blocked for 1 h with 5% Nutrilon (Nutricia) in PBS and incubated overnight at 4°C with primary antibodies H3 (Abcam 1791, 1:2000), LoxP [Citation53, 1:2500], FLAG M2 (Sigma F3165, 1: 2000) or V5 (Invitrogen R960-25, 1:5000) in 2% Nutrilon in Tris-buffered saline-Tween (TBST). The next day membranes were washed four times with TBST before incubating the membrane with the appropriate Odyssey IRDye® 800CW secondary antibody (LI-COR Biosciences) at 1:10,000 dilution in 2% Nutrilon in TBST for 1 h. Membranes were washed four times with TBST before scanning on a LI-COR Odyssey IR Imager (LI-COR Biosciences).
Chromatin immunoprecipitation
Approximately 2 × 107 cells were crosslinked by adding formaldehyde to a final concentration of 1% and gently agitated at room temperature (RT) for 10 min. Glycine was added to a final concentration of 125 mM and cells were agitated for 5 min at RT. Cells were washed three times with ice cold PBS with Protease Inhibitor Cocktail (PIC; Roche). Cells were resuspended in 1 ml of Lysis Buffer 1 (50 mM Tris-HCl, 140 mM NaCl, 1 mM EDTA, 10% glycerol, 0.25% Triton X-100, final pH 7.5) with PIC and rotated for 10 minutes at 4°C. Nuclei were isolated by centrifugation at 2400 rpm for 4 min at 4°C and resuspended in 1 ml Lysis Buffer 2 (10 mM Tris-HCl, 200 mM NaCl, 1 mM EDTA, 0,5 mM EGTA, final pH 8) with PIC and rotated for 5 min at 4°C. Chromatin was isolated by centrifugation at 2400 rpm for 4 min at 4°C and resuspended in 300 μl Lysis Buffer 3 (50 mM Tris-HCl, 140 mM NaCl, 1 mM EDTA, 1% Triton X-100, final pH 8). Chromatin was sheared by sonicating for 10 cycles at high setting (30 s on and 30 s off) using a Bioruptor Pico sonicator (Diagenode) and centrifuged at 13,000 rpm for 10 min to pellet debris. Chromatin was pre-cleared with 40 μl Protein G Dynabeads (Invitrogen) for 1 h at 4°C. Next, chromatin protein concentration was determined with the DC protein assay (Bio-Rad) and 100 μg chromatin was incubated with 5 μg FLAG M2 (F1804; Sigma) or V5 antibody (R96025; Invitrogen) overnight at 4°C. Next, 80 μl Protein G Dynabeads was added and incubated for 4 h at 4°C. Thereafter the beads were washed once with Lysis buffer 3, twice with high salt buffer (50 mM Tris-HCl pH8, 500 mM NaCl, 1 mM EDTA pH8, 1% Triton X- 100, final pH 8), twice with LiCl wash buffer (50 mM Tris-HCl, 250 mM LiCl, 1 mM EDTA, 1% Triton X-100, final pH 8) and once with TE pH 8.0. Dynabeads were centrifuged and resuspended in 150 μl direct elution buffer (10 mM Tris-HCl, 0.3 M NaCl, 5 mM EDTA, 0.5% SDS, pH 8). IPs and inputs (10%) were incubated at 65°C overnight to reverse cross-links and incubated with 10 μg RNase A (10 mg/mL) and 30 μg Proteinase K for 1 h at 55°C. DNA was purified using a PCR purification kit (Qiagen) and was analysed by quantitative PCR using FastStart SYBRgreen Master Mix (Roche) on a LightCycler 480 II (Roche) instrument. Primers used for qPCR are listed in . The qPCR primers for PTGIS intron 1 and the Chr16 intergenic and enhancer region are from Pchelintsev et al. [Citation67]. The primers for GAPDH and MYC promoter and exon regions are from Carvalho et al. [Citation68].
ChIP- and RNA-sequencing
ChIP- and RNA-seq libraries were prepared using the KAPA LTP library preparation kit (KAPA Biosystems). ChIP-seq reads were aligned to the human genome (hg38) using Bowtie [Citation69]. For profiling H3.3 enrichment according to gene expression, genes were divided into three bins of equal size based on RPE1 gene expression. For this, RNA-seq reads were aligned to hg38 using HISAT [Citation70] and gene expression was measured by RPKM. For H3.3 ChIP-seq profiles, gene bodies were scaled to 6kb and H3.3 was plotted on gene bodies, 3kb upstream of the transcription start site (TSS), and 6kb downstream of the transcription end site (TES).
Results
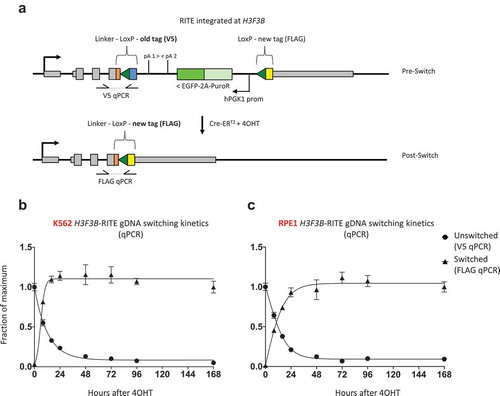
To measure replication-independent histone turnover, we used CRISPR-Cas9 to endogenously tag one of the two H3.3 encoding genes (H3F3B) with a C-terminal RITE-cassette in human chronic myelogenous leukaemia K562 cells and retinal pigment epithelial RPE1 cells (). The RITE-cassette consists of a V5 epitope tag flanked by two tandem LoxP sites with a downstream orphan FLAG tag. Old (V5) and new (FLAG) H3.3 are under the control of the same endogenous promoter but have a different 3ʹUTR; old H3.3 has an ectopic 3ʹUTR while new H3.3 has the endogenous H3F3B 3ʹUTR. In addition, to enable tag switching by DNA recombination, we integrated a 4-hydroxytamoxifen (4OHT)-inducible Cre-ERT2 recombinase at the AAVS1 safe-harbour locus using CRSIPR-Cas9. In this way, exposing cells to 4OHT induces DNA recombination at H3F3B leading to the permanent exchange of a V5-tag on H3.3 for a FLAG-tag, allowing for simultaneous measurement of old and new H3.3 ().
Figure 1. Strategy to knock-in a RITE cassette at H3F3B and Cre-ERT2 at the AAVS1 safe-harbour locus. (a) A RITE cassette (V5→FLAG) is integrated at the C-terminus of H3.3 (H3F3B gene) using CRISPR-Cas9 and a repair plasmid for homologous recombination. Note that the gRNA is targeted to the last intron of H3F3B to prevent off-target cleavage at homologous histone H3 genes. (b) Genotyping PCR to confirm heterozygous integration of the RITE cassette at H3F3B in RPE1 and K562 cells. The asterisk denotes non-specific PCR amplicons. (c) A PGK1-promoter driven Cre-ERT2 is integrated at the AAVS1 safe-harbour locus using a HDR (homology-directed repair) plasmid donor derived from Mali et al. [Citation65]. (d) Genotyping PCR to confirm homozygous integration of Cre-ERT2 is at AAVS1 in RPE1 and K562 cells. The asterisk denotes non-specific PCR amplicons. HA, homology arm; PuroR-2A-EGFP, puromycin resistance gene followed by self-cleaving peptide P2A from porcine teschovirus-1 polyprotein and green fluorescent protein; IRES, internal ribosome entry site; NeoR, neomycin resistance gene.
![Figure 1. Strategy to knock-in a RITE cassette at H3F3B and Cre-ERT2 at the AAVS1 safe-harbour locus. (a) A RITE cassette (V5→FLAG) is integrated at the C-terminus of H3.3 (H3F3B gene) using CRISPR-Cas9 and a repair plasmid for homologous recombination. Note that the gRNA is targeted to the last intron of H3F3B to prevent off-target cleavage at homologous histone H3 genes. (b) Genotyping PCR to confirm heterozygous integration of the RITE cassette at H3F3B in RPE1 and K562 cells. The asterisk denotes non-specific PCR amplicons. (c) A PGK1-promoter driven Cre-ERT2 is integrated at the AAVS1 safe-harbour locus using a HDR (homology-directed repair) plasmid donor derived from Mali et al. [Citation65]. (d) Genotyping PCR to confirm homozygous integration of Cre-ERT2 is at AAVS1 in RPE1 and K562 cells. The asterisk denotes non-specific PCR amplicons. HA, homology arm; PuroR-2A-EGFP, puromycin resistance gene followed by self-cleaving peptide P2A from porcine teschovirus-1 polyprotein and green fluorescent protein; IRES, internal ribosome entry site; NeoR, neomycin resistance gene.](/cms/asset/0b999c1e-c6a5-4987-b0a6-74f71ddd491e/kepi_a_1741777_f0001_oc.jpg)
Figure 2. RITE allows for simultaneous monitoring of old and new H3.3 histones in human cells. Western blot of old (V5) and new H3.3 (FLAG) after Cre-ERT2 activation by 4OHT in (a) K562 and (b) RPE1 cells. An antibody against the LoxP-encoded spacer peptide recognizes both old and new H3.3. Note that the antibody directed against the C-terminus of H3 does not seem to recognize C-terminally tagged H3.3.
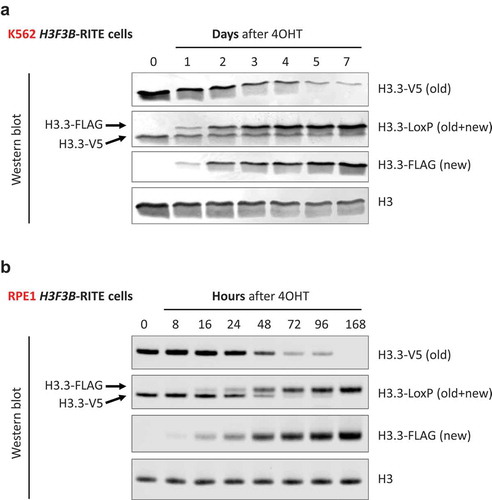
To determine the kinetics of DNA recombination and H3.3 turnover in asynchronous H3F3B-RITE cells, we harvested genomic DNA at several time points after 4OHT exposure. Quantitative PCR indicated that DNA recombination took about 24 h to occur in most of the cells (). In the absence of 4OHT, recombination rates were 1.3% and 3.3% for RPE1 and K562, respectively, confirming the dependence of Cre-ERT2 activity on 4OHT. Global incorporation of new FLAG-tagged H3.3 lagged only a few hours behind DNA recombination suggesting that H3.3 is highly dynamic in asynchronous cells.
Figure 3. Efficiency of Cre-ERT2 mediated H3.3 tag-switching at the DNA level. (a) Overview of H3F3B-RITE locus before and after Cre-LoxP recombination (tag-switching). (b and c) qPCR on genomic DNA to determine tag-switching efficiency in (b) K562 and (c) RPE1 cells. Unswitched and switched qPCR signals were normalized to a control region (MYC exon 3) to normalize for input gDNA. For the unswitched fraction, the qPCR signal at 0 h was set to 1, while for the switched fraction the qPCR signal at 168 h was set to 1, and a sigmoidal curve was fitted to the data points.
H3.3 has been reported to be highly dynamic in active promoter and enhancer regions but relatively stable in heterochromatic regions in non-dividing cells [Citation32–Citation34]. To look at locus-specific H3.3 turnover with the RITE assay, we performed ChIP-qPCR using V5 and FLAG specific antibodies at several time points following 4OHT exposure in K562 cells ()). We examined old H3.3 (V5) and new H3.3 (FLAG) at active genes (GAPDH, MYC, CCNB1 and REEP5), inactive genes (KRT6A and PTGIS), and intergenic loci, including a putative active enhancer and a pericentric heterochromatin (PCH) region ()). As a control, we also examined H3.3-V5 and H3.3-FLAG levels in H3F3B-RITE K562 cells cultured for 7 days after 4OHT exposure, in which H3.3-V5 was been nearly completely replaced by H3.3-FLAG as determined by Western blot (). In the absence of 4OHT, a low level of H3.3-FLAG was already detectable by ChIP-qPCR possibly due to background recombination (–C)) or non-specific immunoprecipitation ()). After 4OHT treatment, H3.3-FLAG accumulated at all loci while H3.3-V5 levels decreased, indicating histone replacement. H3.3 turnover rates [measured by the turnover index as described by Citation51] were comparable between the different loci ()) indicating we could not recapitulate previously reported differences in H3.3 turnover at active promoters and gene bodies/intergenic regions.
Figure 4. Loss of old H3.3 and gain of new H3.3 can be detected by ChIP-qPCR at different loci. (a) Experimental overview. Switching of the H3F3B-RITE cassette (H3.3-V5 to H3.3-FLAG) was induced in K562 cells by treatment with 4OHT and chromatin was harvested at different time points. (b) Overview of the loci analysed by ChIP-qPCR. Chr7 PCH stands for a pericentric heterochromatin locus on chromosome 7. Features are based on ENCODE data for K562. (c) ChIP-qPCR (IP/input) for old (V5) and new H3.3 (FLAG) at selected loci in K562 cells. (d) Turnover index [Citation51] at selected loci. A higher index means more turnover.
![Figure 4. Loss of old H3.3 and gain of new H3.3 can be detected by ChIP-qPCR at different loci. (a) Experimental overview. Switching of the H3F3B-RITE cassette (H3.3-V5 to H3.3-FLAG) was induced in K562 cells by treatment with 4OHT and chromatin was harvested at different time points. (b) Overview of the loci analysed by ChIP-qPCR. Chr7 PCH stands for a pericentric heterochromatin locus on chromosome 7. Features are based on ENCODE data for K562. (c) ChIP-qPCR (IP/input) for old (V5) and new H3.3 (FLAG) at selected loci in K562 cells. (d) Turnover index [Citation51] at selected loci. A higher index means more turnover.](/cms/asset/36d7664f-0deb-4f09-b6a4-5a4d0d8fe258/kepi_a_1741777_f0004_c.jpg)
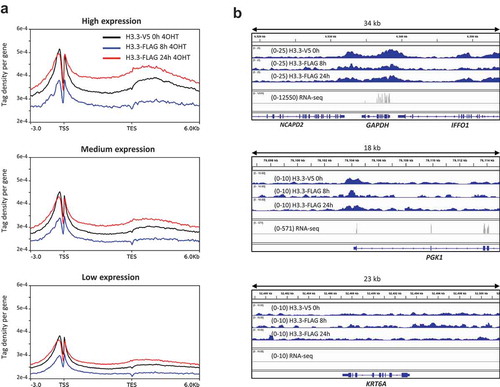
As an alternative way to measure differential H3.3 turnover at active versus inactive genes using RITE, we assessed new H3.3-FLAG levels 8 h and 24 h after 4OHT exposure at a genome-wide level using ChIP-seq in RPE1 cells. Steady-state H3.3-V5 (old H3.3 before the switch) was enriched around the transcription start sites of active genes as previously reported (,b)). At both 8 h and 24 h, new H3.3-FLAG resembled steady-state H3.3 in terms of enrichment pattern. In agreement with the ChIP-qPCR results in K562 cells (), we did not observe the strong enrichment of new H3.3 in the promoters of active genes () reported by others [Citation32–Citation34].
Figure 5. ChIP-seq of old and new H3.3 in RPE1 cells. (a) Distribution profiles of steady-state (old) H3.3-V5, and new H3.3-FLAG 8 h and 24 h after switch induction by 4OHT. Gene bodies are scaled to 6kb. (b) Examples of old and new H3.3 enrichment in a highly expressed gene (GAPDH), a mediumly expressed gene (PGK1) and a silent gene (KRT6A).
Discussion
RITE is a versatile method to monitor protein replacement that has been applied to histones [Citation53–Citation55, Citation57,Citation59–Citation63,Citation31] Ramakrishnan et al., 2016 and non-histone proteins [Citation58,Citation71–Citation73] Menendez-Benito et al. 2012 [Citation74]; in budding and fission yeast. RITE is compatible with pull-down strategies, which enables genome-wide mapping or determining modifications of old and new histones by mass-spectrometry [Citation56]. In addition, RITE can be used in combination with fluorescence microscopy to track the localization, stability, and inheritance of old and new proteins [Citation72–Citation76]. Here, we developed RITE in human cells and applied it to examine replication-independent histone dynamics in human cells.
Using CRISPR-Cas9, we tagged one of the endogenous histone H3.3 genes in RPE1 and K562 cells with a RITE cassette that was able to undergo controlled Cre-mediated DNA recombination. RITE thus enables differential tagging and monitoring of old and new H3.3 in human cells. A drawback of RITE, which relies on expression from the endogenous histone promoter, is that it depends on de novo synthesis of differentially epitope-tagged histones after Cre-mediated recombination. The time resolution of RITE is therefore limited by both the efficiency of Cre recombinase and the time it takes to synthesize new histone proteins. Using RPE1 and K562 cells in which a PGK1 promoter driven Cre-ERT2 gene was integrated at the AAVS1 safe-harbour locus, we found that recombination at H3F3B took about 24 h to occur in most of the cell population after exposure to 4OHT. Because of this unsynchronized tag switching and the lag in new protein synthesis, time resolution is lower in RITE than in CATCH-IT and time-ChIP. This low time resolution limits the ability of RITE to track the dynamics of proteins with high turnover rates. When we examined H3.3 turnover by RITE we observed little difference between regions reported to have high turnover versus those that have low turnover of H3.3. Replication-independent H3.3 turnover differs throughout the genome but H3.3 has fast turnover rates at active enhancers and promoters [Citation32–Citation34,Citation51]. We hypothesize that we were unable to measure differences in H3.3 turnover rates at active and inactive genomic loci due to the limited time resolution of RITE when performing bulk analysis in human cells.
The low time resolution of RITE in human cells precludes its use in tracking highly dynamic histones. Instead, RITE might be more suited for tracking long-lived histone proteins in human cells. In addition, selection markers such as fluorescent proteins or cell sorting compatible surface markers could be used to remove unswitched cells from the population before analysis. RITE is particularly suited for tracking long-lived histones as the tag switch is permanent. In addition, in the current setup RITE might be more compatible with cell-by-cell analysis compared to bulk cell analysis. For example, a similar strategy to RITE has been applied in Drosophila male germ line stem cells to track the fate of tagged histones using live-cell fluorescent microscopy [Citation77]. In addition, RITE was recently used to study the localization of long-lived histones and other proteins in mammalian cells using immunofluorescence microscopy [Citation76]. Thus, cell-by-cell analysis of global histone levels can be used to circumvent inefficient Cre-recombination in RITE in mammalian cells.
RITE is an attractive method to study histone dynamics because it can be used to simultaneously detect and pull-down old and new histones. RITE has been successfully used to study histone dynamics in budding and fission yeast. DNA recombination is presumably more efficient in these organisms due to a smaller genome size. In human cells, a method that relies on tag-switching on the protein level rather than the DNA level might therefore be more suitable for tracking old and new proteins at the same time. Self-labelling tags such as Halo and SNAP tags [e.g. time-ChIP; Citation51] can be used for pulse-chase pull-down of proteins but have only been used so far to track old histones. Tracking both old and new histones will require the development of new orthogonal labelling ligands suitable for pull-down. Alternatively, inhibitable self-cleaving degrons [i.e. small molecule assisted shutoff or SMASh-tag; Citation78] could be useful for chasing old and new histones in parallel. Specifically, it will be interesting to determine if recently described orthogonal SMASh-tags [called stabilizable polypeptide linkers or StaPLs; Citation79] could be applied to studying histone dynamics. For example, by tagging two alleles of H3F3B with orthogonal StaPLs and epitope tags, it might be possible to distinguish old H3.3 (degron inhibited before switch and activated after switch) from new H3.3 (degron activated before switch and inhibited after switch). Similar to RITE, both self-labelling and degron type tags can be engineered at endogenous loci, avoiding the need for inducible histone overexpression.
Author Contributions
TMM and FvL: Conception and design of the studies, drafting and revising the article; TMM, MP-G, and VM: Acquisition, analysis and interpretation of data.
Data access
All raw and processed sequencing data generated in this study have been submitted to the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) under accession numbers GSE146120 (ChIP-seq) and GSE146121 (RNA-seq).
Acknowledgments
The authors thank Tessy Korthout, Deepani Poramba, Ila van Kruijsbergen and other lab members for insightful discussions and valuable suggestions.
Disclosure Statement
The authors declare that no competing interests exist.
Additional information
Funding
References
- Bondarenko VA, Steele LM, Újvári A, et al. Nucleosomes can form a polar barrier to transcript elongation by RNA polymerase II. Mol Cell. 2006;24:469–479.
- de Jong BE, van Noort J. Overcoming chromatin barriers. Elife. 2019;8. DOI:10.7554/eLife.50761
- Li B, Carey M, Workman JL. The role of chromatin during transcription. Cell. 2007;128:707–719.
- Weber CM, Ramachandran S, Henikoff S. Nucleosomes are context-specific, H2A.Z-modulated barriers to RNA polymerase. Mol Cell. 2014;53:819–830.
- Clapier CR, Cairns BR. The biology of chromatin remodeling complexes. Annu Rev Biochem. 2009;78:273–304.
- Cramer P. Organization and regulation of gene transcription. Nature. 2019;573:45–54.
- Smolle M, Workman JL, Venkatesh S. reSETting chromatin during transcription elongation. Epigenetics. 2013;8:10–15.
- Smolle M, Venkatesh S, Gogol MM, et al. Chromatin remodelers Isw1 and Chd1 maintain chromatin structure during transcription by preventing histone exchange. Nat Struct Mol Biol. 2012;19:884–892.
- Venkatesh S, Smolle M, Li H, et al. Set2 methylation of histone H3 lysine 36 suppresses histone exchange on transcribed genes. Nature. 2012;489:452–455.
- Terweij M, van Leeuwen F. Histone exchange: sculpting the epigenome. Front Life Sci. 2013;7:63–79.
- Mei Q, Huang J, Chen W, et al. Regulation of DNA replication-coupled histone gene expression. Oncotarget. 2017;8. DOI:10.18632/oncotarget.21887
- Hamiche A, Shuaib M. Chaperoning the histone H3 family. Biochim Biophys Acta Gene Regul Mech. 2012;1819(3–4):230–237.
- Sauer PV, Gu Y, Liu WH, et al. Mechanistic insights into histone deposition and nucleosome assembly by the chromatin assembly factor-1. Nucleic Acids Res. 2018;46:9907–9917.
- Shibahara K-I, Stillman B. Replication-dependent marking of DNA by PCNA facilitates CAF-1-coupled inheritance of chromatin. Cell. 1999;96:575–585.
- Kimura H, Cook PR. Kinetics of core histones in living human cells. J Cell Biol. 2001;153:1341–1354.
- Thiriet C, Hayes JJ. Histone dynamics during transcription: exchange of H2a/H2b dimers and H3/H4 tetramers during pol II elongation. Results Probl Cell Differ. 2006;41:77–90.
- Ahmad K, Henikoff S. The histone variant H3.3 marks active chromatin by replication-independent nucleosome assembly. Mol Cell. 2002;9:1191–1200.
- Ray-Gallet D, Woolfe A, Vassias I, et al. Dynamics of histone H3 deposition in vivo reveal a nucleosome gap-filling mechanism for H3.3 to maintain chromatin integrity. Mol Cell. 2011;44:928–941.
- Schneiderman JI, Orsi GA, Hughes KT, et al. Nucleosome-depleted chromatin gaps recruit assembly factors for the H3.3 histone variant. Proc Nat Acad Sci. 2012;109:19721–19726.
- Schwartz BE. Transcriptional activation triggers deposition and removal of the histone variant H3.3. Genes Dev. 2005;19:804–814.
- Tagami H, Ray-Gallet D, Almouzni G, et al. Histone H3.1 and H3.3 complexes mediate nucleosome assembly pathways dependent or independent of DNA synthesis. Cell. 2004;116:51–61.
- Goldberg AD, Banaszynski LA, Noh K-M, et al. Distinct factors control histone variant H3.3 localization at specific genomic regions. Cell. 2010;140:678–691.
- Lewis PW, Elsaesser SJ, Noh K-M, et al. Daxx is an H3.3-specific histone chaperone and cooperates with ATRX in replication-independent chromatin assembly at telomeres. Proc Nat Acad Sci. 2010;107:14075–14080.
- Ray-Gallet D, Quivy J-P, Scamps C, et al. HIRA is critical for a nucleosome assembly pathway independent of DNA synthesis. Mol Cell. 2002;9:1091–1100.
- Drane P, Ouararhni K, Depaux A, et al. The death-associated protein DAXX is a novel histone chaperone involved in the replication-independent deposition of H3.3. Genes Dev. 2010;24:1253–1265.
- Maze I, Wenderski W, Noh K-M, et al. Critical role of histone turnover in neuronal transcription and plasticity. Neuron. 2015;87:77–94.
- Tvardovskiy A, Schwämmle V, Kempf SJ, et al. Accumulation of histone variant H3.3 with age is associated with profound changes in the histone methylation landscape. Nucleic Acids Res. 2017;45:9272–9289.
- Dion MF, Kaplan T, Kim M, et al. Dynamics of replication-independent histone turnover in budding yeast. Science. 2007;315:1405–1408.
- Jamai A, Imoberdorf RM, Strubin M. Continuous histone H2B and transcription-dependent histone H3 exchange in yeast cells outside of replication. Mol Cell. 2007;25:345–355.
- Rufiange A, Jacques P-É, Bhat W, et al. Genome-wide replication-independent histone H3 exchange occurs predominantly at promoters and implicates H3K56 acetylation and Asf1. Mol Cell. 2007;27:393–405.
- Radman-Livaja M, Verzijlbergen KF, Weiner A, et al. Patterns and mechanisms of ancestral histone protein inheritance in budding yeast. PLoS Biol. 2011;9:e1001075.
- Huang C, Zhang Z, Xu M, et al. H3.3-H4 Tetramer splitting events feature cell-type specific enhancers. PLoS Genet. 2013;9(6):e1003558.
- Kraushaar DC, Jin W, Maunakea A, et al. Genome-wide incorporation dynamics reveal distinct categories of turnover for the histone variant H3.3. Genome Biol. 2013;14:R121.
- Yildirim O, Hung J-H, Cedeno RJ, et al. A system for genome-wide histone variant dynamics in ES cells reveals dynamic MacroH2A2 replacement at promoters. PLoS Genet. 2014;10:e1004515.
- Hödl M, Basler K. Transcription in the absence of histone H3.3. Curr Biol. 2009;19:1221–1226.
- Jang C-W, Shibata Y, Starmer J, et al. Histone H3.3 maintains genome integrity during mammalian development. Genes Dev. 2015;29:1377–1392.
- Nashun B, Hill PW, Smallwood SA, et al. Continuous histone replacement by Hira is essential for normal transcriptional regulation and de novo DNA methylation during mouse oogenesis. Mol Cell. 2015;60:611–625.
- Fang H-T, Farran CAE, Xing QR, et al. Global H3.3 dynamic deposition defines its bimodal role in cell fate transition. Nat Commun. 2018;9. DOI:10.1038/s41467-018-03904-7
- Kong Q, Banaszynski LA, Geng F, et al. Histone variant H3.3–mediated chromatin remodeling is essential for paternal genome activation in mouse preimplantation embryos. J Biol Chem. 2018;293:3829–3838.
- Delaney K, Mailler J, Wenda JM, et al. Differential expression of histone H3.3 genes and their role in modulating temperature stress response in Caenorhabditis elegans. Genetics. 2018;209:551–565.
- Banaszynski LA, Wen D, Dewell S, et al. Hira-dependent histone H3.3 deposition facilitates PRC2 recruitment at developmental loci in ES cells. Cell. 2013;155:107–120.
- Harada A, Okada S, Konno D, et al. Chd2 interacts with H3.3 to determine myogenic cell fate. Embo J. 2012;31:2994–3007.
- Harada A, Maehara K, Sato Y, et al. Incorporation of histone H3.1 suppresses the lineage potential of skeletal muscle. Nucleic Acids Res. 2014;43:775–786.
- Yuen BTK, Bush KM, Barrilleaux BL, et al. Histone H3.3 regulates dynamic chromatin states during spermatogenesis. Development. 2014;141:3483–3494.
- Jullien J, Astrand C, Szenker E, et al. HIRA dependent H3.3 deposition is required for transcriptional reprogramming following nuclear transfer to Xenopus oocytes. Epigenetics Chromatin. 2012;5. DOI:10.1186/1756-8935-5-17
- Tamura T, Smith M, Kanno T, et al. Inducible deposition of the Histone variant H3.3 in interferon-stimulated genes. J Biol Chem. 2009;284:12217–12225.
- Buschbeck M, Hake SB. Variants of core histones and their roles in cell fate decisions, development and cancer. Nat Rev Mol Cell Biol. 2017;18:299–314.
- Deal RB, Henikoff S. Capturing the dynamic epigenome. Genome Biol. 2010;11:218.
- Ha M, Kraushaar DC, Zhao K. Genome-wide analysis of H3.3 dissociation reveals high nucleosome turnover at distal regulatory regions of embryonic stem cells. Epigenetics Chromatin. 2014;7(1). DOI:10.1186/1756-8935-7-38
- Deal RB, Henikoff JG, Henikoff S. Genome-wide kinetics of nucleosome turnover determined by metabolic labeling of histones. Science. 2010;328:1161–1164.
- Deaton AM, Mariluz G-R, Mieczkowski J, et al. Enhancer regions show high histone H3.3 turnover that changes during differentiation. eLife. 2016;5. DOI:10.7554/eLife.15316
- Terweij M, van Welsem T, van Deventer S, et al. Recombination-Induced Tag Exchange (RITE) cassette series to monitor protein dynamics in saccharomyces cerevisiae. G3. 2013;3:1261–1272.
- Verzijlbergen KF, Menendez-Benito V, van Welsem T, et al. Recombination-induced tag exchange to track old and new proteins. Proc Nat Acad Sci. 2010;107:64–68.
- Verzijlbergen KF, van Welsem T, Sie D, et al. A barcode screen for epigenetic regulators reveals a role for the NuB4/HAT-B histone acetyltransferase complex in histone turnover. PLoS Genet. 2011;7:e1002284.
- Audergon PNCB, Catania S, Kagansky A, et al. Restricted epigenetic inheritance of H3K9 methylation. Science. 2015;348:132–135.
- De Vos D, Frederiks F, Terweij M, et al. Progressive methylation of ageing histones by Dot1 functions as a timer. EMBO Rep. 2011;12:956–962.
- Gan H, Serra-Cardona A, Hua X, et al. The Mcm2-Ctf4-Polα axis facilitates parental histone H3-H4 transfer to lagging strands. Mol Cell. 2018;72. DOI:10.1016/j.molcel.2018.09.001
- Hughes AL, Hughes CE, Henderson KA, et al. Selective sorting and destruction of mitochondrial membrane proteins in aged yeast. eLife. 2016;5. DOI:10.7554/eLife.13943
- Sadeghi L, Prasad P, Ekwall K, et al. The Paf1 complex factors Leo1 and Paf1 promote local histone turnover to modulate chromatin states in fission yeast. EMBO Rep. 2015;16:1673–1687.
- Svensson JP, Shukla M, Menendez-Benito V, et al. A nucleosome turnover map reveals that the stability of histone H4 Lys20 methylation depends on histone recycling in transcribed chromatin. Genome Res. 2015;25:872–883.
- Yu C, Gan H, Serra-Cardona A, et al. A mechanism for preventing asymmetric histone segregation onto replicating DNA strands. Science. 2018;361:1386–1389.
- Ramakrishnan S, Pokhrel S, Palani S, et al. Counteracting h3k4 methylation modulators set1 and jhd2 co-regulate chromatin dynamics and gene transcription. Nat Commun. 2016;7.
- Radman-Livaja M, Quan TK, Valenzuela L, et al. A key role for Chd1 in histone H3 dynamics at the 3′ ends of long genes in yeast. PLoS Genet. 2012;8:e1002811.
- Cong L, Ran FA, Cox D, et al. Multiplex genome engineering using crispr/cas systems. Science. 2013;15:819-823.
- Mali P, Yang L, Esvelt KM, et al. RNA-guided human genome engineering via Cas9. Science. 2013;339:823–826.
- Hockemeyer D, Soldner F, Beard C, et al. Efficient targeting of expressed and silent genes in human escs and ipscs using zinc-finger nucleases. Nat Biotechnol. 2009;27:851-857.
- Pchelintsev NA, Mcbryan T, Rai TS, et al. Placing the HIRA histone chaperone complex in the chromatin landscape. Cell Rep. 2013;3:1012–1019.
- Carvalho S, Raposo AC, Martins FB, et al. Histone methyltransferase SETD2 coordinates FACT recruitment with nucleosome dynamics during transcription. Nucleic Acids Res. 2013;41(5):2881–2893.
- Langmead B, Trapnell C, Pop M, et al. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25.
- Kim D, Langmead B, Salzberg SL. HISAT: a fast spliced aligner with low memory requirements. Nat Methods. 2015;12:357–360.
- Galic H, Vasseur P, Radman-Livaja M. The budding yeast heterochromatic SIR complex resets upon exit from stationary phase. bioRxiv. 2019.
- Thayer NH, Leverich CK, Fitzgibbon MP, et al. Identification of long-lived proteins retained in cells undergoing repeated asymmetric divisions. Proc Nat Acad Sci. 2014;111:14019–14026.
- van Deventer S, Menendez-Benito V, van Leeuwen F, et al. N-terminal acetylation and replicative age affect proteasome localization and cell fitness during aging. J Cell Sci. 2014;128:109–117.
- Menendez-Benito V, van Deventer S, Jimenez-Garcia V, et al. Spatiotemporal analysis of organelle and macromolecular complex inheritance. Proc Nat Acad Sci. 2012;110:175–180.
- Lengefeld J, Yen E, Chen X, et al. Spatial cues and not spindle pole maturation drive the asymmetry of astral microtubules between new and preexisting spindle poles. Mol Biol Cell. 2018;29:10–28.
- Toyama BH, Drigo RAE, Lev-Ram V, et al. Visualization of long-lived proteins reveals age mosaicism within nuclei of postmitotic cells. J Cell Biol. 2018;218:433–444.
- Tran V, Lim C, Xie J, et al. Asymmetric division of Drosophila male germline stem cell shows asymmetric histone distribution. Science. 2012;338:679–682.
- Chung HK, Jacobs CL, Huo Y, et al. Tunable and reversible drug control of protein production via a self-excising degron. Nat Chem Biol. 2015;11:713–720.
- Jacobs CL, Badiee RK, Lin MZ. StaPLs: versatile genetically encoded modules for engineering drug-inducible proteins. Nat Methods. 2018;15:523–526.